
Analysing the Combined Effects of Radiotherapy and Chemokine Receptor 5 Antagonism: Complementary Approaches to Promote T Cell Function and Migration in Oesophageal Adenocarcinoma

 , , , , and
, , , , and
Abstract
1. Introduction
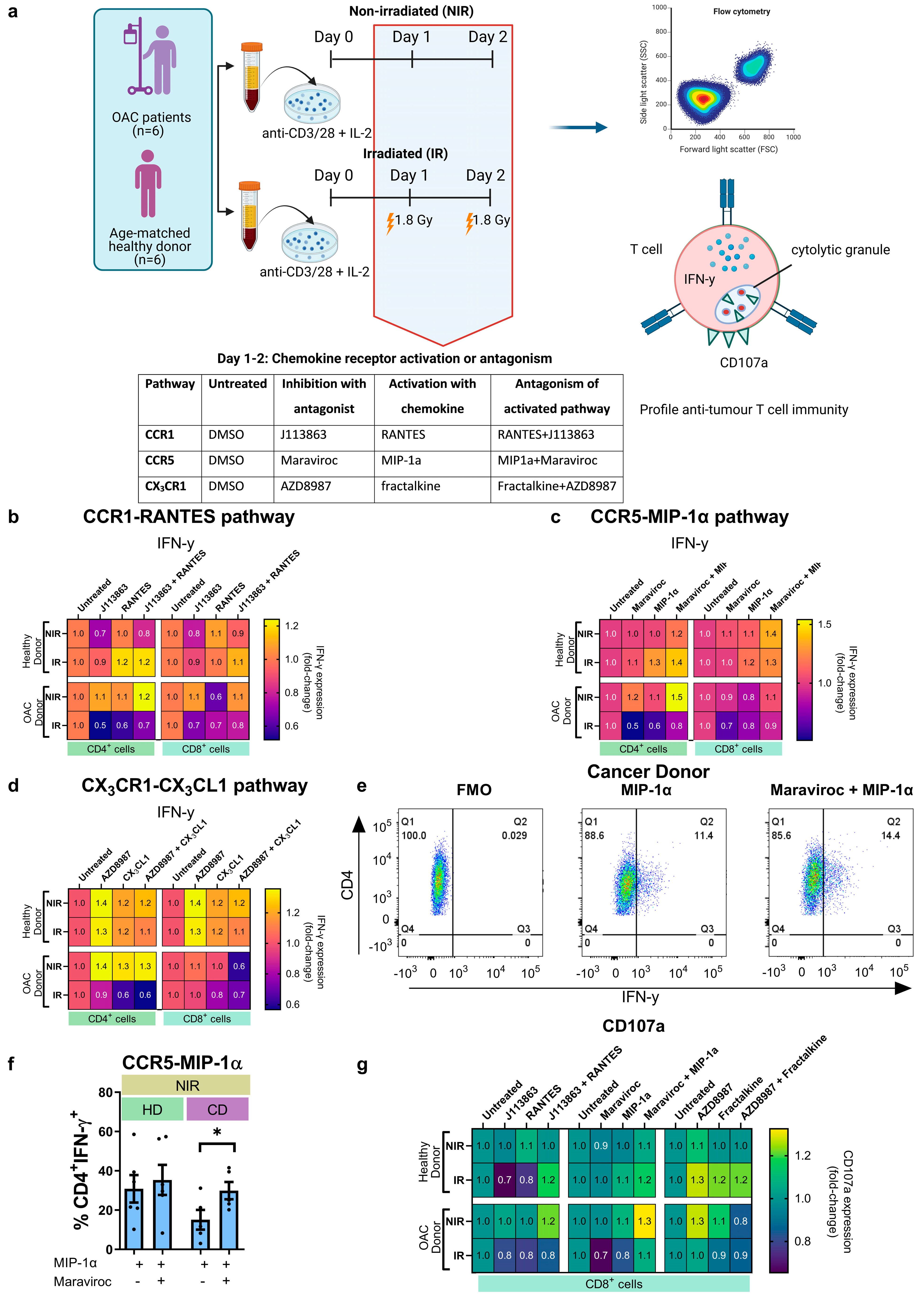
2. Methods
2.1. Ethical Approval
2.2. Specimen Collection
2.3. Generation of Tumour-Conditioned Media
2.4. T Cell Activation
2.5. Chemotaxis Assay
2.6. Flow Cytometry
2.7. Statistical Analysis
3. Results
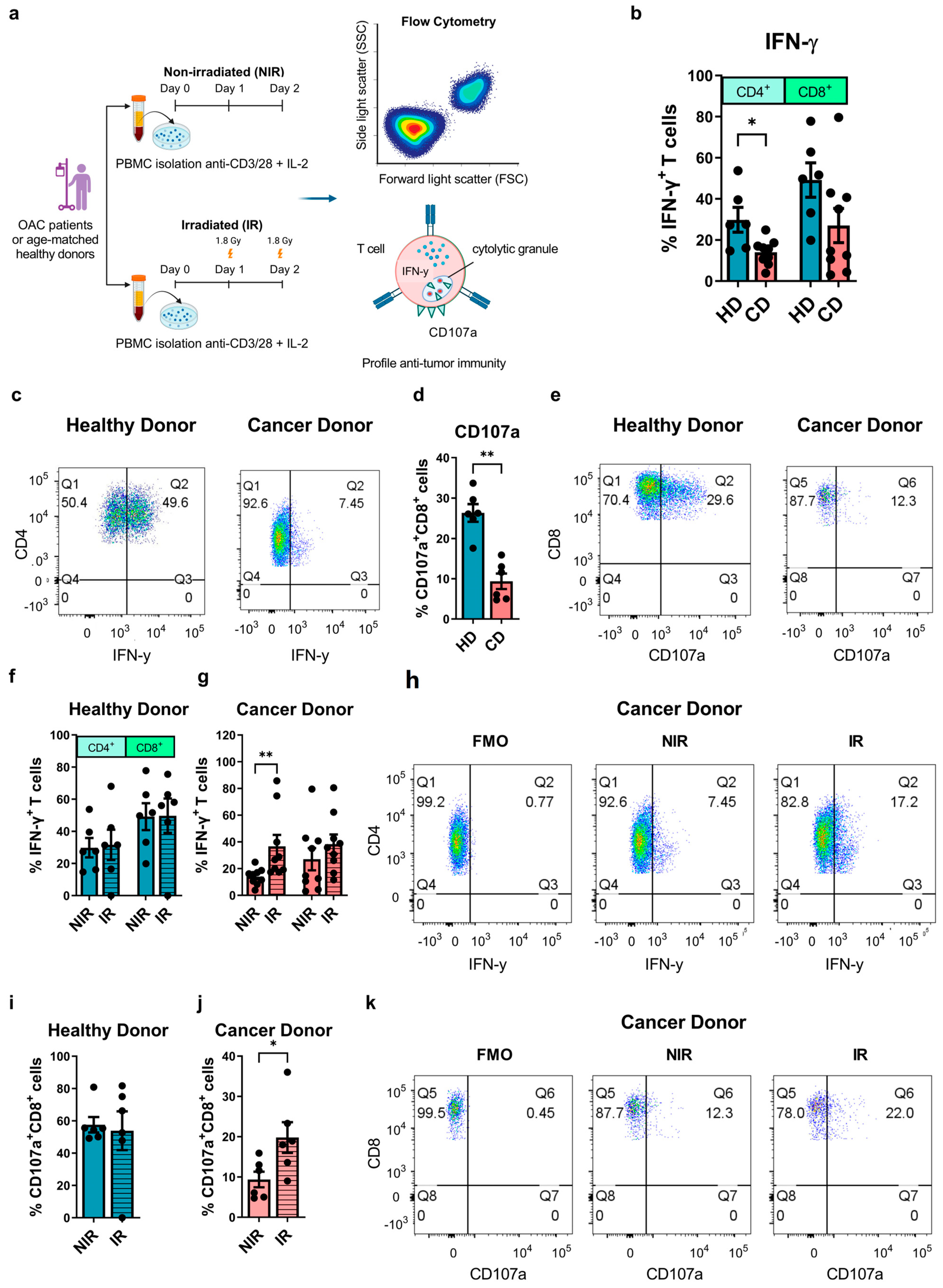
3.1. Clinically Relevant Doses of Radiation Ex Vivo Can Rescue Diminished Effector T Cell Function in OAC Patients
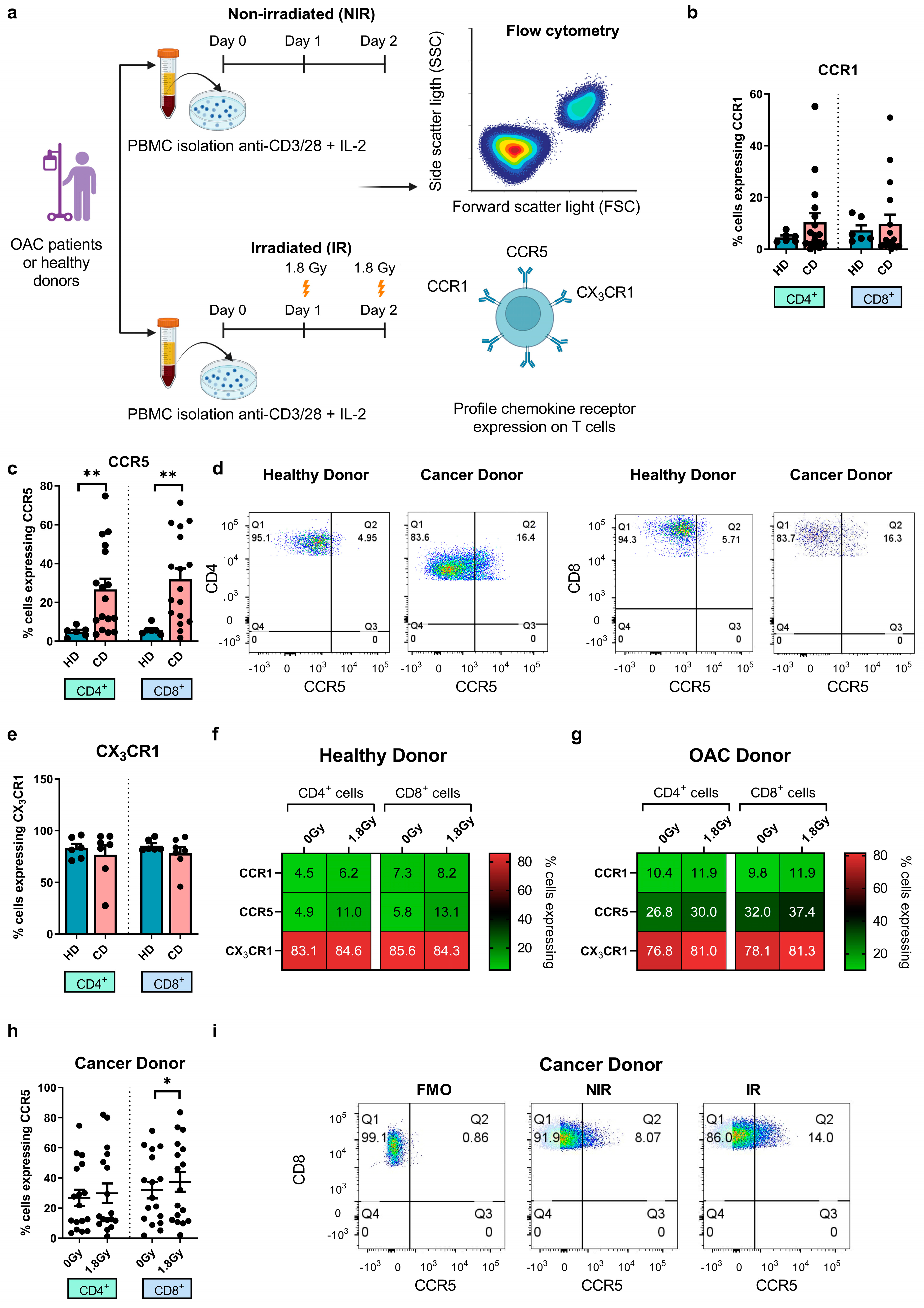
3.2. Significantly Higher Frequencies of Circulating CCR5+ T Cells in OAC Patients Compared with Healthy Controls, While CCR5 Surface Expression Is Upregulated by Clinically Relevant Doses of Radiation
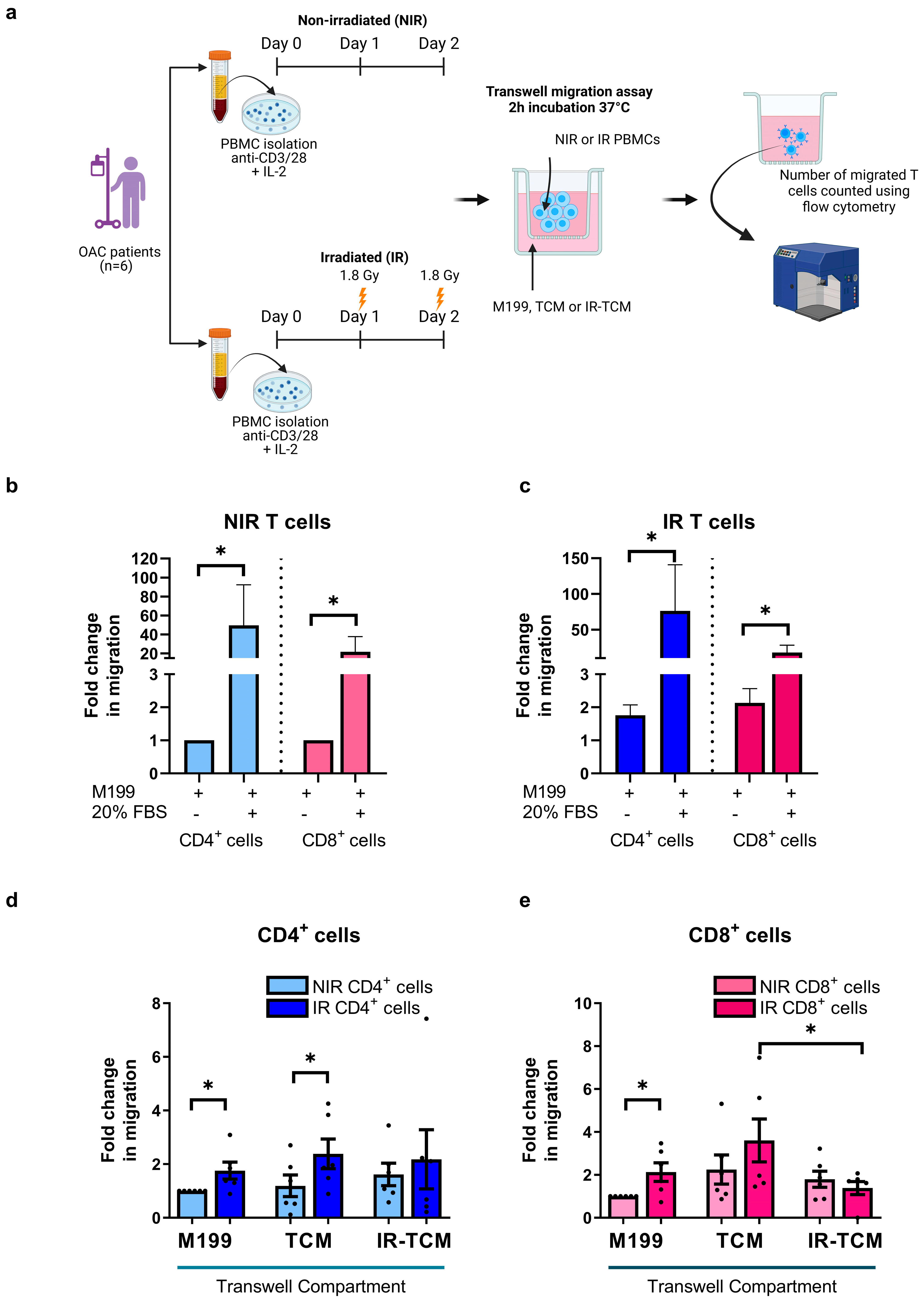
3.3. Irradiation Increased the Migratory Capacity of OAC-Derived T Cells toward OAC Patient-Derived Tumour-Conditioned Media
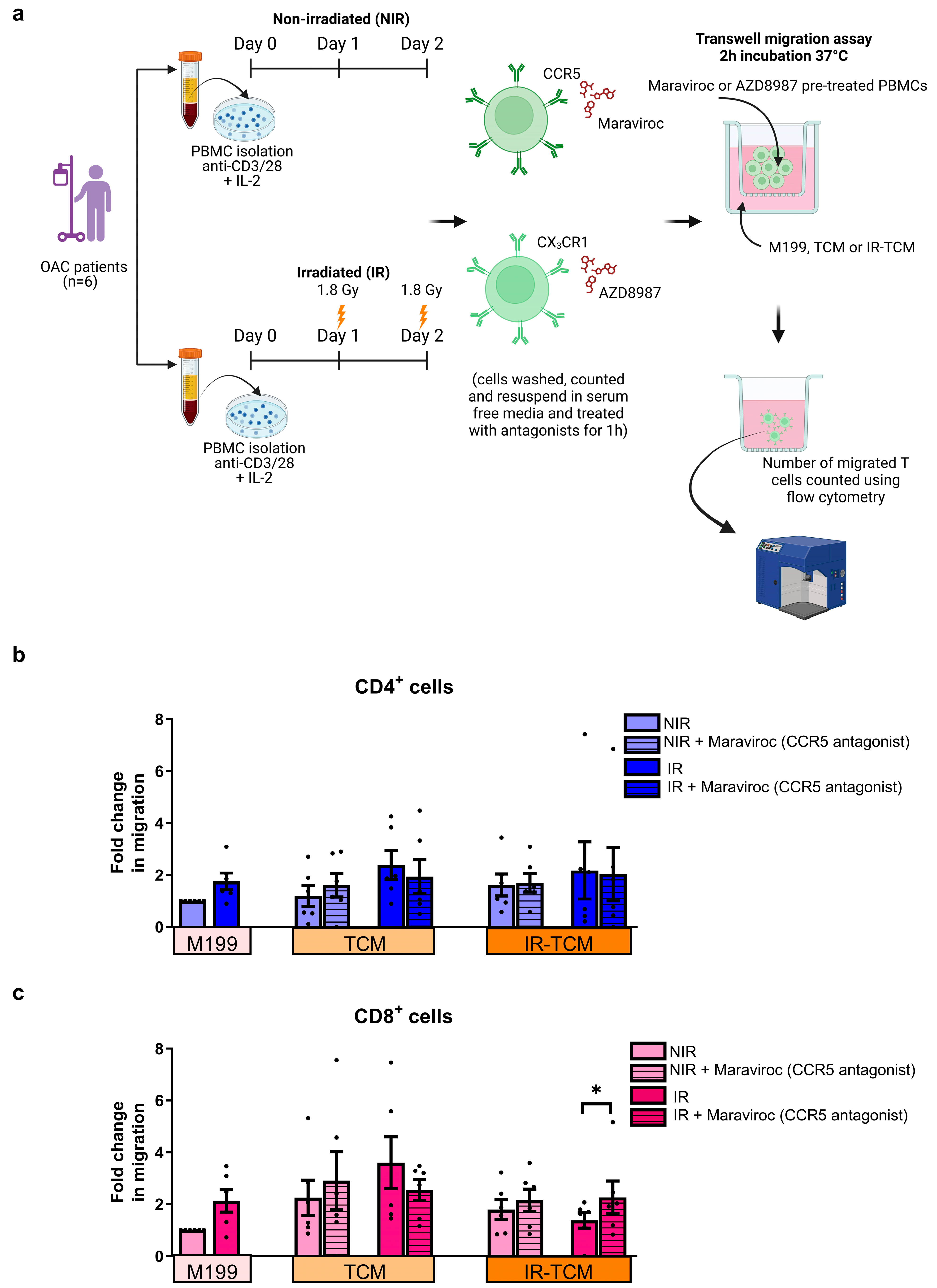
3.4. In Vitro Treatment with CCR5 Antagonist Maraviroc Increased the Migration of Irradiated CD8+ T Cells towards the Irradiated Tumour Compartment and Enhanced Production of IFN-γ by CD4+ T Helper Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, J.; Koulaouzidis, A.; Marlicz, W.; Lok, V.; Chu, C.; Ngai, C.H.; Zhang, L.; Chen, P.; Wang, S.; Yuan, J.; et al. Global Burden, Risk Factors, and Trends of Esophageal Cancer: An Analysis of Cancer Registries from 48 Countries. Cancers 2021, 13, 141. [Google Scholar] [CrossRef] [PubMed]
- Eyck, B.M.; van Lanschot, J.J.B.; Hulshof, M.C.C.M.; van der Wilk, B.J.; Shapiro, J.; van Hagen, P.; van Berge Henegouwen, M.I.; Wijnhoven, B.P.L.; van Laarhoven, H.W.M.; Nieuwenhuijzen, G.; et al. Ten-Year Outcome of Neoadjuvant Chemoradiotherapy Plus Surgery for Esophageal Cancer: The Randomized Controlled CROSS Trial. J. Clin. Oncol. 2021, 39, 1995–2004. [Google Scholar] [CrossRef] [PubMed]
