


Ibrutinib Modulates Proliferation, Migration, Mitochondrial Homeostasis, and Apoptosis in Melanoma Cells

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Cell Lines and Cell Culture
2.2. MTT Assay
2.3. Clonogenic Assay
2.4. Apoptosis Assay
2.5. Lactate Dehydrogenase (LDH) Release
2.6. Mitochondrial Membrane Potential
2.7. Caspase 3/7 Activity
2.8. Cell Proliferation
2.9. Cell Cycle
2.10. Cell Migration
2.11. RNA Extraction and Real Time PCR
2.12. Bioinformatics Analysis
2.13. Statistical Analysis
3. Results
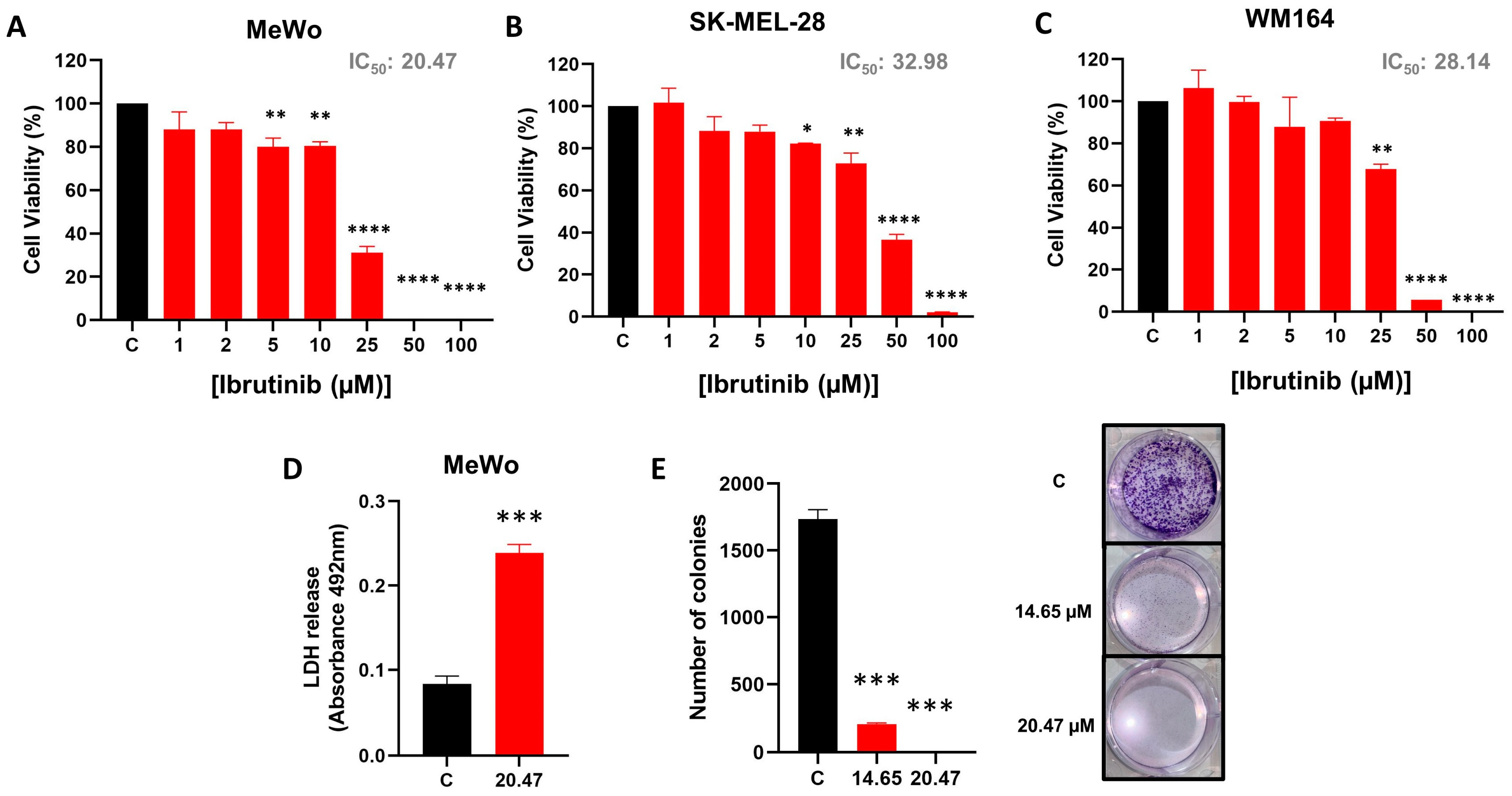
3.1. Ibrutinib Impairs MeWo Cell Line Viability
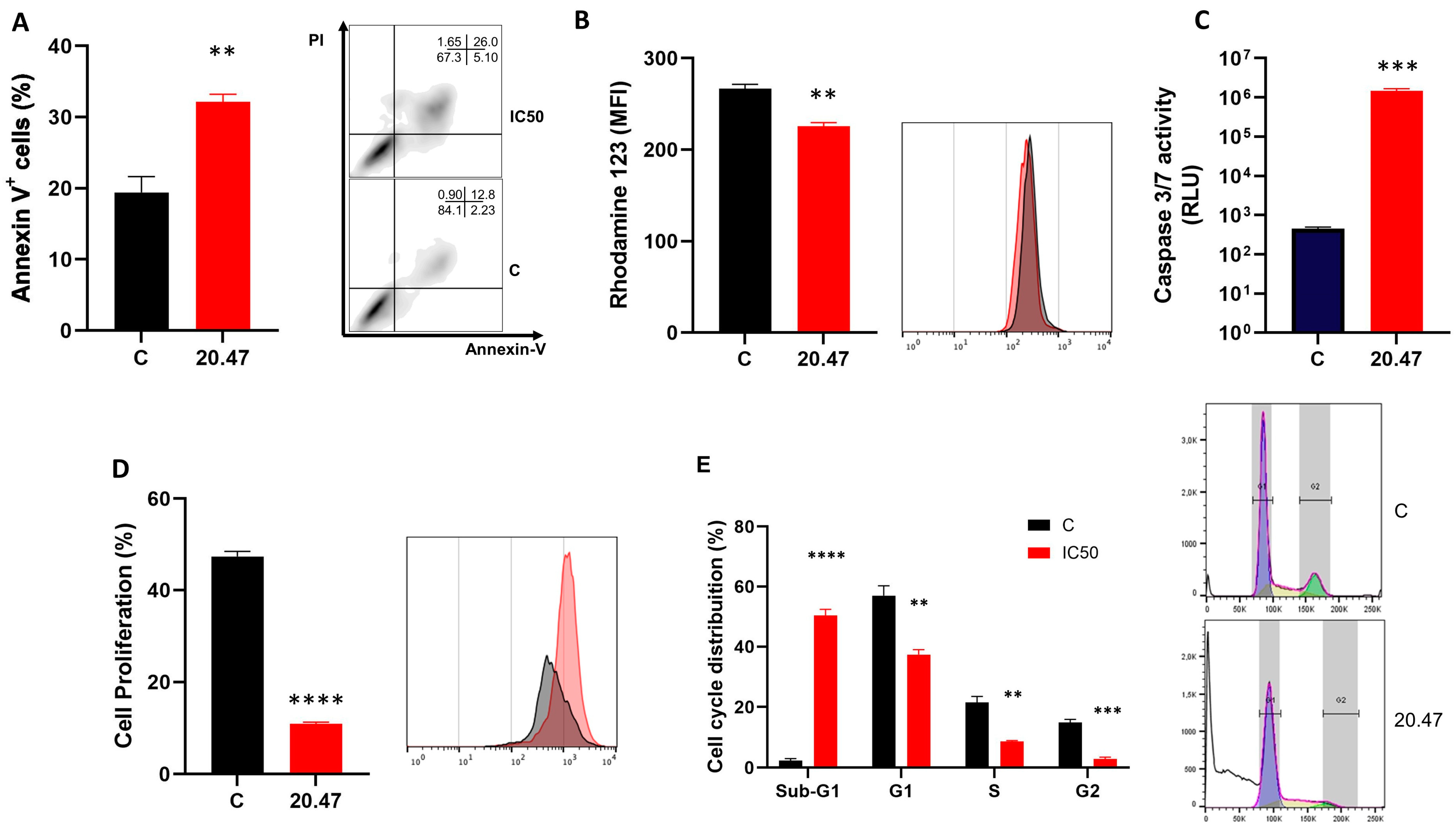
3.2. Ibrutinib Treatment Increases Annexin-V+ Mewo Cells
3.3. Ibrutinib Changes the Mitochondrial Membrane Potential of Melanoma Cells
3.4. Ibrutinib Induces Activation of Caspases 3/7 in Melanoma Cells
3.5. Ibrutinib Inhibits Cell Proliferation of Melanoma Cells
3.6. Ibrutinib Induces Cycle Arrest at the Sub-G1 Phase in Melanoma Cells
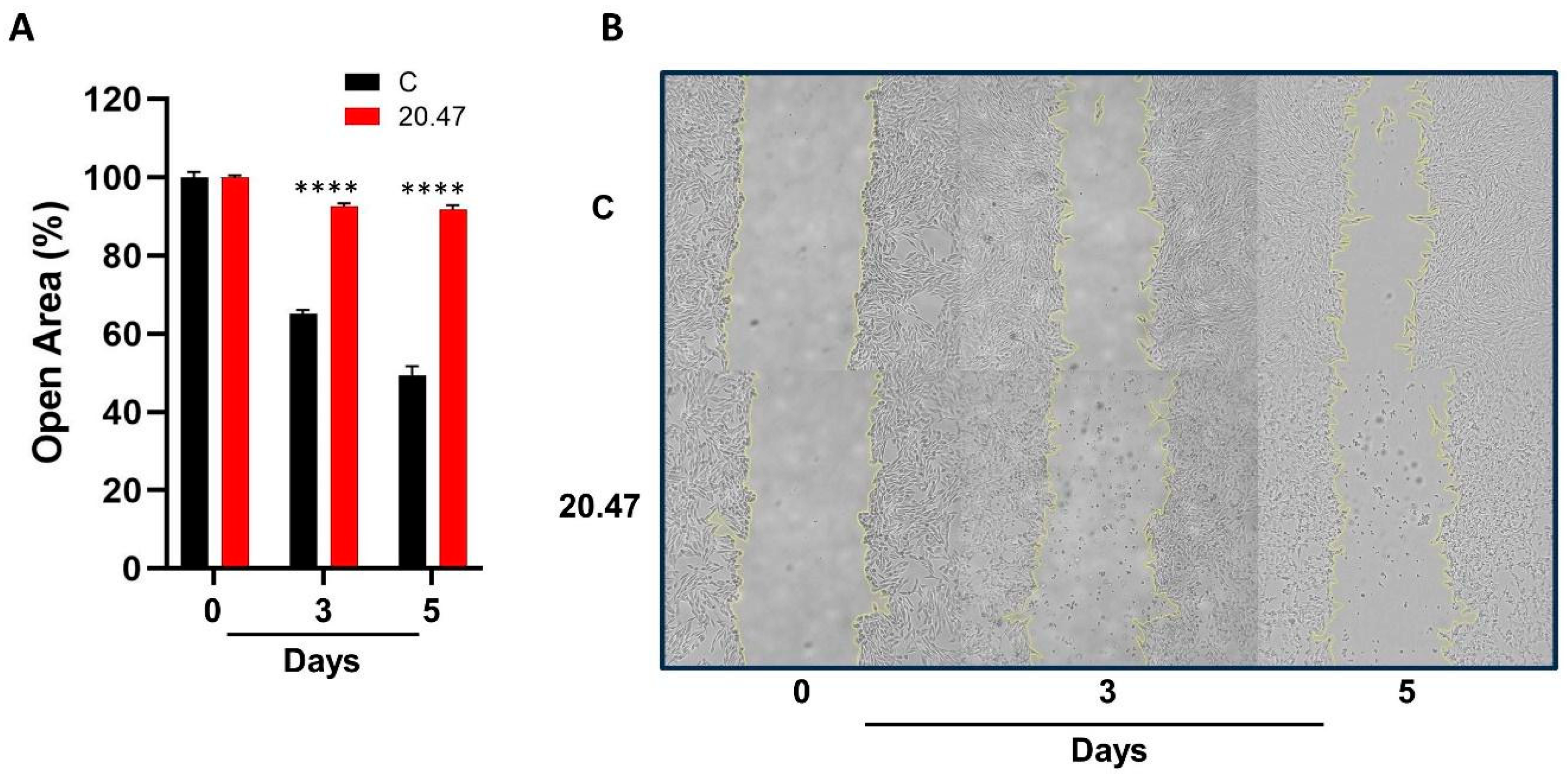
3.7. Ibrutinib Inhibits the Migration of Melanoma Cells
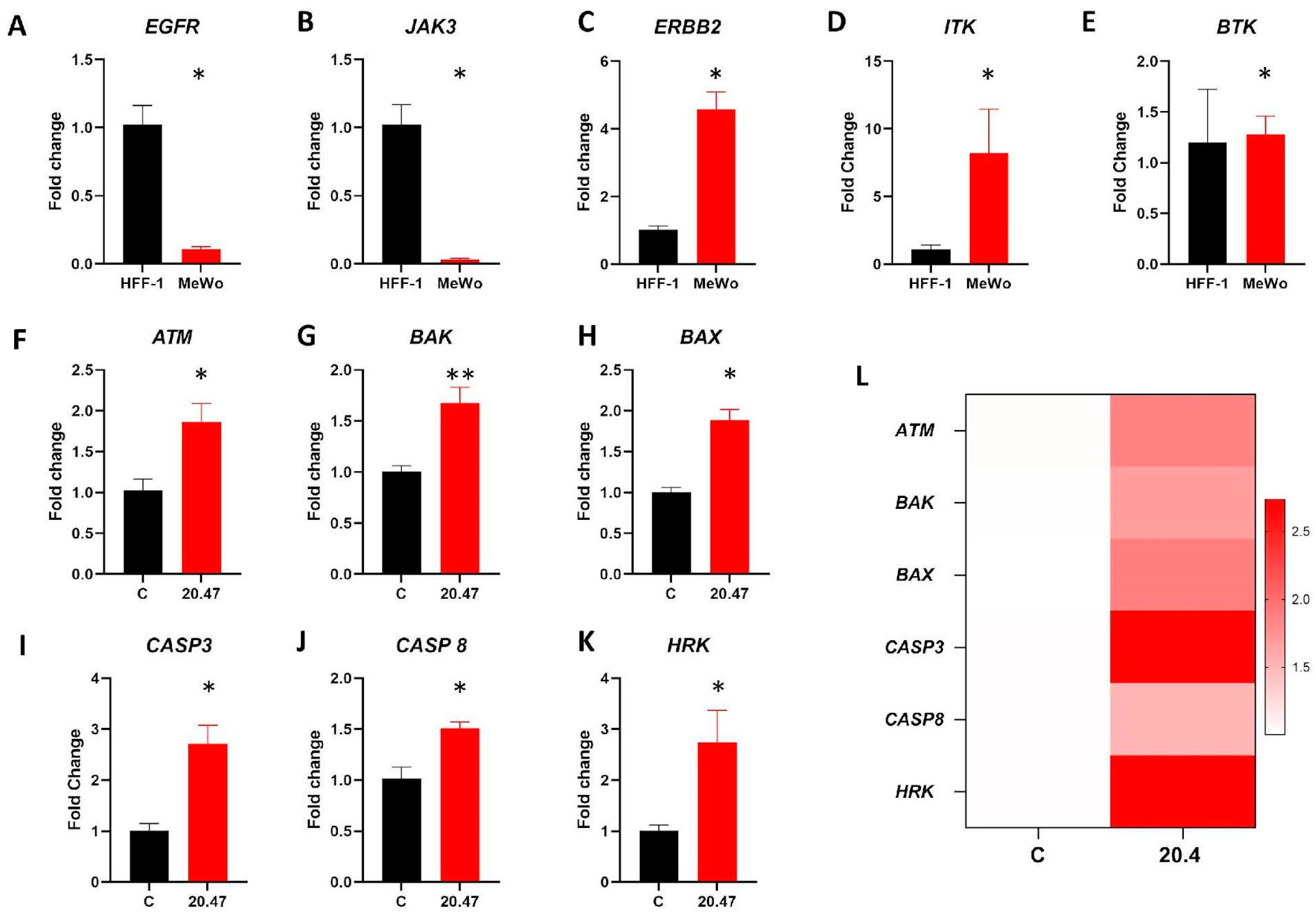
3.8. Real Time PCR
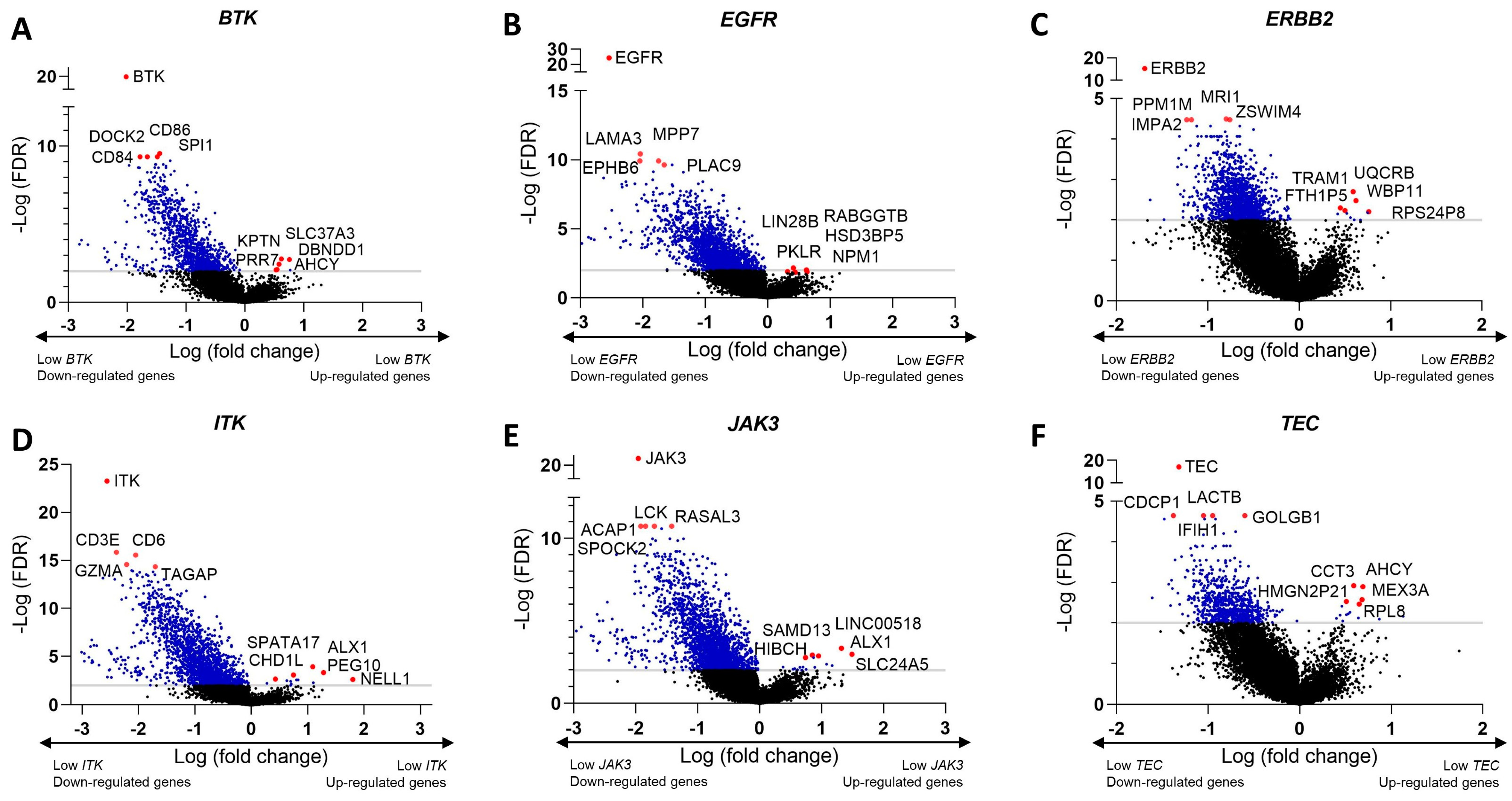
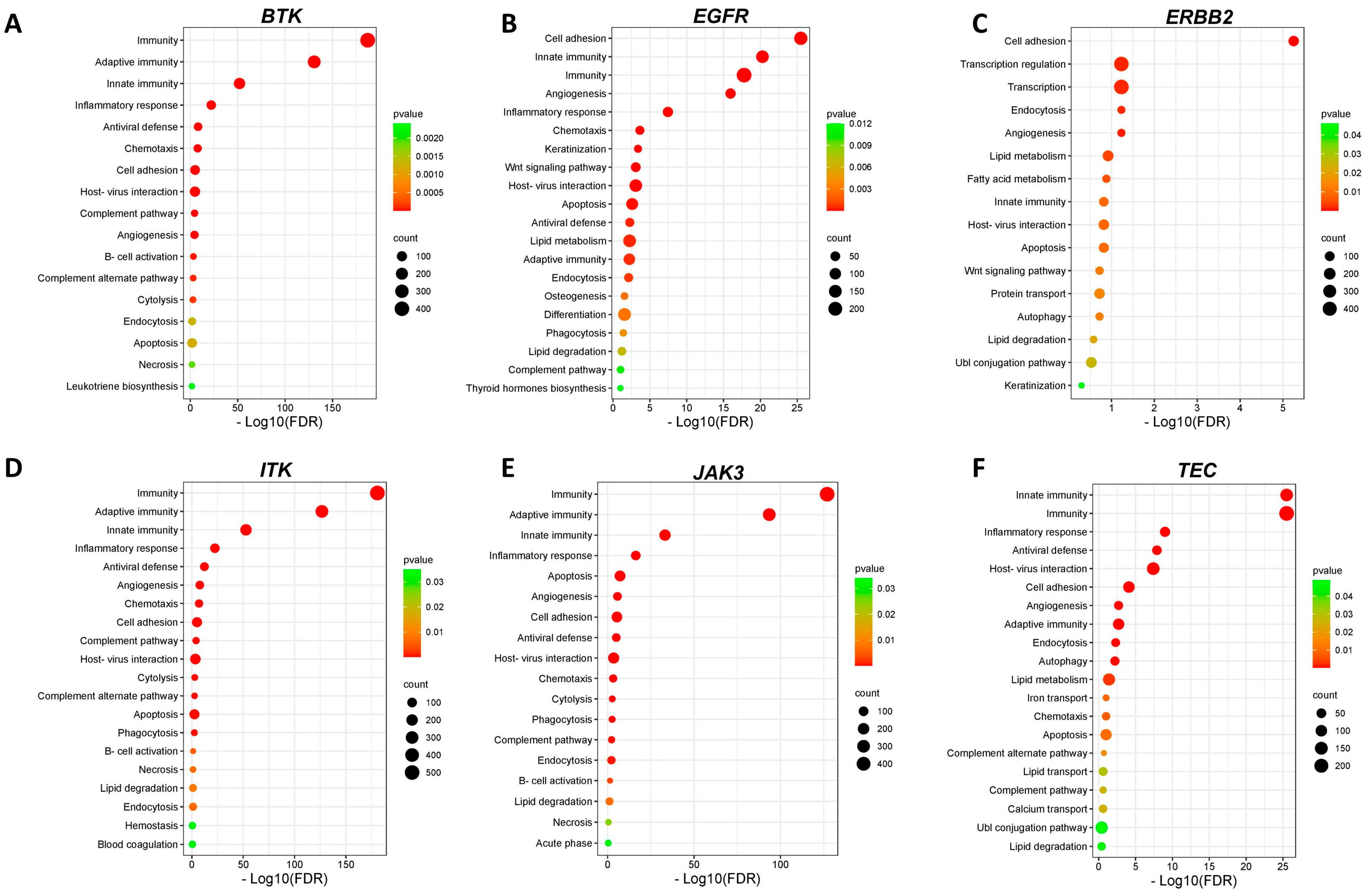
3.9. Identification of Differentially Expressed Genes and Pathways Modulated by Ibrutinib in Clinical Samples of Melanoma
4. Discussion
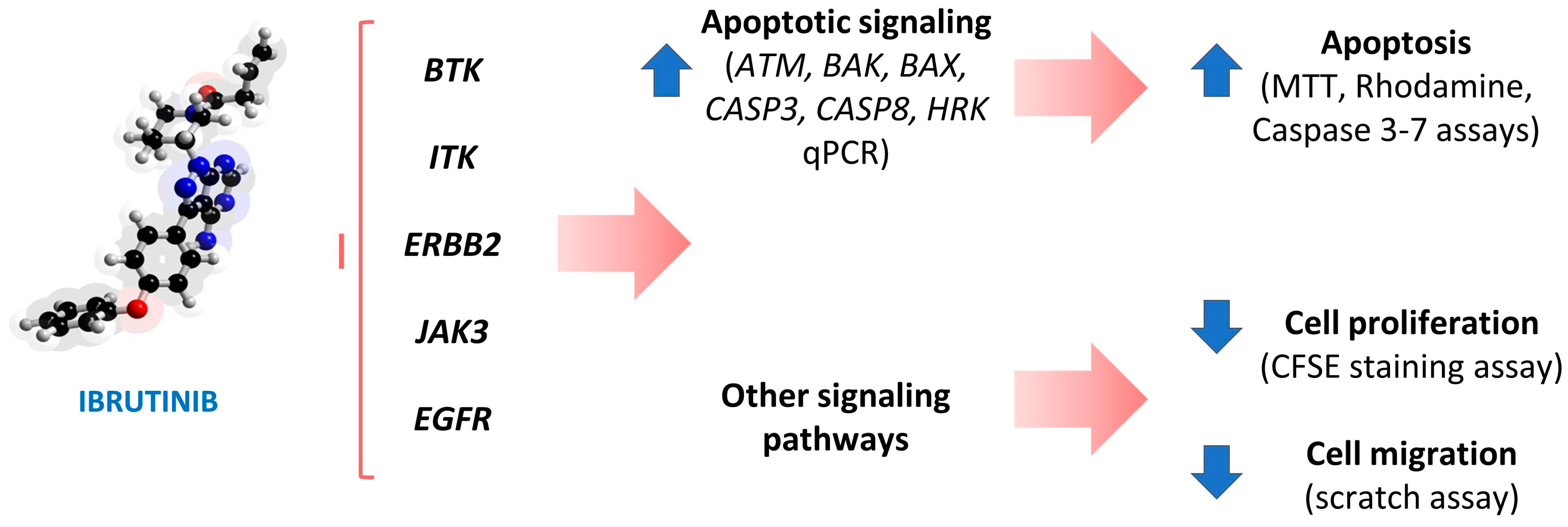
