
MicroRNAs in Atrial Fibrillation: Mechanisms, Vascular Implications, and Therapeutic Potential
 , ,
, ,  ,
, 
Abstract
:1. Introduction
2. MicroRNA-Mediated Fibrotic Mechanisms in Atrial Fibrillation
- Disruption of atrial conduction: Fibrosis can modify the electrical and mechanical properties of the atrial tissue, contributing to the initiation and maintenance of the arrhythmia. One of the key mechanisms through which fibrosis exerts its effect is by disrupting atrial conduction. Fibrosis can create regions of slowed conduction, a conduction block, and conduction heterogeneity in the atrial tissue, which promotes the formation of re-entrant circuits that sustain AFib. The underlying mechanism for this effect is thought to be related to the increased interstitial resistance and reduced intercellular coupling caused by fibrosis. In fibrotic tissue, the gap junctions that allow for rapid cell-to-cell communication become disrupted, leading to a decrease in the velocity and uniformity of the electrical signal propagation. This, in turn, can create areas of slow or blocked conduction that serve as the substrate for the formation of re-entrant circuits, which sustain AFib [3].
- Enhancement of automaticity: Fibrosis can facilitate the emergence of ectopic foci in the atrial tissue by modifying the electrophysiological characteristics of the cardiomyocytes. In particular, fibrosis can induce alterations in the ion channels and calcium handling proteins that govern the generation and propagation of action potentials in the atrial myocytes. These modifications can enhance the automaticity of the cardiomyocytes, rendering them more susceptible to generating spontaneous impulses that give rise to ectopic foci capable of triggering Afib [4].
- Alteration of repolarization: Fibrosis can modify the repolarization properties of the atrial myocytes, resulting in an elongation of the action potential duration and a heterogeneity of repolarization across the atrial tissue. These changes can produce a substrate that promotes the development of arrhythmias by facilitating the formation of re-entrant circuits and ectopic foci. Moreover, fibrosis can increase the susceptibility of the atrial tissue to the effects of triggers, such as adrenergic stimulation or rapid pacing, by altering the electrophysiological properties of the cells. Consequently, these triggers can more readily cause disruptions in the conduction of electrical impulses across the atrial tissue, potentially leading to the onset or perpetuation of Afib [5].
