
Decoding Tumor Angiogenesis for Therapeutic Advancements: Mechanistic Insights


Abstract
:1. Introduction
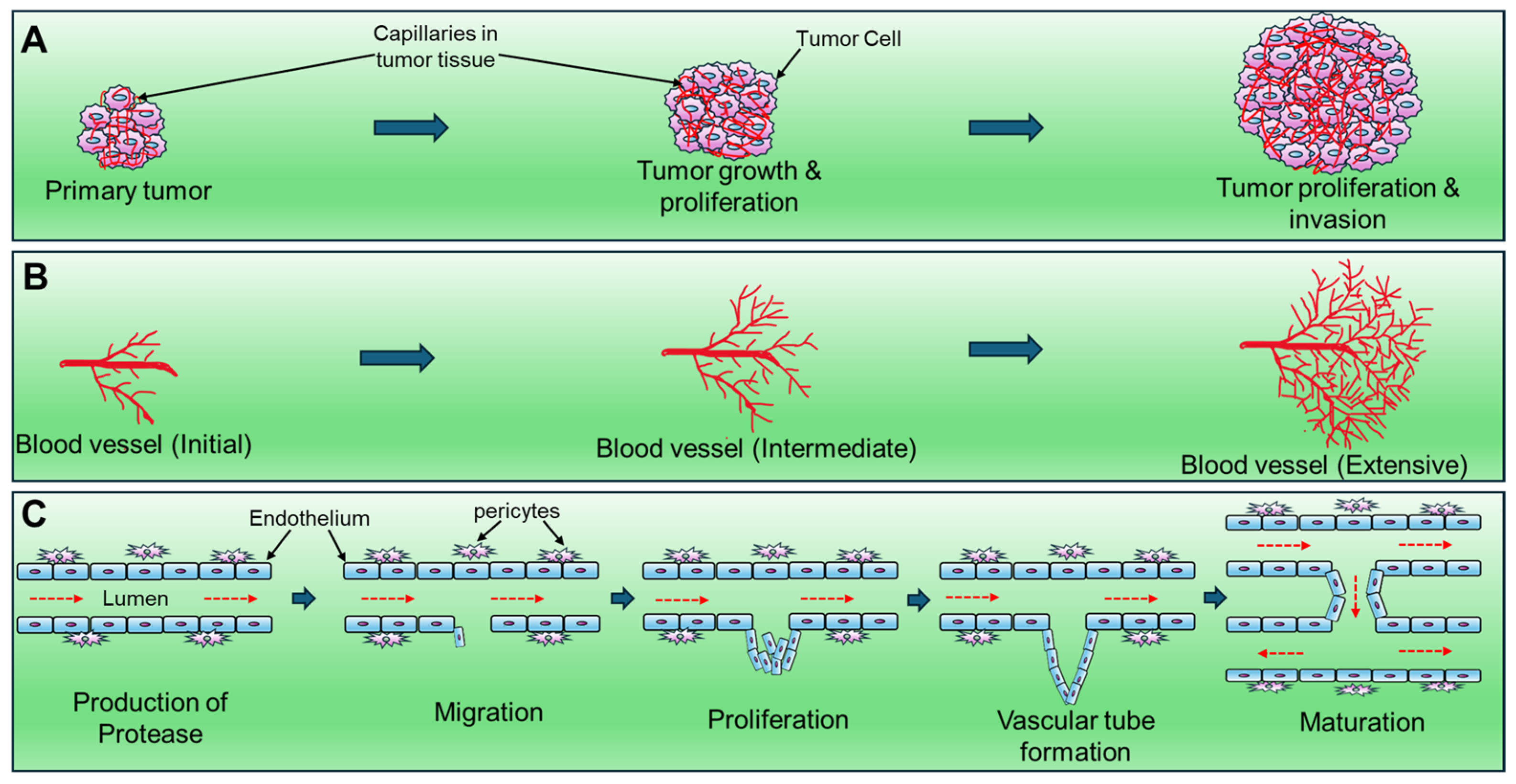
2. Possible Molecular Mechanisms of Tumor Angiogenesis: Angiogenic Switch
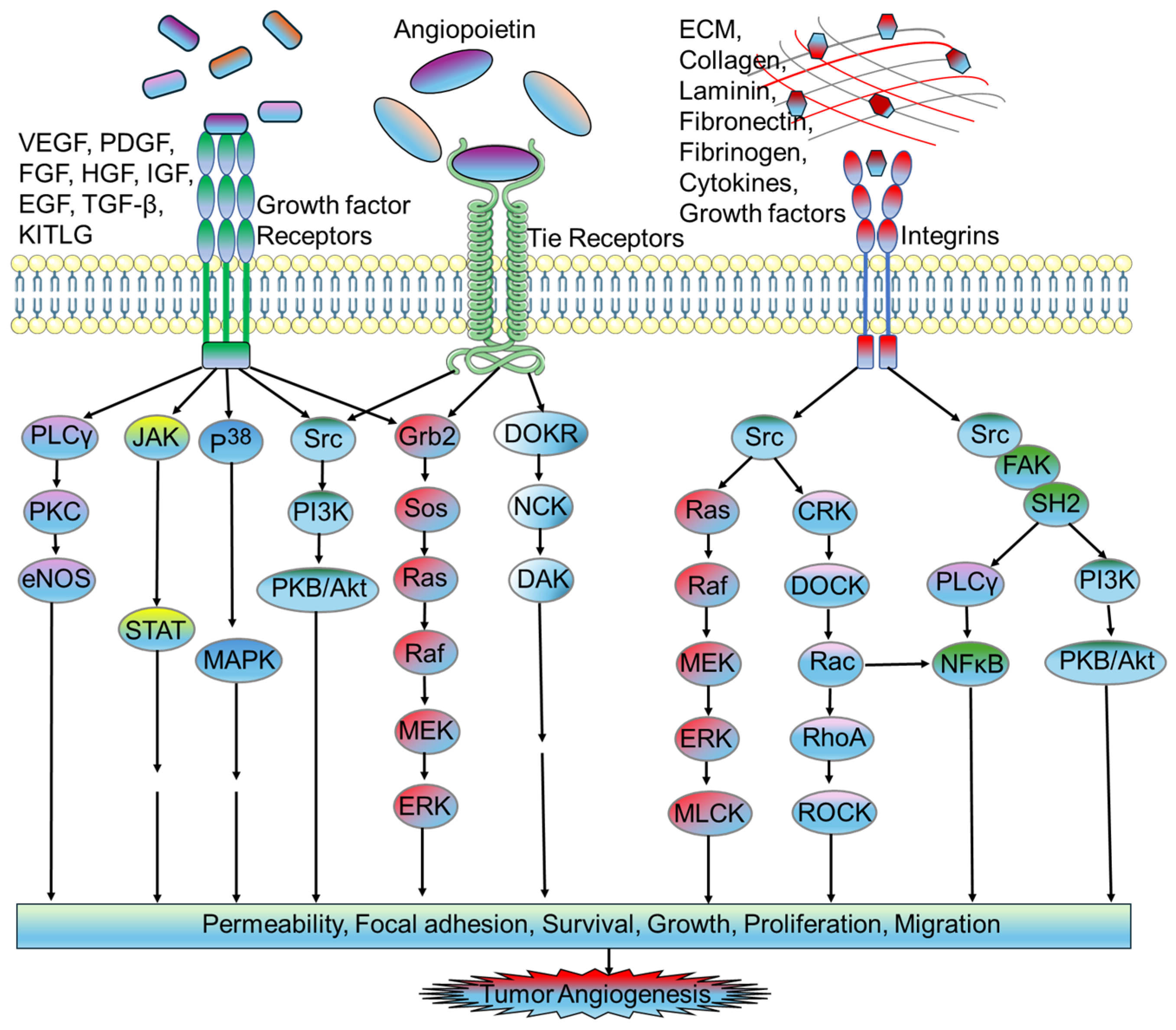
2.1. Proangiogenic Factors in Tumor Microvasculature
2.1.1. Vascular Endothelial Growth Factor (VEGF)
2.1.2. Fibroblast Growth Factor (FGF)
2.1.3. Platelet-Derived Growth Factor (PDGF)
2.1.4. Angiopoietin
2.1.5. Matrix Metalloproteases (MMPs)
2.1.6. Interleukins
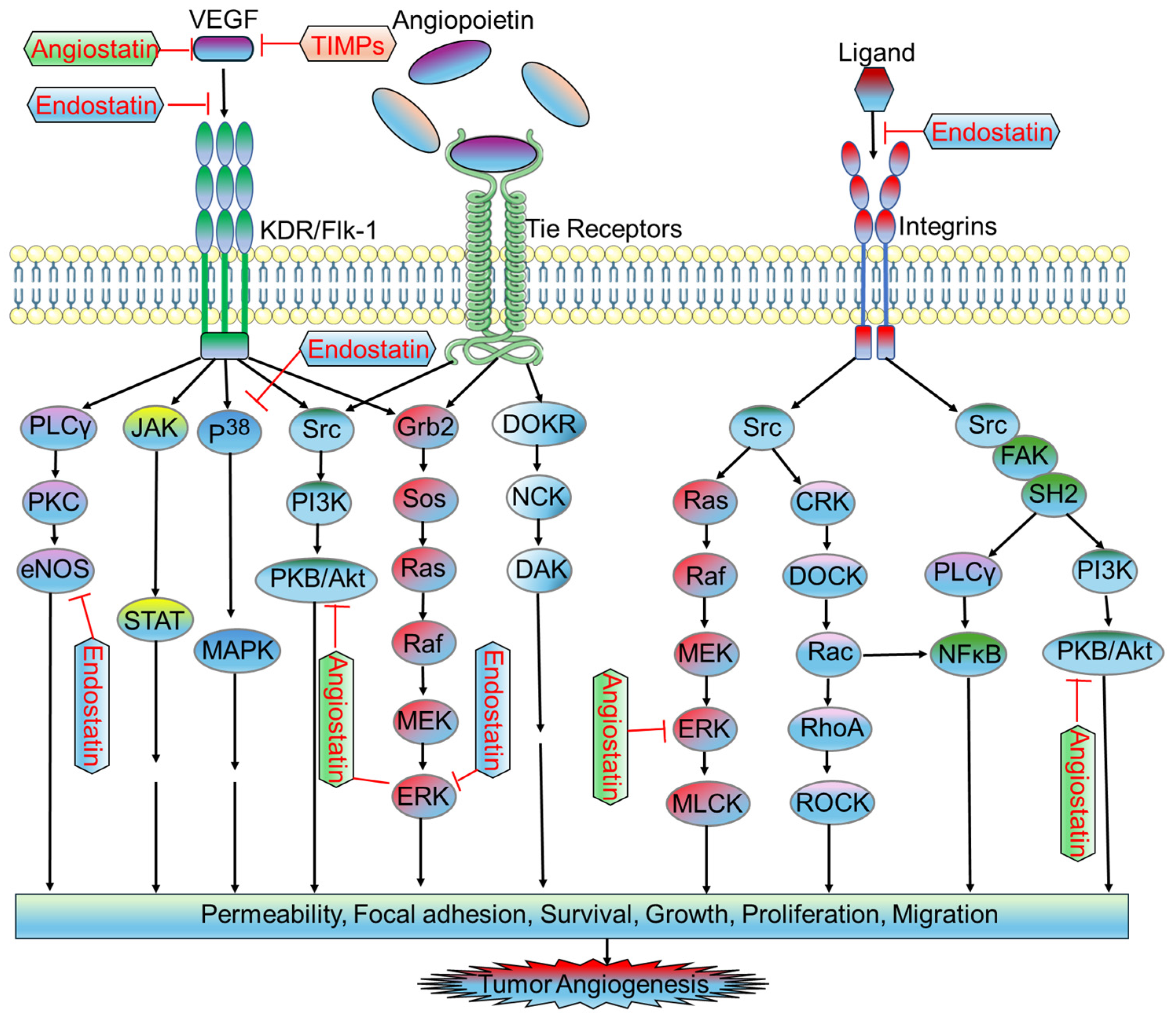
2.2. Antiangiogenic Factors in Tumor Microvasculature
2.2.1. Endostatin
2.2.2. Angiostatin
2.2.3. Tissue Inhibitors of Metalloproteinases (TIMPs)
2.2.4. Interferons and Interleukins
3. Antiangiogenic Therapies
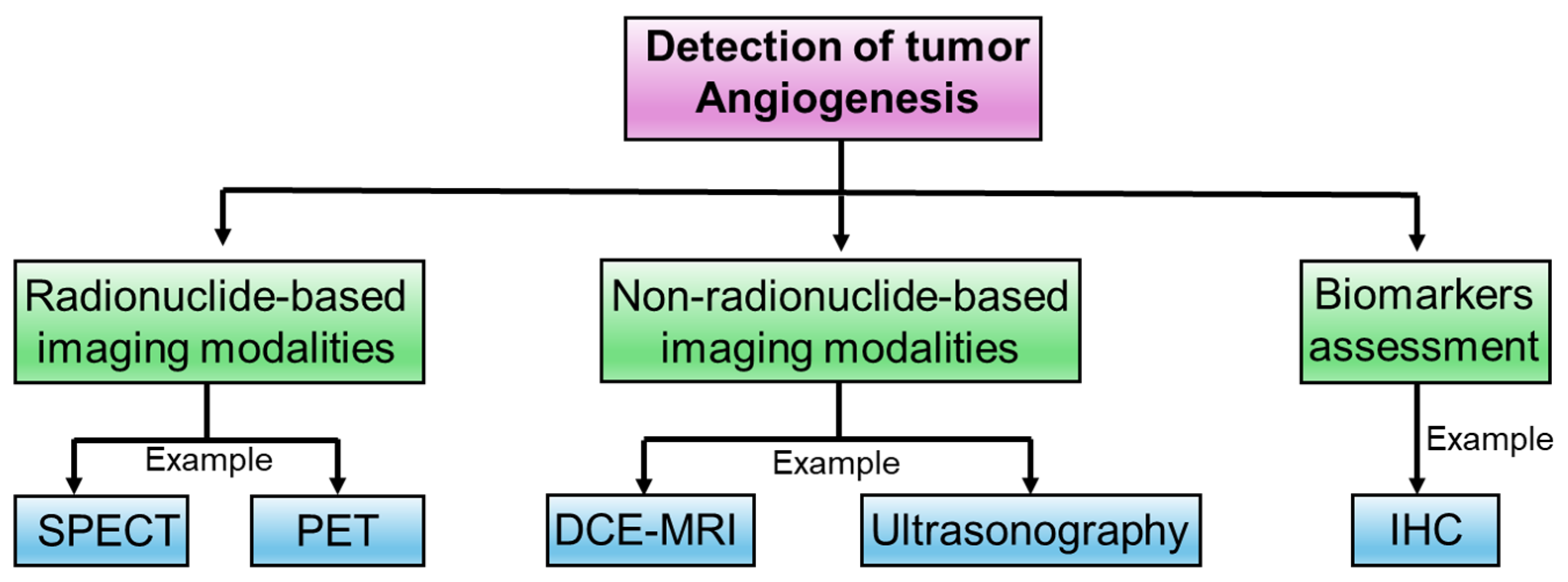
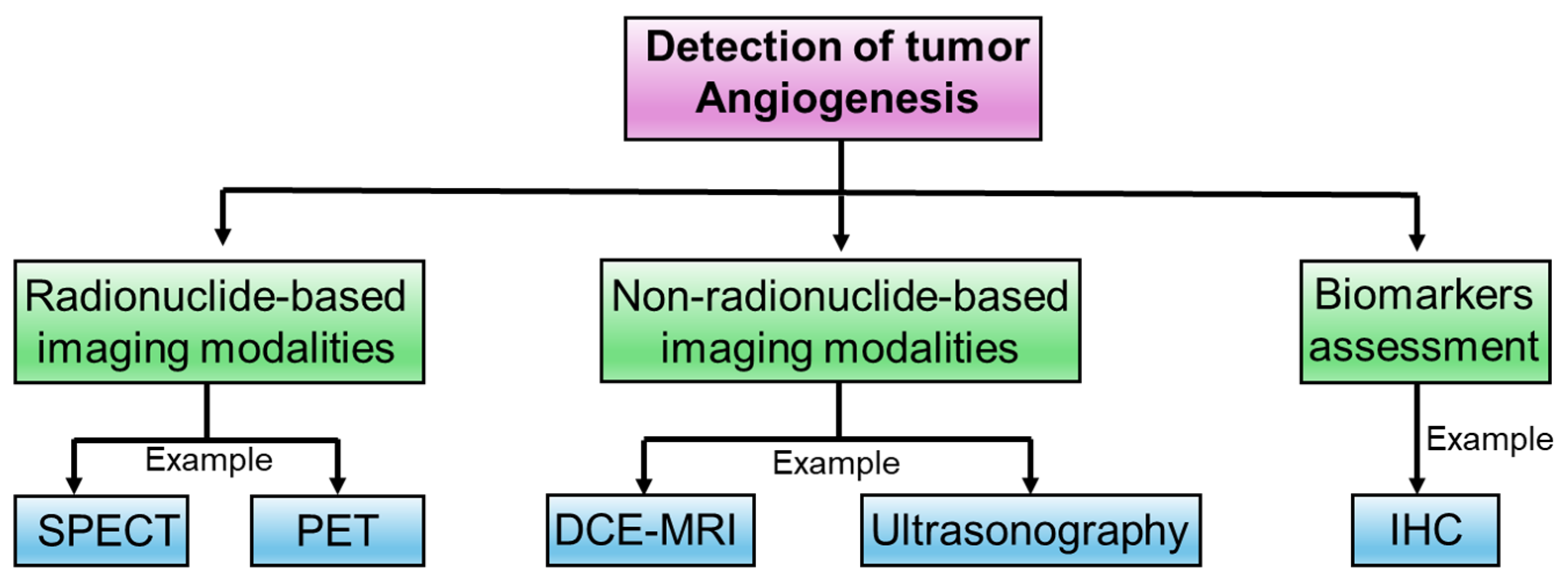
4. Experimental Approaches to Determine Intricacies in Tumor Angiogenesis
4.1. Radionuclide-Based Imaging Modalities
4.2. Non-Radionuclide-Based Imaging Modalities
4.3. Biomarkers Assessment
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Adair, T.H.; Montani, J.P. Integrated Systems Physiology: From Molecule to Function to Disease. In Angiogenesis; Biota Publishing: San Rafael, CA, USA, 2010. [Google Scholar]
- Tahergorabi, Z.; Khazaei, M. A review on angiogenesis and its assays. Iran. J. Basic Med. Sci. 2012, 15, 1110–1126. [Google Scholar] [PubMed]
- Johnson, K.E.; Wilgus, T.A. Vascular Endothelial Growth Factor and Angiogenesis in the Regulation of Cutaneous Wound Repair. Adv. Wound Care 2014, 3, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Watnick, R.S. The role of the tumor microenvironment in regulating angiogenesis. Cold Spring Harb. Perspect. Med. 2012, 2, a006676. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Zhang, S.; Gong, Z.; Li, X.; Cao, K.; Deng, H.; He, Y.; et al. The role of microenvironment in tumor angiogenesis. J. Exp. Clin. Cancer Res. 2020, 39, 204. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Yan, W.; Zhu, L.; Tang, C.; Wang, G. The role of blood flow in vessel remodeling and its regulatory mechanism during developmental angiogenesis. Cell. Mol. Life Sci. 2023, 80, 162. [Google Scholar] [CrossRef] [PubMed]
- Saharinen, P.; Eklund, L.; Alitalo, K. Therapeutic targeting of the angiopoietin-TIE pathway. Nat. Rev. Drug Discov. 2017, 16, 635–661. [Google Scholar] [CrossRef] [PubMed]
- Akwii, R.G.; Mikelis, C.M. Targeting the Angiopoietin/Tie Pathway: Prospects for Treatment of Retinal and Respiratory Disorders. Drugs 2021, 81, 1731–1749. [Google Scholar] [CrossRef] [PubMed]
- Ionescu, C.; Oprea, B.; Ciobanu, G.; Georgescu, M.; Bica, R.; Mateescu, G.O.; Huseynova, F.; Barragan-Montero, V. The Angiogenic Balance and Its Implications in Cancer and Cardiovascular Diseases: An Overview. Medicina 2022, 58, 903. [Google Scholar] [CrossRef] [PubMed]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef]
- Quintero-Fabian, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argaez, V.; Lara-Riegos, J.; Ramirez-Camacho, M.A.; Alvarez-Sanchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370. [Google Scholar] [CrossRef]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuna, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. 2005, 7, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Kong, X.; Qiu, X.; Huang, C.; Wong, P.P. The Emerging Roles of Pericytes in Modulating Tumor Microenvironment. Front. Cell Dev. Biol. 2021, 9, 676342. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Wagner, N.; Wagner, K.D. The Emerging Role of PPAR Beta/Delta in Tumor Angiogenesis. PPAR Res. 2020, 2020, 3608315. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.; Liu, Y.; Xie, C.; Xue, C.; Du, S.; Yao, J. Emerging role of interactions between tumor angiogenesis and cancer stem cells. J. Control. Release 2023, 360, 468–481. [Google Scholar] [CrossRef]
- Folkman, J.; Kalluri, R. Beginning of Angiogenesis Research. In Holland-Frei Cancer Medicine, 6th ed.; Kufe, D.W., Pollock, R.E., Weichselbaum, R.R., Bast, R.C., Jr., Gansler, T.S., Holland, J.F., Frei, E., III, Eds.; BC Decker: Hamilton, ON, USA, 2003. [Google Scholar]
- Ribatti, D. Judah Folkman, a pioneer in the study of angiogenesis. Angiogenesis 2008, 11, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Arbiser, J.; D’Amato, R.J.; D’Amore, P.A.; Ingber, D.E.; Kerbel, R.; Klagsbrun, M.; Lim, S.; Moses, M.A.; Zetter, B.; et al. Forty-year journey of angiogenesis translational research. Sci. Transl. Med. 2011, 3, 114rv113. [Google Scholar] [CrossRef]