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yamaguchi, Y.; Hearing, V.J. Melanocytes and Their Diseases. Cold Spring Harb. Perspect. Med. 2014, 4, a017046. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Fisher, D.E.; Garbe, C.; Gershenwald, J.E.; Grob, J.-J.; Halpern, A.; Herlyn, M.; Marchetti, M.A.; McArthur, G.; Ribas, A.; et al. Melanoma. Nat. Rev. Dis. Primers 2015, 1, 15003. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Bastian, B.C. From Melanocytes to Melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, S.M.; Fisher, D.E. Biology of Melanoma. Hematol. Oncol. Clin. N. Am. 2021, 35, 29–56. [Google Scholar] [CrossRef] [PubMed]
- Bertolotto, C. Melanoma: From Melanocyte to Genetic Alterations and Clinical Options. Scientifica 2013, 2013, 635203. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, W.; Mwamba, R.N.; Grullon, K.; Armstrong, M.; Zhao, P.; Hendren-Santiago, B.; Qin, K.H.; Li, A.J.; Hu, D.A.; Youssef, A.; et al. Melanoma: Molecular Genetics, Metastasis, Targeted Therapies, Immunotherapies, and Therapeutic Resistance. Genes. Dis. 2022, 9, 1608–1623. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cai, Y. Better Prognostic Determination and Feature Characterization of Cutaneous Melanoma through Integrative Genomic Analysis. Aging 2019, 11, 5081–5107. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.E.; Shalin, S.C.; Tackett, A.J. Current State of Melanoma Diagnosis and Treatment. Cancer Biol. Ther. 2019, 20, 1366–1379. [Google Scholar] [CrossRef]