- Runkel, M.; Verst, R.; Spiegelberg, J.; Fichtner-Feigl, S.; Hoeppner, J.; Glatz, T. Perioperative FLOT chemotherapy plus surgery for oligometastatic esophagogastric adenocarcinoma: Surgical outcome and overall survival. BMC Surg. 2021, 21, 35. [Google Scholar] [CrossRef] [PubMed]
- Ajani, J.A.; Harada, K.; Rogers, J.E.; Iwatsuki, M.; Yamashita, K.; Baba, H. Recent advances in treating oesophageal cancer. F1000Research 2020, 9, 1189. [Google Scholar]
- Bang, Y.J.; van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Rogers, J.E.; Xiao, L.; Amlashi, F.G.; Elimova, E.; Blum Murphy, M.A.; Sanders, E.; Shanbhag, N.; Thomas, I.; Ajani, J.A. Ramucirumab and Paclitaxel Administered Every 2 Weeks (mRAINBOW Regimen) in Advanced Gastroesophageal Adenocarcinoma. Oncology 2019, 96, 252–258. [Google Scholar] [CrossRef]
- Hirose, T.; Yamamoto, S.; Kato, K. Pembrolizumab for first-line treatment of advanced unresectable or metastatic esophageal or gastroesophageal junction cancer. Ther. Adv. Gastroenterol. 2023, 16, 17562848221148250. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.J.; Ajani, J.A.; Kuzdzal, J.; Zander, T.; van Cutsem, E.; Piessen, G.; Mendez, G.; Feliciano, J.; Motoyama, S.; Lièvre, A.; et al. Adjuvant Nivolumab in Resected Esophageal or Gastroesophageal Junction Cancer. N. Engl. J. Med. 2021, 384, 1191–1203. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Stratton, M.R.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415. [Google Scholar] [CrossRef]
- Vivaldi, C.; Catanese, S.; Massa, V.; Pecora, I.; Salani, F.; Santi, S.; Lencioni, M.; Vasile, E.; Falcone, A.; Fornaro, L. Immune Checkpoint Inhibitors in Esophageal Cancers: Are We Finally Finding the Right Path in the Mist? Int. J. Mol. Sci. 2020, 21, 1658. [Google Scholar] [CrossRef]
- Shitara, K.; van Cutsem, E.; Bang, Y.J.; Fuchs, C.; Wyrwicz, L.; Lee, K.W.; Kudaba, I.; Garrido, M.; Chung, H.C.; Lee, J.; et al. Efficacy and Safety of Pembrolizumab or Pembrolizumab plus Chemotherapy vs. Chemotherapy Alone for Patients with First-line, Advanced Gastric Cancer: The KEYNOTE-062 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1571–1580. [Google Scholar] [CrossRef] [PubMed]