- Ribatti, D.; Nico, B. Tumor angiogenesis. From bench to bedside. Ital. J. Anat. Embryol. 2016, 121, 12–19. [Google Scholar]
- Ribatti, D.; Pezzella, F. Overview on the Different Patterns of Tumor Vascularization. Cells 2021, 10, 639. [Google Scholar] [CrossRef]
- Karamysheva, A.F. Mechanisms of angiogenesis. Biochemistry 2008, 73, 751–762. [Google Scholar] [CrossRef]
- Luo, Q.; Wang, J.; Zhao, W.; Peng, Z.; Liu, X.; Li, B.; Zhang, H.; Shan, B.; Zhang, C.; Duan, C. Vasculogenic mimicry in carcinogenesis and clinical applications. J. Hematol. Oncol. 2020, 13, 19. [Google Scholar] [CrossRef]
- Morales-Guadarrama, G.; Garcia-Becerra, R.; Mendez-Perez, E.A.; Garcia-Quiroz, J.; Avila, E.; Diaz, L. Vasculogenic Mimicry in Breast Cancer: Clinical Relevance and Drivers. Cells 2021, 10, 1758. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Li, Z.; Wang, W.; Wu, J.; Li, J.; Huang, X.; Zhang, X.; Ye, X. Vasculogenic mimicry score identifies the prognosis and immune landscape of lung adenocarcinoma. Front. Genet. 2023, 14, 1206141. [Google Scholar] [CrossRef] [PubMed]
- Hida, K.; Maishi, N.; Takeda, R.; Hida, Y. The Roles of Tumor Endothelial Cells in Cancer Metastasis. In Metastasis; Sergi, C.M., Ed.; Exon Publications: Brisbane, CA, USA, 2022. [Google Scholar]
- Yao, X.; Zeng, Y. Tumour associated endothelial cells: Origin, characteristics and role in metastasis and anti-angiogenic resistance. Front. Physiol. 2023, 14, 1199225. [Google Scholar] [CrossRef] [PubMed]
- Schito, L.; Rey, S. Hypoxia: Turning vessels into vassals of cancer immunotolerance. Cancer Lett. 2020, 487, 74–84. [Google Scholar] [CrossRef]
- Zheng, W.; Qian, C.; Tang, Y.; Yang, C.; Zhou, Y.; Shen, P.; Chen, W.; Yu, S.; Wei, Z.; Wang, A.; et al. Manipulation of the crosstalk between tumor angiogenesis and immunosuppression in the tumor microenvironment: Insight into the combination therapy of anti-angiogenesis and immune checkpoint blockade. Front. Immunol. 2022, 13, 1035323. [Google Scholar] [CrossRef]
- Madu, C.O.; Wang, S.; Madu, C.O.; Lu, Y. Angiogenesis in Breast Cancer Progression, Diagnosis, and Treatment. J. Cancer 2020, 11, 4474–4494. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.L.; Chen, H.H.; Zheng, L.L.; Sun, L.P.; Shi, L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduct. Target. Ther. 2023, 8, 198. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.K.; Qin, R.Y. Mechanism and its regulation of tumor-induced angiogenesis. World J. Gastroenterol. 2003, 9, 1144–1155. [Google Scholar] [CrossRef]
- Aspritoiu, V.M.; Stoica, I.; Bleotu, C.; Diaconu, C.C. Epigenetic Regulation of Angiogenesis in Development and Tumors Progression: Potential Implications for Cancer Treatment. Front. Cell Dev. Biol. 2021, 9, 689962. [Google Scholar] [CrossRef]
- Ateeq, M.; Broadwin, M.; Sellke, F.W.; Abid, M.R. Extracellular Vesicles’ Role in Angiogenesis and Altering Angiogenic Signaling. Med. Sci. 2024, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Nowak-Sliwinska, P.; Alitalo, K.; Allen, E.; Anisimov, A.; Aplin, A.C.; Auerbach, R.; Augustin, H.G.; Bates, D.O.; van Beijnum, J.R.; Bender, R.H.F.; et al. Consensus guidelines for the use and interpretation of angiogenesis assays. Angiogenesis 2018, 21, 425–532. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Trivedi, R.; Lin, S.Y. Tumor microenvironment: Barrier or opportunity towards effective cancer therapy. J. Biomed. Sci. 2022, 29, 83. [Google Scholar] [CrossRef] [PubMed]
- Benekli, M.; Baumann, H.; Wetzler, M. Targeting signal transducer and activator of transcription signaling pathway in leukemias. J. Clin. Oncol. 2009, 27, 4422–4432. [Google Scholar] [CrossRef] [PubMed]
- Bafico, A.; Aaronson, S.A. Signaling Pathways of Tyrosine Kinase Receptors. In Holland-Frei Cancer Medicine; Kufe, D.W., Pollock, R.E., Weichselbaum, R.R., Bast, R.C., Jr., Gansler, T.S., Holland, J.F., Frei, E., III, Eds.; BC Decker: Hamilton, ON, USA, 2003. [Google Scholar]
- Saman, H.; Raza, S.S.; Uddin, S.; Rasul, K. Inducing Angiogenesis, a Key Step in Cancer Vascularization, and Treatment Approaches. Cancers 2020, 12, 1172. [Google Scholar] [CrossRef]
- Langlois, B.; Saupe, F.; Rupp, T.; Arnold, C.; van der Heyden, M.; Orend, G.; Hussenet, T. AngioMatrix, a signature of the tumor angiogenic switch-specific matrisome, correlates with poor prognosis for glioma and colorectal cancer patients. Oncotarget 2014, 5, 10529–10545. [Google Scholar] [CrossRef]
- D’Andrea, M.R.; Cereda, V.; Coppola, L.; Giordano, G.; Remo, A.; De Santis, E. Propensity for Early Metastatic Spread in Breast Cancer: Role of Tumor Vascularization Features and Tumor Immune Infiltrate. Cancers 2021, 13, 5917. [Google Scholar] [CrossRef]