- Michielin, O.; Atkins, M.B.; Koon, H.B.; Dummer, R.; Ascierto, P.A. Evolving Impact of Long-Term Survival Results on Metastatic Melanoma Treatment. J. Immunother. Cancer 2020, 8, e000948. [Google Scholar] [CrossRef]
- Kozar, I.; Margue, C.; Rothengatter, S.; Haan, C.; Kreis, S. Many Ways to Resistance: How Melanoma Cells Evade Targeted Therapies. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Moser, J.C.; Grossman, K.F. Adjuvant Therapy for Resected High-Risk Melanoma. Semin. Cutan. Med. Surg. 2018, 37, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Scheerens, H.; Li, S.-J.; Schultz, B.E.; Sprengeler, P.A.; Burrill, L.C.; Mendonca, R.V.; Sweeney, M.D.; Scott, K.C.K.; Grothaus, P.G.; et al. Discovery of Selective Irreversible Inhibitors for Bruton’s Tyrosine Kinase. ChemMedChem 2007, 2, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Szklener, K.; Michalski, A.; Żak, K.; Piwoński, M.; Mańdziuk, S. Ibrutinib in the Treatment of Solid Tumors: Current State of Knowledge and Future Directions. Cells 2022, 11, 1338. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Wang, X.; Song, J.; Jin, G. Discovery of Novel Ibrutinib Analogues to Treat Malignant Melanoma. Bioorg. Chem. 2021, 117, 105419. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Petter, R.C.; Kluge, A.F. Targeted Covalent Drugs of the Kinase Family. Curr. Opin. Chem. Biol. 2010, 14, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A New Mathematical Model for Relative Quantification in Real-Time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Schilling, B.; Liu, D.; Sucker, A.; Livingstone, E.; Jerby-Arnon, L.; Zimmer, L.; Gutzmer, R.; Satzger, I.; Loquai, C.; et al. Integrative Molecular and Clinical Modeling of Clinical Outcomes to PD1 Blockade in Patients with Metastatic Melanoma. Nat. Med. 2019, 25, 1916–1927. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Messex, J.K.; Liou, G.-Y. Targeting BTK Signaling in the Microenvironment of Solid Tumors as a Feasible Cancer Therapy Option. Cancers 2021, 13, 2198. [Google Scholar] [CrossRef]
- Krishnamurthy, K.; Urioste, S.N.; Cusnir, M.; Schwartz, M.; Alghamdi, S.; Sriganeshan, V.; Poppiti, R. Analysis of Genetic Alterations in Cutaneous Malignant Melanomas Unveils Unique Loco-Regional Variations and Novel Predictors of Metastatic Potential. Am. J. Dermatopathol. 2021, 43, e185–e189. [Google Scholar] [CrossRef]
- Hamida, R.S.; Albasher, G.; Bin-Meferij, M.M. Oxidative Stress and Apoptotic Responses Elicited by -Synthesized Silver Nanoparticles against Different Cancer Cell Lines. Cancers 2020, 12, 209. [Google Scholar] [CrossRef]
- Sun, F.-D.; Wang, P.-C.; Shang, J.; Zou, S.-H.; Du, X. Ibrutinib Presents Antitumor Activity in Skin Cancer and Induces Autophagy. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 561–566. [Google Scholar] [PubMed]