- Davern, M.; Donlon, N.E.; Power, R.; Hayes, C.; King, R.; Dunne, M.R.; Reynolds, J.V. The tumour immune microenvironment in oesophageal cancer. Br. J. Cancer 2021, 125, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.X.; Fu, L. The immune landscape of esophageal cancer. Cancer Commun. 2019, 39, 79. [Google Scholar] [CrossRef] [PubMed]
- Ozga, A.J.; Chow, M.T.; Luster, A.D. Chemokines and the immune response to cancer. Immunity 2021, 54, 859–874. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559. [Google Scholar] [CrossRef]
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944. [Google Scholar] [CrossRef]
- Zhang, Y.; Guan, X.Y.; Jiang, P. Cytokine and Chemokine Signals of T-Cell Exclusion in Tumors. Front. Immunol. 2020, 11, 594609. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, M.E.; Conroy, M.J.; Clarke, N.E.; Gilmartin, N.T.; Feighery, R.; MacCarthy, F.; O’Toole, D.; Ravi, N.; Reynolds, J.V.; O’Sullivan, J.; et al. Altered T Cell Migratory Capacity in the Progression from Barrett Oesophagus to Oesophageal Adenocarcinoma. Cancer Microenviron. 2019, 12, 57–66. [Google Scholar] [CrossRef]
- Wang, L.; Jiang, J.; Chen, Y.; Jia, Q.; Chu, Q. The roles of CC chemokines in response to radiation. Radiat. Oncol. 2022, 17, 63. [Google Scholar] [CrossRef]
- Donlon, N.E.; Davern, M.; O’Connell, F.; Sheppard, A.; Heeran, A.; Bhardwaj, A.; Butler, C.; Narayanasamy, R.; Donohoe, C.; Phelan, J.J.; et al. Impact of radiotherapy on the immune landscape in oesophageal adenocarcinoma. World J. Gastroenterol. 2022, 28, 2302. [Google Scholar] [CrossRef]
- Gough, M.J.; Crittenden, M.R. The paradox of radiation and T cells in tumors. Neoplasia 2022, 31, 100808. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Martin, A.; Mira, E.; Manes, S. CCR5 as a Potential Target in Cancer Therapy: Inhibition or Stimulation? Anticancer Agents Med. Chem. 2012, 12, 1045–1057. [Google Scholar] [CrossRef]