- Jeong, J.H.; Ojha, U.; Lee, Y.M. Pathological angiogenesis and inflammation in tissues. Arch. Pharm. Res. 2021, 44, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Peach, C.J.; Mignone, V.W.; Arruda, M.A.; Alcobia, D.C.; Hill, S.J.; Kilpatrick, L.E.; Woolard, J. Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. [Google Scholar] [CrossRef]
- Yadav, L.; Puri, N.; Rastogi, V.; Satpute, P.; Sharma, V. Tumour Angiogenesis and Angiogenic Inhibitors: A Review. J. Clin. Diagn. Res. 2015, 9, XE01–XE05. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. VEGFR and type-V RTK activation and signaling. Cold Spring Harb. Perspect. Biol. 2013, 5, a009092. [Google Scholar] [CrossRef] [PubMed]
- Ceci, C.; Atzori, M.G.; Lacal, P.M.; Graziani, G. Role of VEGFs/VEGFR-1 Signaling and its Inhibition in Modulating Tumor Invasion: Experimental Evidence in Different Metastatic Cancer Models. Int. J. Mol. Sci. 2020, 21, 1388. [Google Scholar] [CrossRef] [PubMed]
- Ziello, J.E.; Jovin, I.S.; Huang, Y. Hypoxia-Inducible Factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J. Biol. Med. 2007, 80, 51–60. [Google Scholar]
- Rodriguez, D.; Watts, D.; Gaete, D.; Sormendi, S.; Wielockx, B. Hypoxia Pathway Proteins and Their Impact on the Blood Vasculature. Int. J. Mol. Sci. 2021, 22, 9191. [Google Scholar] [CrossRef]
- Wu, M.H.; Yuan, S.Y.; Granger, H.J. The protein kinase MEK1/2 mediate vascular endothelial growth factor- and histamine-induced hyperpermeability in porcine coronary venules. J. Physiol. 2005, 563, 95–104. [Google Scholar] [CrossRef]
- Wang, X.; Bove, A.M.; Simone, G.; Ma, B. Molecular Bases of VEGFR-2-Mediated Physiological Function and Pathological Role. Front. Cell Dev. Biol. 2020, 8, 599281. [Google Scholar] [CrossRef] [PubMed]
- Suehiro, J.; Kanki, Y.; Makihara, C.; Schadler, K.; Miura, M.; Manabe, Y.; Aburatani, H.; Kodama, T.; Minami, T. Genome-wide approaches reveal functional vascular endothelial growth factor (VEGF)-inducible nuclear factor of activated T cells (NFAT) c1 binding to angiogenesis-related genes in the endothelium. J. Biol. Chem. 2014, 289, 29044–29059. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, N.; Mihalcioiu, C.; Rabbani, S.A. Multifaceted Role of the Urokinase-Type Plasminogen Activator (uPA) and Its Receptor (uPAR): Diagnostic, Prognostic, and Therapeutic Applications. Front. Oncol. 2018, 8, 24. [Google Scholar] [CrossRef]
- Song, Y.Y.; Liang, D.; Liu, D.K.; Lin, L.; Zhang, L.; Yang, W.Q. The role of the ERK signaling pathway in promoting angiogenesis for treating ischemic diseases. Front. Cell Dev. Biol. 2023, 11, 1164166. [Google Scholar] [CrossRef]
- Ntellas, P.; Mavroeidis, L.; Gkoura, S.; Gazouli, I.; Amylidi, A.L.; Papadaki, A.; Zarkavelis, G.; Mauri, D.; Karpathiou, G.; Kolettas, E.; et al. Old Player-New Tricks: Non Angiogenic Effects of the VEGF/VEGFR Pathway in Cancer. Cancers 2020, 12, 3145. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Zhou, Z.; Chen, Z.; Xu, G.; Chen, Y. Fibroblast Growth Factor Receptors (FGFRs): Structures and Small Molecule Inhibitors. Cells 2019, 8, 614. [Google Scholar] [CrossRef] [PubMed]
- Zahra, F.T.; Sajib, M.S.; Mikelis, C.M. Role of bFGF in Acquired Resistance upon Anti-VEGF Therapy in Cancer. Cancers 2021, 13, 1422. [Google Scholar] [CrossRef] [PubMed]
- Khodabakhsh, F.; Merikhian, P.; Eisavand, M.R.; Farahmand, L. Crosstalk between MUC1 and VEGF in angiogenesis and metastasis: A review highlighting roles of the MUC1 with an emphasis on metastatic and angiogenic signaling. Cancer Cell Int. 2021, 21, 200. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Bao, S.D. Roles of main pro- and anti-angiogenic factors in tumor angiogenesis. World J. Gastroenterol. 2004, 10, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Li, Y.; Zhang, S.; Wang, X.; Dou, H.; Yu, X.; Zhang, Z.; Yang, S.; Xiao, M. Extracellular matrix remodeling in tumor progression and immune escape: From mechanisms to treatments. Mol. Cancer 2023, 22, 48. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed]
- Mohan, C.D.; Hari, S.; Preetham, H.D.; Rangappa, S.; Barash, U.; Ilan, N.; Nayak, S.C.; Gupta, V.K.; Basappa; Vlodavsky, I.; et al. Targeting Heparanase in Cancer: Inhibition by Synthetic, Chemically Modified, and Natural Compounds. iScience 2019, 15, 360–390. [Google Scholar] [CrossRef] [PubMed]
- Ohkawara, H.; Ikeda, K.; Ogawa, K.; Takeishi, Y. Membrane Type 1-Matrix Metalloproteinase (Mt1-Mmp) Identified as a Multifunctional Regulator of Vascular Responses. Fukushima J. Med. Sci. 2015, 61, 91–100. [Google Scholar] [CrossRef]