- Tan, B.; Huang, Y.; Zhang, B.; Lin, N. The Effect of Ibrutinib on Radiosensitivity in Pancreatic Cancer Cells by Targeting EGFR/AKT/mTOR Signaling Pathway. Biomed. Pharmacother. 2020, 128, 110133. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhang, J.; Shi, D.; Wu, Y.; Liu, R.; Liu, T.; Xu, J.; Yao, X.; Fang, J. Targeting Thioredoxin Reductase by Ibrutinib Promotes Apoptosis of SMMC-7721 Cells. J. Pharmacol. Exp. Ther. 2019, 369, 212–222. [Google Scholar] [CrossRef]
- Prabaharan, C.B.; Yang, A.B.; Chidambaram, D.; Rajamanickam, K.; Napper, S.; Sakharkar, M.K. Ibrutinib as a Potential Therapeutic Option for HER2 Overexpressing Breast Cancer—The Role of STAT3 and p21. Investig. New Drugs 2020, 38, 909–921. [Google Scholar] [CrossRef]
- Hanif, A.; Ibrahim, A.H.; Ismail, S.; Al-Rawi, S.S.; Ahmad, J.N.; Hameed, M.; Mustufa, G.; Tanwir, S. Cytotoxicity against A549 Human Lung Cancer Cell Line via the Mitochondrial Membrane Potential and Nuclear Condensation Effects of Briq., a Perennial Herb. Molecules 2023, 28, 2812. [Google Scholar] [CrossRef]
- Gupta, P.S.P.; Kaushik, K.; Johnson, P.; Krishna, K.; Nandi, S.; Mondal, S.; Nikhil Kumar Tej, J.; Somoskoi, B.; Cseh, S. Effect of Different Vitrification Protocols on Post Thaw Viability and Gene Expression of Ovine Preantral