- Mechanical stiffness and stretch-induced remodeling: Fibrosis can modify the mechanical properties of the atrial tissue, reducing its compliance and increasing its susceptibility to stretch-induced remodeling. This can predispose the tissue to atrial dilatation and hypertension, both of which are established risk factors for atrial fibrillation. Additionally, fibrosis can instigate a pro-inflammatory and pro-fibrotic milieu within the atrial tissue, amplifying the fibrotic process and fueling the development of Afib [6].
3. MicroRNAs in Atrial Fibrillation’s Inflammatory Pathways
4. Oxidative Stress and MicroRNA Interplay in Atrial Fibrillation
5. MicroRNAs and Their Vascular Implications in Atrial Fibrillation
6. MicroRNAs as Emerging Biomarkers and Therapeutic Facilitators of Atrial Fibrillation; Future Perspectives
6.1. miRNAs as Potential Biomarkers of Atrial Fibrillation
6.2. miRNAs as Biomarkers of the Risk of Major Adverse Thrombotic Cardiovascular Events in Atrial Fibrillation
6.3. miRNAs as Potential Therapeutic Agents in Atrial Fibrillation Management
7. Conclusions
Funding
Conflicts of Interest
References
- Di Carlo, A.; Bellino, L.; Consoli, D.; Mori, F.; Zaninelli, A.; Baldereschi, M.; Cattarinussi, A.; D’Alfonso, M.G.; Gradia, C.; Sgherzi, B.; et al. Prevalence of atrial fibrillation in the Italian elderly population and projections from 2020 to 2060 for Italy and the European Union: The FAI Project. Europace 2019, 21, 1468–1475. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S. How does fibrosis promote atrial fibrillation persistence: In silico findings, clinical observations, and experimental data. Cardiovasc. Res. 2016, 110, 295–297. [Google Scholar] [CrossRef] [PubMed]