- Yang, X.; Liaw, L.; Prudovsky, I.; Brooks, P.C.; Vary, C.; Oxburgh, L.; Friesel, R. Fibroblast growth factor signaling in the vasculature. Curr. Atheroscler. Rep. 2015, 17, 509. [Google Scholar] [CrossRef]
- Santolla, M.F.; Maggiolini, M. The FGF/FGFR System in Breast Cancer: Oncogenic Features and Therapeutic Perspectives. Cancers 2020, 12, 3029. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ponten, A.; Aase, K.; Karlsson, L.; Abramsson, A.; Uutela, M.; Backstrom, G.; Hellstrom, M.; Bostrom, H.; Li, H.; et al. PDGF-C is a new protease-activated ligand for the PDGF alpha-receptor. Nat. Cell Biol. 2000, 2, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Bergsten, E.; Uutela, M.; Li, X.; Pietras, K.; Ostman, A.; Heldin, C.H.; Alitalo, K.; Eriksson, U. PDGF-D is a specific, protease-activated ligand for the PDGF beta-receptor. Nat. Cell Biol. 2001, 3, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Khan, F.; Upadhyay, T.K.; Seungjoon, M.; Park, M.N.; Kim, B. New insights about the PDGF/PDGFR signaling pathway as a promising target to develop cancer therapeutic strategies. Biomed. Pharmacother. 2023, 161, 114491. [Google Scholar] [CrossRef] [PubMed]
- Uutela, M.; Lauren, J.; Bergsten, E.; Li, X.; Horelli-Kuitunen, N.; Eriksson, U.; Alitalo, K. Chromosomal location, exon structure, and vascular expression patterns of the human PDGFC and PDGFD genes. Circulation 2001, 103, 2242–2247. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed]
- Furuhashi, M.; Sjoblom, T.; Abramsson, A.; Ellingsen, J.; Micke, P.; Li, H.; Bergsten-Folestad, E.; Eriksson, U.; Heuchel, R.; Betsholtz, C.; et al. Platelet-derived growth factor production by B16 melanoma cells leads to increased pericyte abundance in tumors and an associated increase in tumor growth rate. Cancer Res. 2004, 64, 2725–2733. [Google Scholar] [CrossRef] [PubMed]
- Gladh, H.; Folestad, E.B.; Muhl, L.; Ehnman, M.; Tannenberg, P.; Lawrence, A.L.; Betsholtz, C.; Eriksson, U. Mice Lacking Platelet-Derived Growth Factor D Display a Mild Vascular Phenotype. PLoS ONE 2016, 11, e0152276. [Google Scholar] [CrossRef] [PubMed]
- Raica, M.; Cimpean, A.M. Platelet-Derived Growth Factor (PDGF)/PDGF Receptors (PDGFR) Axis as Target for Antitumor and Antiangiogenic Therapy. Pharmaceuticals 2010, 3, 572–599. [Google Scholar] [CrossRef]
- Thomas, M.; Augustin, H.G. The role of the Angiopoietins in vascular morphogenesis. Angiogenesis 2009, 12, 125–137. [Google Scholar] [CrossRef]
- Yu, X.; Ye, F. Role of Angiopoietins in Development of Cancer and Neoplasia Associated with Viral Infection. Cells 2020, 9, 457. [Google Scholar] [CrossRef] [PubMed]
- Marcola, M.; Rodrigues, C.E. Endothelial progenitor cells in tumor angiogenesis: Another brick in the wall. Stem Cells Int. 2015, 2015, 832649. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Li, S.; Zhang, Z.; Wen, X.; Quan, W.; Tian, Q.; Gao, C.; Su, W.; Zhang, J.; Jiang, R. Progesterone-mediated angiogenic activity of endothelial progenitor cell and angiogenesis in traumatic brain injury rats were antagonized by progesterone receptor antagonist. Cell Prolif. 2017, 50, e12362. [Google Scholar] [CrossRef] [PubMed]
- Lobov, I.B.; Brooks, P.C.; Lang, R.A. Angiopoietin-2 displays VEGF-dependent modulation of capillary structure and endothelial cell survival in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 11205–11210. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Kim, H.G.; So, J.N.; Kim, J.H.; Kwak, H.J.; Koh, G.Y. Angiopoietin-1 regulates endothelial cell survival through the phosphatidylinositol 3’-Kinase/Akt signal transduction pathway. Circ. Res. 2000, 86, 24–29. [Google Scholar] [CrossRef]
- Babaei, S.; Teichert-Kuliszewska, K.; Zhang, Q.; Jones, N.; Dumont, D.J.; Stewart, D.J. Angiogenic actions of angiopoietin-1 require endothelium-derived nitric oxide. Am. J. Pathol. 2003, 162, 1927–1936. [Google Scholar] [CrossRef] [PubMed]
- Moliere, S.; Jaulin, A.; Tomasetto, C.L.; Dali-Youcef, N. Roles of Matrix Metalloproteinases and Their Natural Inhibitors in Metabolism: Insights into Health and Disease. Int. J. Mol. Sci. 2023, 24, 649. [Google Scholar] [CrossRef]
- Wang, N.; Jain, R.K.; Batchelor, T.T. New Directions in Anti-Angiogenic Therapy for Glioblastoma. Neurotherapeutics 2017, 14, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.E.; Kirkpatrick, N.D.; Huang, Y.; Farrar, C.T.; Marijt, K.A.; Kloepper, J.; Datta, M.; Amoozgar, Z.; Seano, G.; Jung, K.; et al. Dual inhibition of Ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, 4470–4475. [Google Scholar] [CrossRef]