Follicles. Theriogenology 2022, 178, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, X.; Hong, Y.; Wang, S.; Chen, P.; Gu, A.; Guo, X.; Zhao, P. Ibrutinib, a Bruton’s Tyrosine Kinase Inhibitor, Exhibits Antitumoral Activity and Induces Autophagy in Glioblastoma. J. Exp. Clin. Cancer Res. 2017, 36, 96. [Google Scholar] [CrossRef]
- Herman, S.E.M.; Mustafa, R.Z.; Gyamfi, J.A.; Pittaluga, S.; Chang, S.; Chang, B.; Farooqui, M.; Wiestner, A. Ibrutinib Inhibits BCR and NF-κB Signaling and Reduces Tumor Proliferation in Tissue-Resident Cells of Patients with CLL. Blood 2014, 123, 3286–3295. [Google Scholar] [CrossRef]
- Sivina, M.; Kreitman, R.J.; Arons, E.; Ravandi, F.; Burger, J.A. The Bruton Tyrosine Kinase Inhibitor Ibrutinib (PCI-32765) Blocks Hairy Cell Leukaemia Survival, Proliferation and B Cell Receptor Signalling: A New Therapeutic Approach. Br. J. Haematol. 2014, 166, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Misek, S.A.; Newbury, P.A.; Chekalin, E.; Paithankar, S.; Doseff, A.I.; Chen, B.; Gallo, K.A.; Neubig, R.R. Ibrutinib Blocks YAP1 Activation and Reverses BRAF Inhibitor Resistance in Melanoma Cells. Mol. Pharmacol. 2022, 101, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Ling, L.; Qi, L.; Chong, Y.; Xue, L. Bruton’s Tyrosine Kinase (BTK) Inhibitor (Ibrutinib) -Suppressed Migration and Invasion of Prostate Cancer. Onco. Targets. Ther. 2020, 13, 4113–4122. [Google Scholar] [CrossRef]