- Allessie, M.A.; de Groot, N.M.; Houben, R.P.; Schotten, U.; Boersma, E.; Smeets, J.L.; Crijns, H.J. Electropathological substrate of long-standing persistent atrial fibrillation in patients with structural heart disease: Longitudinal dissociation. Circ. Arrhythmia Electrophysiol. 2010, 3, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Platonov, P.G. Atrial fibrosis: An obligatory component of arrhythmia mechanisms in atrial fibrillation? J. Geriatr. Cardiol. 2017, 14, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Quah, J.X.; Dharmaprani, D.; Tiver, K.; Lahiri, A.; Hecker, T.; Perry, R.; Selvanayagam, J.B.; Joseph, M.X.; McGavigan, A.; Ganesan, A. Atrial fibrosis and substrate based characterization in atrial fibrillation: Time to move forwards. J. Cardiovasc. Electrophysiol. 2021, 32, 1147–1160. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Garcia-Elias, A.; Benito, B.; Nattel, S. The effects of cardiac stretch on atrial fibroblasts: Analysis of the evidence and potential role in atrial fibrillation. Cardiovasc. Res. 2022, 118, 440–460. [Google Scholar] [CrossRef]
- Ye, Q.; Liu, Q.; Ma, X.; Bai, S.; Chen, P.; Zhao, Y.; Bai, C.; Liu, Y.; Liu, K.; Xin, M.; et al. MicroRNA-146b-5p promotes atrial fibrosis in atrial fibrillation by repressing TIMP4. J. Cell. Mol. Med. 2021, 25, 10543–10553. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Yan, S.; Zhao, X.; Han, X.; Fang, N.; Zhang, Y.; Dai, C.; Li, W.; Yu, H.; Gao, Y.; et al. Atrial myocyte-derived exosomal microRNA contributes to atrial fibrosis in atrial fibrillation. J. Transl. Med. 2022, 20, 407. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S. Molecular and Cellular Mechanisms of Atrial Fibrosis in Atrial Fibrillation. JACC Clin. Electrophysiol. 2017, 3, 425–435. [Google Scholar] [CrossRef]
- Lai, Y.J.; Tsai, F.C.; Chang, G.J.; Chang, S.H.; Huang, C.C.; Chen, W.J.; Yeh, Y.H. miR-181b targets semaphorin 3A to mediate TGF-β-induced endothelial-mesenchymal transition related to atrial fibrillation. J. Clin. Invest. 2022, 132, e142548. [Google Scholar] [CrossRef]
- Cao, F.; Li, Z.; Ding, W.M.; Yan, L.; Zhao, Q.Y. LncRNA PVT1 regulates atrial fibrosis via miR-128-3p-SP1-TGF-β1-Smad axis in atrial fibrillation. Mol. Med. 2019, 25, 7. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Xiao, Z.; Guo, H.; Fang, X.; Liang, J.; Zhu, J.; Yang, J.; Li, H.; Pan, R.; Yuan, S.; et al. Novel role of the clustered miR-23b-3p and miR-27b-3p in enhanced expression of fibrosis-associated genes by targeting TGFBR3 in atrial fibroblasts. J. Cell. Mol. Med. 2019, 23, 3246–3256. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.F.; Chen, Y.J.; Lin, Y.J.; Chen, S.A. Inflammation and the pathogenesis of atrial fibrillation. Nat. Rev. Cardiol. 2015, 12, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Li, G.; Li, L.; Korantzopoulos, P. Association between C-reactive protein and recurrence of atrial fibrillation after successful electrical cardioversion: A meta-analysis. J. Am. Coll. Cardiol. 2007, 49, 1642–1648. [Google Scholar] [CrossRef] [PubMed]
- Tilly, M.J.; Geurts, S.; Zhu, F.; Bos, M.M.; Ikram, M.A.; de Maat, M.P.M.; de Groot, N.M.S.; Kavousi, M. Autoimmune diseases and new-onset atrial fibrillation: A UK Biobank study. Europace 2023, 25, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Sagris, M.; Vardas, E.P.; Theofilis, P.; Antonopoulos, A.S.; Oikonomou, E.; Tousoulis, D. Atrial Fibrillation: Pathogenesis, Predisposing Factors, and Genetics. Int. J. Mol. Sci. 2021, 23, 6. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.S.; Chou, C.C.; Tan, A.Y.; Zhou, S.; Fishbein, M.C.; Hwang, C.; Karagueuzian, H.S.; Lin, S.F. The mechanisms of atrial fibrillation. J. Cardiovasc. Electrophysiol. 2006, 17 (Suppl. S3), S2–S7. [Google Scholar] [CrossRef] [PubMed]
- Lip, G.Y.; Patel, J.V.; Hughes, E.; Hart, R.G. High-sensitivity C-reactive protein and soluble CD40 ligand as indices of inflammation and platelet activation in 880 patients with nonvalvular atrial fibrillation: Relationship to stroke risk factors, stroke risk stratification schema, and prognosis. Stroke 2007, 38, 1229–1237. [Google Scholar] [CrossRef]
- Rahimi, K.; Emberson, J.; McGale, P.; Majoni, W.; Merhi, A.; Asselbergs, F.W.; Krane, V.; Macfarlane, P.W. Effect of statins on atrial fibrillation: Collaborative meta-analysis of published and unpublished evidence from randomised controlled trials. BMJ 2011, 342, d1250. [Google Scholar] [CrossRef]
- Arroyo, A.B.; Fernández-Pérez, M.P.; Del Monte, A.; Águila, S.; Méndez, R.; Hernández-Antolín, R.; García-Barber, N.; de Los Reyes-García, A.M.; González-Jiménez, P.; Arcas, M.I.; et al. miR-146a is a pivotal regulator of neutrophil extracellular trap formation promoting thrombosis. Haematologica 2021, 106, 1636–1646. [Google Scholar] [CrossRef]