- Qi, S.; Deng, S.; Lian, Z.; Yu, K. Novel Drugs with High Efficacy against Tumor Angiogenesis. Int. J. Mol. Sci. 2022, 23, 6934. [Google Scholar] [CrossRef]
- Lian, G.Y.; Wang, Q.M.; Mak, T.S.; Huang, X.R.; Yu, X.Q.; Lan, H.Y. Inhibition of tumor invasion and metastasis by targeting TGF-beta-Smad-MMP2 pathway with Asiatic acid and Naringenin. Mol. Ther. Oncolytics 2021, 20, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wei, P.K. Interleukin-8: A potent promoter of angiogenesis in gastric cancer. Oncol. Lett. 2016, 11, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef] [PubMed]
- Niland, S.; Riscanevo, A.X.; Eble, J.A. Matrix Metalloproteinases Shape the Tumor Microenvironment in Cancer Progression. Int. J. Mol. Sci. 2021, 23, 146. [Google Scholar] [CrossRef] [PubMed]
- Geindreau, M.; Bruchard, M.; Vegran, F. Role of Cytokines and Chemokines in Angiogenesis in a Tumor Context. Cancers 2022, 14, 2446. [Google Scholar] [CrossRef]
- Cohen, T.; Nahari, D.; Cerem, L.W.; Neufeld, G.; Levi, B.Z. Interleukin 6 induces the expression of vascular endothelial growth factor. J. Biol. Chem. 1996, 271, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Bellail, A.C.; Van Meir, E.G. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro Oncol. 2005, 7, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.L.; Failla, C.M. Interleukin role in the regulation of endothelial cell pathological activation. Vasc. Biol. 2021, 3, R96–R105. [Google Scholar] [CrossRef]
- Yang, B.; Kang, H.; Fung, A.; Zhao, H.; Wang, T.; Ma, D. The role of interleukin 17 in tumour proliferation, angiogenesis, and metastasis. Mediat. Inflamm. 2014, 2014, 623759. [Google Scholar] [CrossRef]
- Schinocca, C.; Rizzo, C.; Fasano, S.; Grasso, G.; La Barbera, L.; Ciccia, F.; Guggino, G. Role of the IL-23/IL-17 Pathway in Rheumatic Diseases: An Overview. Front. Immunol. 2021, 12, 637829. [Google Scholar] [CrossRef] [PubMed]
- Pastor-Fernandez, G.; Mariblanca, I.R.; Navarro, M.N. Decoding IL-23 Signaling Cascade for New Therapeutic Opportunities. Cells 2020, 9, 2044. [Google Scholar] [CrossRef] [PubMed]
- Walia, A.; Yang, J.F.; Huang, Y.H.; Rosenblatt, M.I.; Chang, J.H.; Azar, D.T. Endostatin’s emerging roles in angiogenesis, lymphangiogenesis, disease, and clinical applications. Biochim. Biophys. Acta 2015, 1850, 2422–2438. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.H.; Dong, X.S.; Wang, X.S. Effects of endostatin on expression of vascular endothelial growth factor and its receptors and neovascularization in colonic carcinoma implanted in nude mice. World J. Gastroenterol. 2004, 10, 3361–3364. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhang, Y.; Zhong, Y.; Ye, Y.; Hu, X.; Gu, L.; Xiong, X. Inflammation-Mediated Angiogenesis in Ischemic Stroke. Front. Cell Neurosci. 2021, 15, 652647. [Google Scholar] [CrossRef] [PubMed]
- Rehn, M.; Veikkola, T.; Kukk-Valdre, E.; Nakamura, H.; Ilmonen, M.; Lombardo, C.; Pihlajaniemi, T.; Alitalo, K.; Vuori, K. Interaction of endostatin with integrins implicated in angiogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 1024–1029. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, P.; Heikkila, P.; Sorsa, T.; Luostarinen, J.; Heljasvaara, R.; Stenman, U.H.; Pihlajaniemi, T.; Salo, T. Endostatin inhibits human tongue carcinoma cell invasion and intravasation and blocks the activation of matrix metalloprotease-2, -9, and -13. J. Biol. Chem. 2003, 278, 22404–22411. [Google Scholar] [CrossRef]
- Mohajeri, A.; Sanaei, S.; Kiafar, F.; Fattahi, A.; Khalili, M.; Zarghami, N. The Challenges of Recombinant Endostatin in Clinical Application: Focus on the Different Expression Systems and Molecular Bioengineering. Adv. Pharm. Bull. 2017, 7, 21–34. [Google Scholar] [CrossRef]
- Li, T.; Kang, G.; Wang, T.; Huang, H. Tumor angiogenesis and anti-angiogenic gene therapy for cancer. Oncol. Lett. 2018, 16, 687–702. [Google Scholar] [CrossRef]
- Wang, T.; Ma, F.; Qian, H.L. Defueling the cancer: ATP synthase as an emerging target in cancer therapy. Mol. Ther. Oncolytics 2021, 23, 82–95. [Google Scholar] [CrossRef]
- Wajih, N.; Sane, D.C. Angiostatin selectively inhibits signaling by hepatocyte growth factor in endothelial and smooth muscle cells. Blood 2003, 101, 1857–1863. [Google Scholar] [CrossRef]
- Usatyuk, P.V.; Fu, P.; Mohan, V.; Epshtein, Y.; Jacobson, J.R.; Gomez-Cambronero, J.; Wary, K.K.; Bindokas, V.; Dudek, S.M.; Salgia, R.; et al. Role of c-Met/phosphatidylinositol 3-kinase (PI3k)/Akt signaling in hepatocyte growth factor (HGF)-mediated lamellipodia formation, reactive oxygen species (ROS) generation, and motility of lung endothelial cells. J. Biol. Chem. 2014, 289, 13476–13491. [Google Scholar] [CrossRef] [PubMed]