- Conroy, M.J.; Maher, S.G.; Melo, A.M.; Doyle, S.L.; Foley, E.; Reynolds, J.V.; Long, A.; Lysaght, J. Identifying a novel role for fractalkine (CX3CL1) in memory CD8+ T cell accumulation in the omentum of obesity-associated cancer patients. Front. Immunol. 2018, 9, 402547. [Google Scholar] [CrossRef]
- Gerlach, C.; Moseman, E.A.; Loughhead, S.M.; Alvarez, D.; Zwijnenburg, A.J.; Waanders, L.; Garg, R.; de la Torre, J.C.; von Andrian, U.H. The Chemokine Receptor CX3CR1 Defines Three Antigen-Experienced CD8 T Cell Subsets with Distinct Roles in Immune Surveillance and Homeostasis. Immunity 2016, 45, 1270–1284. [Google Scholar] [CrossRef]
- Schaller, M.A.; Kallal, L.E.; Lukacs, N.W. A key role for CC chemokine receptor 1 in T-cell-mediated respiratory inflammation. Am. J. Pathol. 2008, 172, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.T.; Li, K.K.; Hepburn, E.; Weston, C.J.; Curbishley, S.M.; Reynolds, G.M.; Hejmadi, R.K.; Bicknell, R.; Eksteen, B.; Ismail, T.; et al. The effects of CCR5 inhibition on regulatory T-cell recruitment to colorectal cancer. Br. J. Cancer 2015, 112, 319–328. [Google Scholar] [CrossRef]
- González-Martín, A.; Mira, E.; Mañes, S. CCR5 in cancer immunotherapy: More than an “attractive” receptor for T cells. Oncoimmunology 2012, 1, 106. [Google Scholar] [CrossRef]
- Jiao, X.; Nawab, O.; Patel, T.; Kossenkov, A.V.; Halama, N.; Jaeger, D.; Pestell, R.G. Recent advances targeting CCR5 for cancer and its role in immuno-oncology. Cancer Res. 2019, 79, 4801–4807. [Google Scholar] [CrossRef] [PubMed]
- Donlon, N.E.; Davern, M.; Sheppard, A.; Power, R.; O’connell, F.; Heeran, A.B.; King, R.; Hayes, C.; Bhardwaj, A.; Phelan, J.J.; et al. The Prognostic Value of the Lymph Node in Oesophageal Adenocarcinoma; Incorporating Clinicopathological and Immunological Profiling. Cancers 2021, 13, 4005. [Google Scholar] [CrossRef]