- Huang, Z.; Chen, X.J.; Qian, C.; Dong, Q.; Ding, D.; Wu, Q.F.; Li, J.; Wang, H.F.; Li, W.H.; Xie, Q.; et al. Signal Transducer and Activator of Transcription 3/MicroRNA-21 Feedback Loop Contributes to Atrial Fibrillation by Promoting Atrial Fibrosis in a Rat Sterile Pericarditis Model. Circ. Arrhythmia Electrophysiol. 2016, 9, e003396. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, L.; Hu, J.; Song, L. MicroRNA Regulatory Network Revealing the Mechanism of Inflammation in Atrial Fibrillation. Med. Sci. Monit. 2015, 21, 3505–3513. [Google Scholar] [CrossRef] [PubMed]
- Galenko, O.; Jacobs, V.; Knight, S.; Taylor, M.; Cutler, M.J.; Muhlestein, J.B.; Carlquist, J.L.; Knowlton, K.U.; Jared Bunch, T. The role of microRNAs in the development, regulation, and treatment of atrial fibrillation. J. Interv. Card. Electrophysiol. 2019, 55, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Menezes Junior, A.D.S.; Ferreira, L.C.; Barbosa, L.J.V.; Silva, D.M.E.; Saddi, V.A.; Silva, A. Circulating MicroRNAs as Specific Biomarkers in Atrial Fibrillation: A Meta-Analysis. Noncoding RNA 2023, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Lin, S.; Li, X.; Guo, D.; Wang, Y.; Hu, Y. Serum miR-222 is independently associated with atrial fibrillation in patients with degenerative valvular heart disease. BMC Cardiovasc. Disord. 2021, 21, 98. [Google Scholar] [CrossRef] [PubMed]
- Sovari, A.A.; Dudley, S.C., Jr. Reactive oxygen species-targeted therapeutic interventions for atrial fibrillation. Front. Physiol. 2012, 3, 311. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.Y.; Zhang, J.; Zhang, Y.; Chen, H.; Liu, D.; Ping, P.; Weiss, J.N.; Cai, H. Oxidative stress in atrial fibrillation: An emerging role of NADPH oxidase. J. Mol. Cell. Cardiol. 2013, 62, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Pool, L.; Wijdeveld, L.; de Groot, N.M.S.; Brundel, B. The Role of Mitochondrial Dysfunction in Atrial Fibrillation: Translation to Druggable Target and Biomarker Discovery. Int. J. Mol. Sci. 2021, 22, 8463. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Santulli, G.; Reiken, S.R.; Yuan, Q.; Osborne, B.W.; Chen, B.X.; Marks, A.R. Mitochondrial oxidative stress promotes atrial fibrillation. Sci. Rep. 2015, 5, 11427. [Google Scholar] [CrossRef]