- Simon-Gracia, L.; Hunt, H.; Teesalu, T. Peritoneal Carcinomatosis Targeting with Tumor Homing Peptides. Molecules 2018, 23, 1190. [Google Scholar] [CrossRef] [PubMed]
- Wahl, M.L.; Moser, T.L.; Pizzo, S.V. Angiostatin and anti-angiogenic therapy in human disease. Recent. Prog. Horm. Res. 2004, 59, 73–104. [Google Scholar] [CrossRef]
- Burwick, N.R.; Wahl, M.L.; Fang, J.; Zhong, Z.; Moser, T.L.; Li, B.; Capaldi, R.A.; Kenan, D.J.; Pizzo, S.V. An Inhibitor of the F1 subunit of ATP synthase (IF1) modulates the activity of angiostatin on the endothelial cell surface. J. Biol. Chem. 2005, 280, 1740–1745. [Google Scholar] [CrossRef]
- Gupta, N.; Nodzenski, E.; Khodarev, N.N.; Yu, J.; Khorasani, L.; Beckett, M.A.; Kufe, D.W.; Weichselbaum, R.R. Angiostatin effects on endothelial cells mediated by ceramide and RhoA. EMBO Rep. 2001, 2, 536–540. [Google Scholar] [CrossRef]
- Bourboulia, D.; Jensen-Taubman, S.; Rittler, M.R.; Han, H.Y.; Chatterjee, T.; Wei, B.; Stetler-Stevenson, W.G. Endogenous angiogenesis inhibitor blocks tumor growth via direct and indirect effects on tumor microenvironment. Am. J. Pathol. 2011, 179, 2589–2600. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Stetler-Stevenson, W.G. Overexpression of tissue inhibitors of metalloproteinase 2 up-regulates NF-kappaB activity in melanoma cells. J. Mol. Signal 2009, 4, 4. [Google Scholar] [CrossRef]
- Qi, J.H.; Anand-Apte, B. Tissue inhibitor of metalloproteinase-3 (TIMP3) promotes endothelial apoptosis via a caspase-independent mechanism. Apoptosis 2015, 20, 523–534. [Google Scholar] [CrossRef]
- English, W.R.; Ireland-Zecchini, H.; Baker, A.H.; Littlewood, T.D.; Bennett, M.R.; Murphy, G. Tissue Inhibitor of Metalloproteinase-3 (TIMP-3) induces FAS dependent apoptosis in human vascular smooth muscle cells. PLoS ONE 2018, 13, e0195116. [Google Scholar] [CrossRef]
- Wang, X.; Khalil, R.A. Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv. Pharmacol. 2018, 81, 241–330. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Avila, G.; Sommer, B.; Mendoza-Posada, D.A.; Ramos, C.; Garcia-Hernandez, A.A.; Falfan-Valencia, R. Matrix metalloproteinases participation in the metastatic process and their diagnostic and therapeutic applications in cancer. Crit. Rev. Oncol. Hematol. 2019, 137, 57–83. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Qin, H.; Benveniste, E.N. Transcriptional suppression of matrix metalloproteinase-9 gene expression by IFN-gamma and IFN-beta: Critical role of STAT-1alpha. J. Immunol. 2001, 167, 5150–5159. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, S.; Shinohara, H.; Kanayama, H.O.; Bruns, C.J.; Bucana, C.D.; Ellis, L.M.; Davis, D.W.; Fidler, I.J. Suppression of angiogenesis and therapy of human colon cancer liver metastasis by systemic administration of interferon-alpha. Neoplasia 2001, 3, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Umrath, F.; Cen, W.; Salgado, A.J.; Reinert, S.; Alexander, D. Pre-Conditioning with IFN-gamma and Hypoxia Enhances the Angiogenic Potential of iPSC-Derived MSC Secretome. Cells 2022, 11, 988. [Google Scholar] [CrossRef]
- Ning, Y.; Manegold, P.C.; Hong, Y.K.; Zhang, W.; Pohl, A.; Lurje, G.; Winder, T.; Yang, D.; LaBonte, M.J.; Wilson, P.M.; et al. Interleukin-8 is associated with proliferation, migration, angiogenesis and chemosensitivity in vitro and in vivo in colon cancer cell line models. Int. J. Cancer 2011, 128, 2038–2049. [Google Scholar] [CrossRef]
- Yehya, A.H.S.; Asif, M.; Petersen, S.H.; Subramaniam, A.V.; Kono, K.; Majid, A.; Oon, C.E. Angiogenesis: Managing the Culprits behind Tumorigenesis and Metastasis. Medicina 2018, 54, 8. [Google Scholar] [CrossRef]
- Meadows, K.L.; Hurwitz, H.I. Anti-VEGF therapies in the clinic. Cold Spring Harb. Perspect. Med. 2012, 2, a006577. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef]
- Zeng, Y.; Fu, B.M. Resistance Mechanisms of Anti-angiogenic Therapy and Exosomes-Mediated Revascularization in Cancer. Front. Cell Dev. Biol. 2020, 8, 610661. [Google Scholar] [CrossRef]
- Montemagno, C.; Pages, G. Resistance to Anti-angiogenic Therapies: A Mechanism Depending on the Time of Exposure to the Drugs. Front. Cell Dev. Biol. 2020, 8, 584. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Park, H.J. Single photon emission computed tomography (SPECT) or positron emission tomography (PET) imaging for radiotherapy planning in patients with lung cancer: A meta-analysis. Sci. Rep. 2020, 10, 14864. [Google Scholar] [CrossRef] [PubMed]