- Zheng, X.; Ding, N.; Song, Y.; Feng, L.; Zhu, J. Different Sensitivity of Germinal Center B Cell-like Diffuse Large B Cell Lymphoma Cells towards Ibrutinib Treatment. Cancer Cell Int. 2014, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Gong, W.; Liu, S.; Li, Q.; Guo, M.; Wang, J.; Wang, S.; Chen, N.; Wang, Y.; Liu, Q.; et al. Ibrutinib Targets microRNA-21 in Multiple Myeloma Cells by Inhibiting NF-κB and STAT3. Tumour Biol. 2018, 40, 1010428317731369. [Google Scholar] [CrossRef] [PubMed]
- Moschos, S.J.; Eroglu, Z.; Khushalani, N.I.; Kendra, K.L.; Ansstas, G.; In, G.K.; Wang, P.; Liu, G.; Collichio, F.A.; Googe, P.B.; et al. Targeting the IL-2 Inducible Kinase in Melanoma; a Phase 2 Study of Ibrutinib in Systemic Treatment-Refractory Distant Metastatic Cutaneous Melanoma: Preclinical Rationale, Biology, and Clinical Activity (NCI9922). Melanoma Res. 2021, 31, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Wang, X.; Jin, G. Conjugate of Ibrutinib with a TLR7 Agonist Suppresses Melanoma Progression and Enhances Antitumor Immunity. Int. J. Biol. Sci. 2022, 18, 166–179. [Google Scholar] [CrossRef]
- Albuquerque, L.F.F.; Lins, F.V.; Bispo, E.C.I.; Borges, E.N.; Silva, M.T.; Gratieri, T.; Cunha-Filho, M.; Alonso, A.; Carvalho, J.L.; Saldanha-Araujo, F.; et al. Ibrutinib Topical Delivery for Melanoma Treatment: The Effect of Nanostructured Lipid Carriers’ Composition on the Controlled Drug Skin Deposition. Colloids Surf. B Biointerfaces 2024, 237, 113875. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lins, F.V.; Bispo, E.C.I.; Rodrigues, N.S.; Silva, M.V.S.; Carvalho, J.L.; Gelfuso, G.M.; Saldanha-Araujo, F. Ibrutinib Modulates Proliferation, Migration, Mitochondrial Homeostasis, and Apoptosis in Melanoma Cells. Biomedicines 2024, 12, 1012. https://doi.org/10.3390/biomedicines12051012
Lins FV, Bispo ECI, Rodrigues NS, Silva MVS, Carvalho JL, Gelfuso GM, Saldanha-Araujo F. Ibrutinib Modulates Proliferation, Migration, Mitochondrial Homeostasis, and Apoptosis in Melanoma Cells. Biomedicines. 2024; 12(5):1012. https://doi.org/10.3390/biomedicines12051012
Chicago/Turabian StyleLins, Fernanda Vitelli, Elizabete Cristina Iseke Bispo, Naomí Souza Rodrigues, Maria Victória Souto Silva, Juliana Lott Carvalho, Guilherme Martins Gelfuso, and Felipe Saldanha-Araujo. 2024. "Ibrutinib Modulates Proliferation, Migration, Mitochondrial Homeostasis, and Apoptosis in Melanoma Cells" Biomedicines 12, no. 5: 1012. https://doi.org/10.3390/biomedicines12051012
APA StyleLins, F. V., Bispo, E. C. I., Rodrigues, N. S., Silva, M. V. S., Carvalho, J. L., Gelfuso, G. M., & Saldanha-Araujo, F. (2024). Ibrutinib Modulates Proliferation, Migration, Mitochondrial Homeostasis, and Apoptosis in Melanoma Cells. Biomedicines, 12(5), 1012. https://doi.org/10.3390/biomedicines12051012