- Purohit, A.; Rokita, A.G.; Guan, X.; Chen, B.; Koval, O.M.; Voigt, N.; Neef, S.; Sowa, T.; Gao, Z.; Luczak, E.D.; et al. Oxidized Ca(2+)/calmodulin-dependent protein kinase II triggers atrial fibrillation. Circulation 2013, 128, 1748–1757. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, S.; Geng, Y.; Xue, J.; Wang, Z.; Xie, X.; Wang, J.; Zhang, S.; Hou, Y. MicroRNA profiling of atrial fibrillation in canines: miR-206 modulates intrinsic cardiac autonomic nerve remodeling by regulating SOD1. PLoS ONE 2015, 10, e0122674. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Zhang, W. MicroRNA-423-5p mediates H2O2-induced apoptosis in cardiomyocytes through O-GlcNAc transferase. Mol. Med. Rep. 2016, 14, 857–864. [Google Scholar] [CrossRef]
- Song, Y.; Wei, C.; Wang, J. Upregulation of miR-143-3p attenuates oxidative stress-mediated cell ferroptosis in cardiomyocytes with atrial fibrillation by degrading glutamic-oxaloacetic transaminase 1. Biocell 2021, 45, 733–744. [Google Scholar] [CrossRef]
- Yamac, A.H.; Kucukbuzcu, S.; Ozansoy, M.; Gok, O.; Oz, K.; Erturk, M.; Yilmaz, E.; Ersoy, B.; Zeybek, R.; Goktekin, O.; et al. Altered expression of micro-RNA 199a and increased levels of cardiac SIRT1 protein are associated with the occurrence of atrial fibrillation after coronary artery bypass graft surgery. Cardiovasc. Pathol. 2016, 25, 232–236. [Google Scholar] [CrossRef]
- Wei, X.J.; Han, M.; Yang, F.Y.; Wei, G.C.; Liang, Z.G.; Yao, H.; Ji, C.W.; Xie, R.S.; Gong, C.L.; Tian, Y. Biological significance of miR-126 expression in atrial fibrillation and heart failure. Braz. J. Med. Biol. Res. 2015, 48, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cheng, Y.; Zhang, S.; Lin, Y.; Yang, J.; Zhang, C. A necessary role of miR-221 and miR-222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ. Res. 2009, 104, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Huang, M.; Li, Z.; Jia, F.; Ghosh, Z.; Lijkwan, M.A.; Fasanaro, P.; Sun, N.; Wang, X.; Martelli, F.; et al. MicroRNA-210 as a novel therapy for treatment of ischemic heart disease. Circulation 2010, 122, S124–S131. [Google Scholar] [CrossRef]