- Crisan, G.; Moldovean-Cioroianu, N.S.; Timaru, D.G.; Andries, G.; Cainap, C.; Chis, V. Radiopharmaceuticals for PET and SPECT Imaging: A Literature Review over the Last Decade. Int. J. Mol. Sci. 2022, 23, 5023. [Google Scholar] [CrossRef] [PubMed]
- Dijkgraaf, I.; Boerman, O.C. Molecular imaging of angiogenesis with SPECT. Eur. J. Nucl. Med. Mol. Imaging 2010, 37 (Suppl. 1), S104–S113. [Google Scholar] [CrossRef] [PubMed]
- Niccoli Asabella, A.; Di Palo, A.; Altini, C.; Ferrari, C.; Rubini, G. Multimodality Imaging in Tumor Angiogenesis: Present Status and Perspectives. Int. J. Mol. Sci. 2017, 18, 1864. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.; Chen, X. PET Imaging of Angiogenesis. PET Clin. 2009, 4, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Niu, G.; Fang, X.; Chen, X. Preclinical molecular imaging of tumor angiogenesis. Q. J. Nucl. Med. Mol. Imaging 2010, 54, 291–308. [Google Scholar] [PubMed]
- McKnight, B.N.; Viola-Villegas, N.T. (89) Zr-ImmunoPET companion diagnostics and their impact in clinical drug development. J. Labelled Comp. Radiopharm. 2018, 61, 727–738. [Google Scholar] [CrossRef] [PubMed]
- van de Watering, F.C.; Rijpkema, M.; Perk, L.; Brinkmann, U.; Oyen, W.J.; Boerman, O.C. Zirconium-89 labeled antibodies: A new tool for molecular imaging in cancer patients. Biomed. Res. Int. 2014, 2014, 203601. [Google Scholar] [CrossRef]
- Hong, H.; Chen, F.; Zhang, Y.; Cai, W. New radiotracers for imaging of vascular targets in angiogenesis-related diseases. Adv. Drug Deliv. Rev. 2014, 76, 2–20. [Google Scholar] [CrossRef]
- Bai, J.W.; Qiu, S.Q.; Zhang, G.J. Molecular and functional imaging in cancer-targeted therapy: Current applications and future directions. Signal Transduct. Target. Ther. 2023, 8, 89. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Sun, X.; Shen, B. Contrast agents in dynamic contrast-enhanced magnetic resonance imaging. Oncotarget 2017, 8, 43491–43505. [Google Scholar] [CrossRef]
- Klibanov, A.L.; Hossack, J.A. Ultrasound in Radiology: From Anatomic, Functional, Molecular Imaging to Drug Delivery and Image-Guided Therapy. Investig. Radiol. 2015, 50, 657–670. [Google Scholar] [CrossRef]
- Guerraty, M.; Bhargava, A.; Senarathna, J.; Mendelson, A.A.; Pathak, A.P. Advances in translational imaging of the microcirculation. Microcirculation 2021, 28, e12683. [Google Scholar] [CrossRef]
- Turkbey, B.; Kobayashi, H.; Ogawa, M.; Bernardo, M.; Choyke, P.L. Imaging of tumor angiogenesis: Functional or targeted? Am. J. Roentgenol. 2009, 193, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Xuan Leong, K.; Palhares, D.; Czarnota, G.J. Radiation combined with ultrasound and microbubbles: A potential novel strategy for cancer treatment. Z. Med. Phys. 2023, 33, 407–426. [Google Scholar] [CrossRef]
- Biswal, N.C.; Gamelin, J.K.; Yuan, B.; Backer, M.V.; Backer, J.M.; Zhu, Q. Fluorescence imaging of vascular endothelial growth factor in tumors for mice embedded in a turbid medium. J. Biomed. Opt. 2010, 15, 016012. [Google Scholar] [CrossRef]
- Loveless, M.E.; Whisenant, J.G.; Wilson, K.; Lyshchik, A.; Sinha, T.K.; Gore, J.C.; Yankeelov, T.E. Coregistration of ultrasonography and magnetic resonance imaging with a preliminary investigation of the spatial colocalization of vascular endothelial growth factor receptor 2 expression and tumor perfusion in a murine tumor model. Mol. Imaging 2009, 8, 187–198. [Google Scholar] [CrossRef] [PubMed]
- De Bruyne, S.; Van Damme, N.; Smeets, P.; Ferdinande, L.; Ceelen, W.; Mertens, J.; Van de Wiele, C.; Troisi, R.; Libbrecht, L.; Laurent, S.; et al. Value of DCE-MRI and FDG-PET/CT in the prediction of response to preoperative chemotherapy with bevacizumab for colorectal liver metastases. Br. J. Cancer 2012, 106, 1926–1933. [Google Scholar] [CrossRef]
- Kim, Y.E.; Joo, B.; Park, M.S.; Shin, S.J.; Ahn, J.B.; Kim, M.J. Dynamic Contrast-Enhanced Magnetic Resonance Imaging as a Surrogate Biomarker for Bevacizumab in Colorectal Cancer Liver Metastasis: A Single-Arm, Exploratory Trial. Cancer Res. Treat. 2016, 48, 1210–1221. [Google Scholar] [CrossRef]
- Hylton, N. Dynamic contrast-enhanced magnetic resonance imaging as an imaging biomarker. J. Clin. Oncol. 2006, 24, 3293–3298. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Dey, M.K.; Devireddy, R.; Gartia, M.R. Biomarkers in Cancer Detection, Diagnosis, and Prognosis. Sensors 2023, 24, 37. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.R.; Wu, X.L.; Sun, Y.L. Therapeutic targets and biomarkers of tumor immunotherapy: Response versus non-response. Signal Transduct. Target. Ther. 2022, 7, 331. [Google Scholar] [CrossRef]