- Mylod, E.; Melo, A.M.; Donlon, N.E.; Davern, M.; Bhardwaj, A.; Reynolds, J.V.; Lysaght, J.; Conroy, M.J. Fractalkine Elicits Chemotactic, Phenotypic, and Functional Effects on CX3CR1+CD27− NK Cells in Obesity-Associated Cancer. J. Immunol. 2021, 207, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Conroy, M.J.; Galvin, K.C.; Kavanagh, M.E.; Mongan, A.M.; Doyle, S.L.; Gilmartin, N.; O’Farrelly, C.; Reynolds, J.V.; Lysaght, J. CCR1 antagonism attenuates T cell trafficking to omentum and liver in obesity-associated cancer. Immunol. Cell Biol. 2016, 94, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Arberas, H.; Guardo, A.C.; Bargalló, M.E.; Maleno, M.J.; Calvo, M.; Blanco, J.L.; García, F.; Gatell, J.M.; Plana, M. In vitro effects of the CCR5 inhibitor maraviroc on human T cell function. J. Antimicrob. Chemother. 2013, 68, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Davern, M.; Donlon, N.E.; O’ Connell, F.; Sheppard, A.D.; Hayes, C.; King, R.; Temperley, H.; Butler, C.; Bhardwaj, A.; Moore, J. Cooperation between chemotherapy and immune checkpoint blockade to enhance anti-tumour T cell immunity in oesophageal adenocarcinoma. Transl. Oncol. 2022, 20, 101406. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Herrero, S.; Sordo-Bahamonde, C.; Gonzalez, S.; López-Soto, A. CD107a Degranulation Assay to Evaluate Immune Cell Antitumor Activity. Methods Mol. Biol. 2019, 1884, 119–130. [Google Scholar] [PubMed]
- Mylod, E.; O’connell, F.; Donlon, N.E.; Butler, C.; Reynolds, J.V.; Lysaght, J.; Conroy, M.J. The Omentum in Obesity-Associated Cancer: A Hindrance to Effective Natural Killer Cell Migration towards Tumour Which Can Be Overcome by CX3CR1 Antagonism. Cancers 2021, 14, 64. [Google Scholar] [CrossRef] [PubMed]
- Mylod, E.; O’Connell, F.; Donlon, D.E.; Davern, M.; Marion, C.; Butler, C.; Reynolds, J.V.; Lysaght, J.; Conroy, M.J. Real-time ex vivo monitoring of NK cell migration toward obesity-associated oesophageal adenocarcinoma following modulation of CX3CR1. Sci. Rep. 2024, 14, 4017. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, S.; Wang, B.; Kawashima, N.; Braunstein, S.; Badura, M.; Cameron, T.O.; Babb, J.S.; Schneider, R.J.; Formenti, S.C.; Dustin, M.L.; et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J. Immunol. 2008, 181, 3099–3107. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Asea, A.; Multhoff, G.; Pockley, A.G. Radiation-induced effects and the immune system in cancer. Front. Oncol. 2012, 2, 191. [Google Scholar] [CrossRef]