- Nso, N.; Bookani, K.R.; Metzl, M.; Radparvar, F. Role of inflammation in atrial fibrillation: A comprehensive review of current knowledge. J. Arrhythmia 2021, 37, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Raitoharju, E.; Lyytikäinen, L.P.; Levula, M.; Oksala, N.; Mennander, A.; Tarkka, M.; Klopp, N.; Illig, T.; Kähönen, M.; Karhunen, P.J.; et al. miR-21, miR-210, miR-34a, and miR-146a/b are up-regulated in human atherosclerotic plaques in the Tampere Vascular Study. Atherosclerosis 2011, 219, 211–217. [Google Scholar] [CrossRef]
- da Silva, A.M.G.; de Araújo, J.N.G.; de Freitas, R.C.C.; Silbiger, V.N. Circulating MicroRNAs as Potential Biomarkers of Atrial Fibrillation. BioMed Res. Int. 2017, 2017, 7804763. [Google Scholar] [CrossRef]
- Rivera-Caravaca, J.M.; Teruel-Montoya, R.; Roldán, V.; Cifuentes-Riquelme, R.; Crespo-Matas, J.A.; de Los Reyes-García, A.M.; Águila, S.; Fernández-Pérez, M.P.; Reguilón-Gallego, L.; Zapata-Martínez, L.; et al. Pilot Study on the Role of Circulating miRNAs for the Improvement of the Predictive Ability of the 2MACE Score in Patients with Atrial Fibrillation. J. Clin. Med. 2020, 9, 3645. [Google Scholar] [CrossRef] [PubMed]
- Roldán, V.; Arroyo, A.B.; Salloum-Asfar, S.; Manzano-Fernández, S.; García-Barberá, N.; Marín, F.; Vicente, V.; González-Conejero, R.; Martínez, C. Prognostic role of MIR146A polymorphisms for cardiovascular events in atrial fibrillation. Thromb. Haemost. 2014, 112, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, K.; Niehues, P.; Neupane, B.; Maleck, C.; Sharif-Yakan, A.; Emrani, M.; Zink, M.D.; Napp, A.; Marx, N.; Gramlich, M. MicroRNA-21 mediated cross-talk between cardiomyocytes and fibroblasts in patients with atrial fibrillation. Front. Cardiovasc. Med. 2023, 10, 1056134. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhao, Z.; Li, G. The Therapeutic Potential of MicroRNAs in Atrial Fibrillation. Mediat. Inflamm. 2020, 2020, 3053520. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Yang, B.; Nattel, S. MicroRNAs and atrial fibrillation: Mechanisms and translational potential. Nat. Rev. Cardiol. 2015, 12, 80–90. [Google Scholar] [CrossRef]