- Wang, R.C.; Wang, Z. Precision Medicine: Disease Subtyping and Tailored Treatment. Cancers 2023, 15, 3837. [Google Scholar] [CrossRef] [PubMed]
- Sha, D.; He, Y.J. Expression and clinical significance of VEGF and its receptors Flt-1 and KDR in nasopharyngeal carcinoma. Ai Zheng 2006, 25, 229–234. [Google Scholar]
- Yadav, K.; Lim, J.; Choo, J.; Ow, S.G.W.; Wong, A.; Lee, M.; Chan, C.W.; Hartman, M.; Lim, S.E.; Ngoi, N.; et al. Immunohistochemistry study of tumor vascular normalization and anti-angiogenic effects of sunitinib versus bevacizumab prior to dose-dense doxorubicin/cyclophosphamide chemotherapy in HER2-negative breast cancer. Breast Cancer Res. Treat. 2022, 192, 131–142. [Google Scholar] [CrossRef]
- Gauthier, A.; Ho, M. Role of sorafenib in the treatment of advanced hepatocellular carcinoma: An update. Hepatol. Res. 2013, 43, 147–154. [Google Scholar] [CrossRef]
- LeCouter, J.; Zlot, C.; Tejada, M.; Peale, F.; Ferrara, N. Bv8 and endocrine gland-derived vascular endothelial growth factor stimulate hematopoiesis and hematopoietic cell mobilization. Proc. Natl. Acad. Sci. USA 2004, 101, 16813–16818. [Google Scholar] [CrossRef]
- Zhong, C.; Qu, X.; Tan, M.; Meng, Y.G.; Ferrara, N. Characterization and regulation of bv8 in human blood cells. Clin. Cancer Res. 2009, 15, 2675–2684. [Google Scholar] [CrossRef]
- Pircher, A.; Hilbe, W.; Heidegger, I.; Drevs, J.; Tichelli, A.; Medinger, M. Biomarkers in tumor angiogenesis and anti-angiogenic therapy. Int. J. Mol. Sci. 2011, 12, 7077–7099. [Google Scholar] [CrossRef]
- Haibe, Y.; Kreidieh, M.; El Hajj, H.; Khalifeh, I.; Mukherji, D.; Temraz, S.; Shamseddine, A. Resistance Mechanisms to Anti-angiogenic Therapies in Cancer. Front. Oncol. 2020, 10, 221. [Google Scholar] [CrossRef] [PubMed]
- Werner, N.; Kosiol, S.; Schiegl, T.; Ahlers, P.; Walenta, K.; Link, A.; Bohm, M.; Nickenig, G. Circulating endothelial progenitor cells and cardiovascular outcomes. N. Engl. J. Med. 2005, 353, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Erdbruegger, U.; Haubitz, M.; Woywodt, A. Circulating endothelial cells: A novel marker of endothelial damage. Clin. Chim. Acta 2006, 373, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Farinacci, M.; Krahn, T.; Dinh, W.; Volk, H.D.; Dungen, H.D.; Wagner, J.; Konen, T.; von Ahsen, O. Circulating endothelial cells as biomarker for cardiovascular diseases. Res. Pract. Thromb. Haemost. 2019, 3, 49–58. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targets | Inhibition Sites | Drugs | Mechanisms |
|---|---|---|---|
| VEGF signaling | Anti VEGF mAb | Bevacizumab, Aflibercept | Directly neutralizes the VEGF proteins |
| Inhibitors of VEGF receptors | Sunitinib, Sorafenib | Bind to VEGF receptors | |
| Inhibitors of VEGF signal transduction pathway | LY294002, Wortmannin, FARA-A, Rapamycin, Temsirolimus, Everolimus, Tipifarnib, Lonafarnib. | Blocking autophosphorylation of VEGF receptors | |
| VEGF antisense |
| ||
| EGFR signaling | Inhibitor of EGFR | Cetuximab, Panitumumab, Necitumumab | Block EGFR formation |
| RTK signaling | Tyrosine kinase inhibitor | Sunitinib, Sorafenib | Activity against VEGFR, PDGFR, Flt-3, C-kit & RET, CSF 1R |
| PI3K/Akt/mTOR signaling pathway | Inhibitors of PI3K pathway | LY294002 and wortmannin | |
| Inhibitors of mTOR | Temsirolimus (CCI-779) and Everolimus (RAD001) | ||
| MAPK signalling | Antiangiogenic therapy | Tipifarnib (R115777), Lonafarnib (SCH66336) | MAPK-Farnesyltransferase Rho and Ras |
| Other drugs | Methionine aminopeptidase inhibitor | Tnp-470 | Prevents endothelial activation and arrest cell cycle |
| Thalidomide | Inhibits TNF-α synthesis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaur, G.; Roy, B. Decoding Tumor Angiogenesis for Therapeutic Advancements: Mechanistic Insights. Biomedicines 2024, 12, 827. https://doi.org/10.3390/biomedicines12040827
Kaur G, Roy B. Decoding Tumor Angiogenesis for Therapeutic Advancements: Mechanistic Insights. Biomedicines. 2024; 12(4):827. https://doi.org/10.3390/biomedicines12040827
Chicago/Turabian StyleKaur, Geetika, and Bipradas Roy. 2024. "Decoding Tumor Angiogenesis for Therapeutic Advancements: Mechanistic Insights" Biomedicines 12, no. 4: 827. https://doi.org/10.3390/biomedicines12040827
APA StyleKaur, G., & Roy, B. (2024). Decoding Tumor Angiogenesis for Therapeutic Advancements: Mechanistic Insights. Biomedicines, 12(4), 827. https://doi.org/10.3390/biomedicines12040827