- Lin, W.; Xu, Y.; Chen, X.; Liu, J.; Weng, Y.; Zhuang, Q.; Lin, F.; Huang, Z.; Wu, S.; Ding, J.; et al. Radiation-induced small extracellular vesicles as ‘carriages’ promote tumor antigen release and trigger antitumor immunity. Theranostics 2020, 10, 4871–4884. [Google Scholar] [CrossRef]
- Storozynsky, Q.; Hitt, M.M. The Impact of Radiation-Induced DNA Damage on cGAS-STING-Mediated Immune Responses to Cancer. Int. J. Mol. Sci. 2020, 21, 8877. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S.; Coleman, C.N.; Formenti, S.C. Radiotherapy: Changing the Game in Immunotherapy. Trends Cancer 2016, 2, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Jarosz-Biej, M.; Smolarczyk, R.; Cichoń, T.; Kułach, N. Tumor Microenvironment as A “Game Changer” in Cancer Radiotherapy. Int. J. Mol. Sci. 2019, 20, 3212. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Mukherjee, S.; Sinha, D.; Abdisalaam, S.; Krishnan, S.; Asaithamby, A. Immunomodulatory Effects of Radiotherapy. Int. J. Mol. Sci. 2020, 21, 8151. [Google Scholar] [CrossRef]
- Colton, M.; Cheadle, E.J.; Honeychurch, J.; Illidge, T.M. Reprogramming the tumour microenvironment by radiotherapy: Implications for radiotherapy and immunotherapy combinations. Radiat. Oncol. 2020, 15, 254. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Ogino, I.; Shigenaga, D.; Hata, M. Impact of Regional Lymph Node Irradiation on Reducing Lymph Node Recurrence in Esophageal Cancer Patients. Cancer Diagn. Progn. 2022, 2, 223. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, H.A.; Villar, R.C. Radiotherapy and immune response: The systemic effects of a local treatment. Clinics 2018, 73, e557s. [Google Scholar] [CrossRef] [PubMed]