- Jia, X.; Zheng, S.; Xie, X.; Zhang, Y.; Wang, W.; Wang, Z.; Zhang, Y.; Wang, J.; Gao, M.; Hou, Y. MicroRNA-1 Accelerates the Shortening of Atrial Effective Refractory Period by Regulating KCNE1 and KCNB2 Expression: An Atrial Tachypacing Rabbit Model. PLoS ONE 2014, 8, e85639. [Google Scholar] [CrossRef]
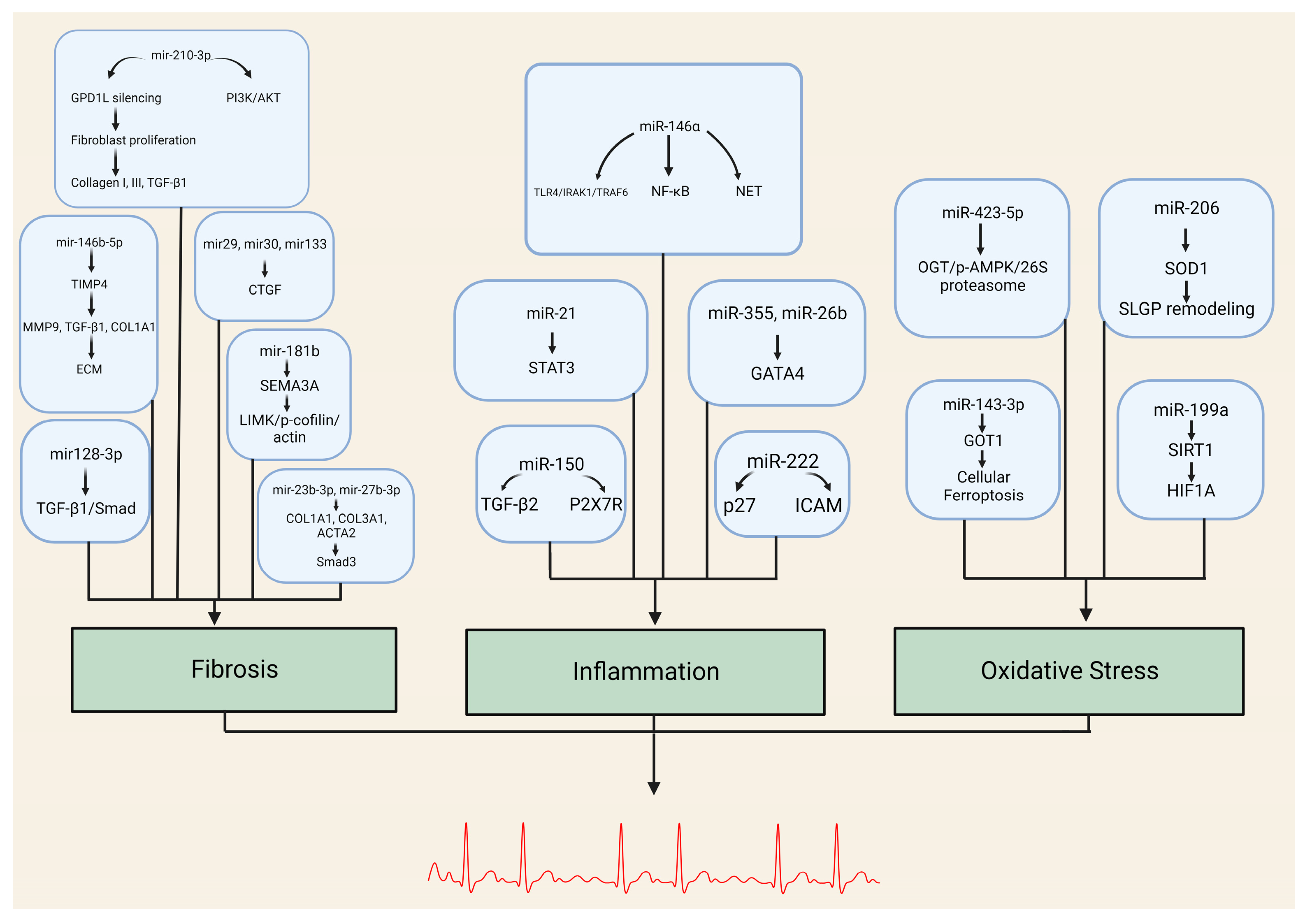


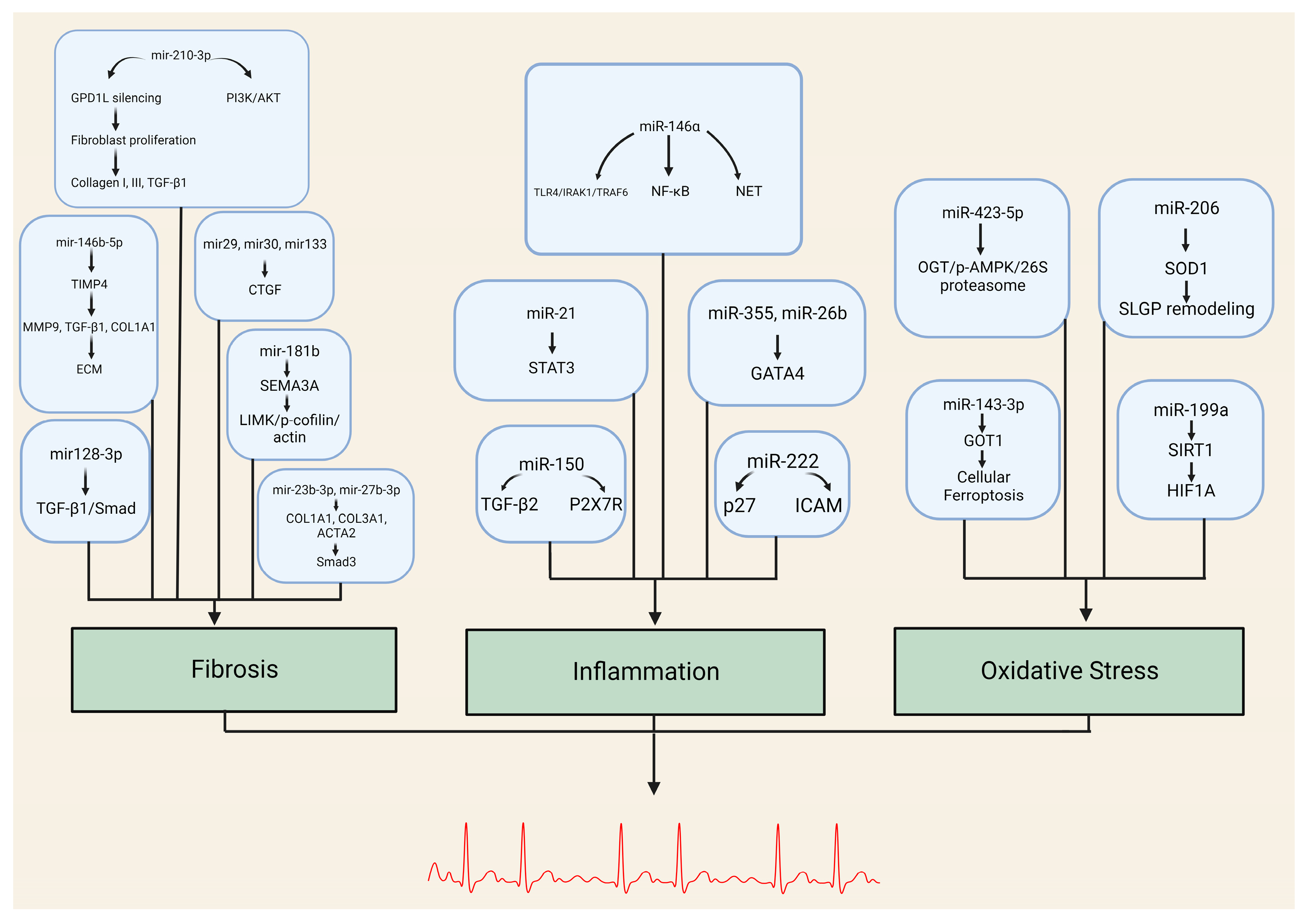{kind=link}
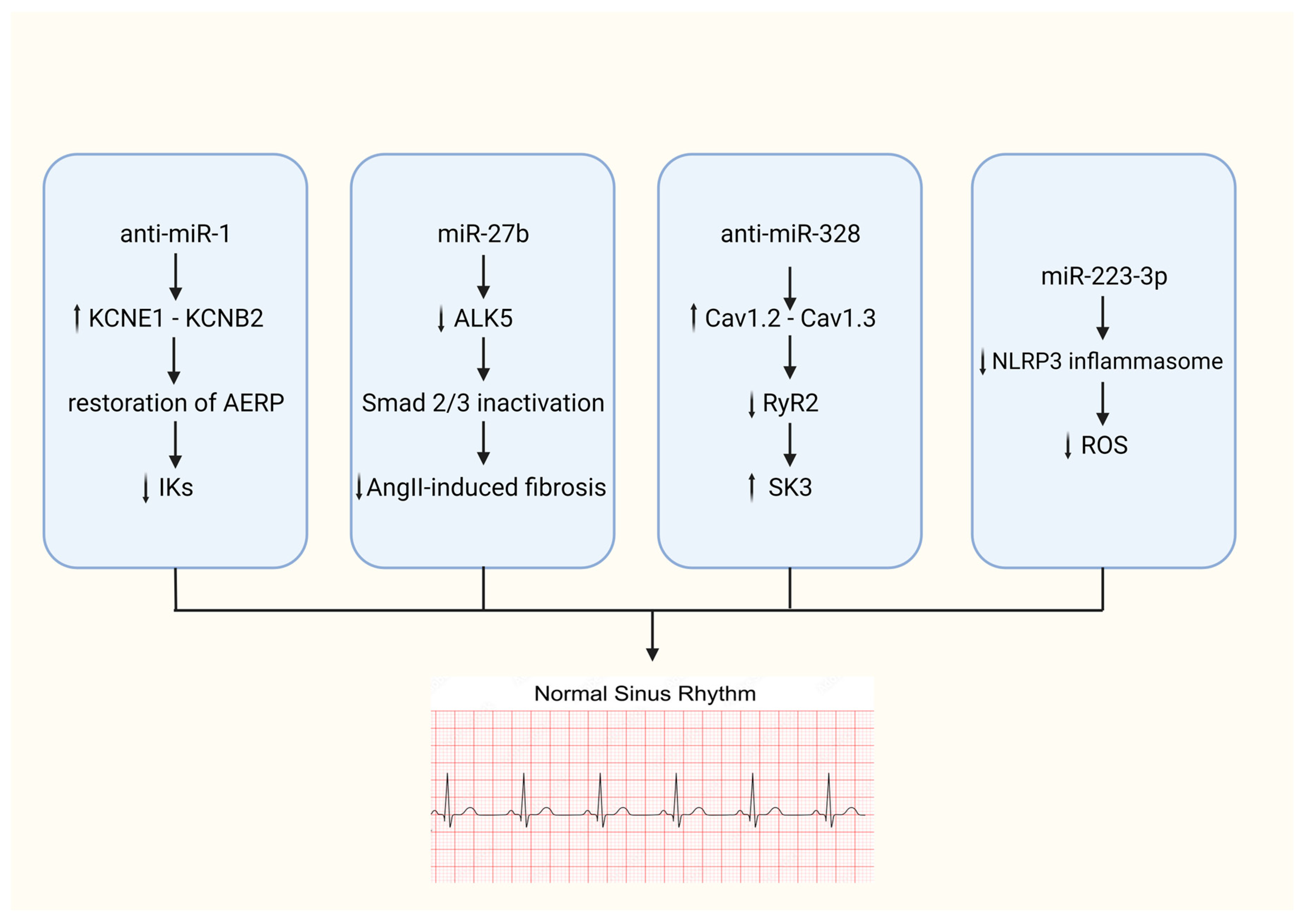{kind=link}
| miRNA | Target Genes/Pathways |
|---|---|
| miR-146b-5p | TIMP4, MMP9, TGFB1, COL1A1 |
| miR-210-3p | GPD1L, PI3K/AKT pathway |
| miR-29, miR-30, miR-133, miR-21 | Connective tissue growth factor (CTGF) |
| miR-23b-3p, miR-27b-3p | COL1A1, COL3A1, ACTA2 TGFBR3/Smad3 pathway |
| miR-128-3p | PVT1, Sp1 TGF-β1/Smad pathway |
| miR-26 | CNJ2/IK TRPC3 |
| miR-181b | Sema3A Sema3A/LIMK/p-cofilin/actin axis |
| miRNA | Target Genes/Pathways | Role in Inflammation |
|---|---|---|
| miR-146a | TLR4, IRAK1, TRAF6 NF-κB pathway | Negative regulator of innate inflammatory responses NET formation |
| miR-21 | STAT3 pathway | Promotes inflammation-associated fibrosis |
| miR-355, miR-26b | GATA4 | Alters fibroblast expression (cellular senescence) Affects atrial conduction through calcium homeostasis |
| miR-150 | TGF-β EGRR2, P2X7R | Modulates inflammatory response system by targeting pro-inflammatory ATP receptor and multiple regulatory genes |
| miR-222 | Associated with DVHD P27 ICAM-1 | Role in inflammation-mediated vascular remodeling Downregulation of IRF-2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vardas, E.P.; Theofilis, P.; Oikonomou, E.; Vardas, P.E.; Tousoulis, D. MicroRNAs in Atrial Fibrillation: Mechanisms, Vascular Implications, and Therapeutic Potential. Biomedicines 2024, 12, 811. https://doi.org/10.3390/biomedicines12040811
Vardas EP, Theofilis P, Oikonomou E, Vardas PE, Tousoulis D. MicroRNAs in Atrial Fibrillation: Mechanisms, Vascular Implications, and Therapeutic Potential. Biomedicines. 2024; 12(4):811. https://doi.org/10.3390/biomedicines12040811
Chicago/Turabian StyleVardas, Emmanouil P., Panagiotis Theofilis, Evangelos Oikonomou, Panos E. Vardas, and Dimitris Tousoulis. 2024. "MicroRNAs in Atrial Fibrillation: Mechanisms, Vascular Implications, and Therapeutic Potential" Biomedicines 12, no. 4: 811. https://doi.org/10.3390/biomedicines12040811