- Merrick, A.; Errington, F.; Milward, K.; O’Donnell, D.; Harrington, K.; Bateman, A.; Pandha, H.; Vile, R.; Morrison, E.; Selby, P.; et al. Immunosuppressive effects of radiation on human dendritic cells: Reduced IL-12 production on activation and impairment of naive T-cell priming. Br. J. Cancer 2005, 92, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, C.E.; Gasparoto, T.H.; Pinheiro, C.R.; Garlet, G.P.; Lara, V.S.; Campanelli, A.P. CCR5-Dependent Homing of T Regulatory Cells to the Tumor Microenvironment Contributes to Skin Squamous Cell Carcinoma Development. Mol. Cancer Ther. 2017, 16, 2871–2881. [Google Scholar] [CrossRef]
- Cinier, J.; Hubert, M.; Besson, L.; di Roio, A.; Rodriguez, C.; Lombardi, V.; Caux, C.; Ménétrier-Caux, C. Recruitment and Expansion of Tregs Cells in the Tumor Environment-How to Target Them? Cancers 2021, 13, 1850. [Google Scholar] [CrossRef]
- Hemmatazad, H.; Berger, M.D. CCR5 is a potential therapeutic target for cancer. Expert Opin. Ther. Targets 2021, 25, 311–327. [Google Scholar] [CrossRef] [PubMed]
- Davern, M.; Gaughan, C.; O’Connell, F.; Moran, B.; Mylod, E.; Sheppard, A.D.; Ramjit, S.; Kung, J.Y.-T.; Phelan, J.J.; Davey, M.G.; et al. PD-1 blockade attenuates surgery-mediated immunosuppression and boosts Th1 immunity perioperatively in oesophagogastric junctional adenocarcinoma. Front. Immunol. 2023, 14, 1150754. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Mishra, M.K.; Eltoum, I.E.A.; Bae, S.; Lillard, J.W.; Singh, R. CCR5/CCL5 axis interaction promotes migratory and invasiveness of pancreatic cancer cells. Sci. Rep. 2018, 8, 1323. [Google Scholar] [CrossRef] [PubMed]
- Casagrande, N.; Borghese, C.; Visser, L.; Mongiat, M.; Colombatti, A.; Aldinucci, D. CCR5 antagonism by maraviroc inhibits hodgkin lymphoma microenvironment interactions and xenograft growth. Haematologica 2019, 104, 564. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Cohort for Blood Samples | Cancer Cohort for Tumour Samples | Healthy Donor Cohort for Blood Samples | |
|---|---|---|---|
| Patient Demographic Table | n = 9 | n = 6 | n = 6 |
| Age (years) | (51–75) 63.2 | (48–75) 61.0 | (55–61) 57.8 |
| Sex ratio (M:F) | 7:2 | 5:1 | 5:1 |
| Diagnosis (no. patients) | OAC (n = 9) | OAC (n = 6) | Non-cancer (n = 6) |
| Clinical tumour stage (no. patients) | |||
| T0 | 0 | 0 | |
| T1 | 1 | 0 | |
| T2 | 2 | 2 | |
| T3 | 6 | 4 | |
| T4 | 0 | 0 | |
| Clinical nodal status (no. patients) | |||
| Negative | 4 | 2 | |
| Positive | 5 | 4 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davern, M.; O’ Donovan, C.; Donlon, N.E.; Mylod, E.; Gaughan, C.; Bhardwaj, A.; Sheppard, A.D.; Bracken-Clarke, D.; Butler, C.; Ravi, N.; et al. Analysing the Combined Effects of Radiotherapy and Chemokine Receptor 5 Antagonism: Complementary Approaches to Promote T Cell Function and Migration in Oesophageal Adenocarcinoma. Biomedicines 2024, 12, 819. https://doi.org/10.3390/biomedicines12040819
Davern M, O’ Donovan C, Donlon NE, Mylod E, Gaughan C, Bhardwaj A, Sheppard AD, Bracken-Clarke D, Butler C, Ravi N, et al. Analysing the Combined Effects of Radiotherapy and Chemokine Receptor 5 Antagonism: Complementary Approaches to Promote T Cell Function and Migration in Oesophageal Adenocarcinoma. Biomedicines. 2024; 12(4):819. https://doi.org/10.3390/biomedicines12040819
Chicago/Turabian StyleDavern, Maria, Cillian O’ Donovan, Noel E. Donlon, Eimear Mylod, Caoimhe Gaughan, Anshul Bhardwaj, Andrew D. Sheppard, Dara Bracken-Clarke, Christine Butler, Narayanasamy Ravi, and et al. 2024. "Analysing the Combined Effects of Radiotherapy and Chemokine Receptor 5 Antagonism: Complementary Approaches to Promote T Cell Function and Migration in Oesophageal Adenocarcinoma" Biomedicines 12, no. 4: 819. https://doi.org/10.3390/biomedicines12040819
APA StyleDavern, M., O’ Donovan, C., Donlon, N. E., Mylod, E., Gaughan, C., Bhardwaj, A., Sheppard, A. D., Bracken-Clarke, D., Butler, C., Ravi, N., Donohoe, C. L., Reynolds, J. V., Lysaght, J., & Conroy, M. J. (2024). Analysing the Combined Effects of Radiotherapy and Chemokine Receptor 5 Antagonism: Complementary Approaches to Promote T Cell Function and Migration in Oesophageal Adenocarcinoma. Biomedicines, 12(4), 819. https://doi.org/10.3390/biomedicines12040819