
Optimizing Recombinant Cas9 Expression: Insights from E. coli BL21(DE3) Strains for Enhanced Protein Purification and Genome Editing

 , ,
, ,
Abstract

1. Introduction
2. Materials
2.1. Biological Materials
- Plasmid—pET-28b-Cas9-His (Addgene, Watertown, MA, USA, cat. no. 47327)
2.2. Competent Cells
- BL21(DE3)-pLysS (Millipore Sigma, Burlington, MA, USA, cat. no. 69451-3)
- BL21(DE3)-Star (ThermoFisher, Waltham, MA, USA, cat. no. C601003)
- Rosetta2 (Millipore Sigma, cat. no. 71402-3)
- BL21(DE3) cells (Millipore Sigma, cat. no. 69450-3)
2.3. Reagents
- Tris-HCl (VWR, Radnor, PA, USA, cat. no. 71003-494)
- KCl (VWR, cat. no. 71003-522)
- Imidazole (MP Biomedicals, Santa Ana, CA, USA, SKU: 02102033-CF)
- Glycerol (Fisher Scientific, Hampton, NH, USA, cat. no. BP229-4)
- HEPES (VWR, cat. no. VWRB30487)
- DTT (VWR, cat. no. 97061-338)
- LB Broth, Lennox (Fisher Scientific, cat. no. BP1427-2)
- LB Agar, Miller (Fisher Scientific, cat. no. BP1425-500)
- Kanamycin Sulphate (VWR, cat. no. 75856-686)
- Chloramphenicol (VWR, cat. no. TCC2255)
- IPTG (ThermoFisher, cat. no. 15529019)
- SDS (Biorad, Hercules, CA, USA, cat. no. 1610301)
- 30% Acrylamide solution (Biorad, cat. no. 1610156)
- Ammonium persulfate (Biorad, cat. no. 1610700)
- TEMED (Biorad, cat. no. 1610801)
- Phenylmethylsulfonyl fluoride (PMSF) (Millipore Sigma, cat. no. 10837091001)
- Methanol (VWR, cat. no. EM-MX0475-1)
- Ethanol (VWR, cat. no. 89125-188)
- Acetic Acid (VWR, cat. no. BDH3094)
- HisPrep FF 16/10 (Cytiva, Marlborough, MA, USA, cat. no. 28936551)
- Deionized water (facilitated by the University of Arkansas)
- Pierce Bradford protein assay kit (ThermoFisher, cat. no. 23200)
- Qiagen QIAprep Spin Miniprep Kit (Qiagen, Germantown, MD, USA, cat. no. 27104)
- 2x Laemmli Sample Buffer (Biorad, cat. no. 1610737)
- β-Mercaptoethanol (BME) (Millipore Sigma, cat. no. 444203)
- SDS-PAGE Protein ladder (GenScript, Piscataway, NJ, USA, cat. no. M00624)
- Western Blot Protein Ladder (Biorad, cat. no. 1610373)
- Cas9 Monoclonal Antibody (Thermofisher, cat. no. MA1-201)
- Rabbit anti-mouse IgG Secondary Antibody, HRP (ThermoFisher, cat. no. 61-6520)
- Tris base (Genesee Scientific, El Cajon, CA, USA, cat. no. 18-144)
- EDTA-free Protease Inhibitor cocktail (Roche, Indianapolis, IN, USA, cat. no. 11873580001)
- lysozyme (Millipore Sigma, cat. no. L6876)
- Clarity Western ECL Substrate (Biorad, cat. no. 1705061)
- NaCl (VWR, cat. no. EM7760)
- Tween 20 (Millipore Sigma, cat. no. P9416)
- Great Value Instant Nonfat Dry Milk (Walmart, Bentonville, AR, USA)
- DNeasy Blood & Tissue Kit (Qiagen, cat. no. 69504)
- Q5® High-Fidelity DNA Polymerase (New England Biolabs, Ipswich, MA, USA, cat. no. M0491)
- 10 X NEBuffer™ r3.1 (New England Biolabs, cat. no. B6003)
- QIAquick PCR purification (Qiagen, cat. no. 28104)
- Proteinase K (New England Biolabs, cat. no. P8107S)
- Agarose (Genesee Scientific, cat. no. 20-102GP)
- 1 kB Plus DNA Ladder (New England Biolabs, cat. no. N3200L)
- Glacial Acetic Acid (Millipore Sigma, cat. no. PHR1748)
- EDTA, Disodium Salt, Dihydrate, Crystal (Avantor, Radnor, PA, USA, cat. no. 4040-04)
- Coomasie blue stain (Biorad, cat. no. 1610803)
- 10 X SDS-PAGE running buffer (ThermoFisher, cat. no. NP0001)
- Trans-Blot Turbo transfer packs (Biorad, cat. no. 1704159EDU)
- Out glass plt w/1mm (Biorad, cat. no. 1653311)
- Inner glass plate (Biorad, cat. no. 1653308)
- Mini-PROTEAN Casting Stand (Biorad, cat. no. 1653303)
- Casting frame (Biorad, cat. no. 1653304)
- Mini-PROTEAN Comb 10W 1.0 mm 44 µL (Biorad, cat. no. 1653311)
- Roller (Biorad, cat. no. 1651279)
- Gel releasers (Biorad, cat. no. 1653320)
2.4. Equipment
- 1 L and 500 mL Erlenmeyer Flasks (Chemglass Life Sciences, Vineland, NJ, USA)
- Temperature-Controlled Shaking Incubator (New Brunswick Scientific, Edison, NJ, USA)
- Temperature-controlled incubator (Fisher Scientific, cat. no. 13-762-721)
- Mechanical Device to Disrupt E. coli Cells:
- Options: Ultrasonicator (Qsonica Sonicators, Newtown, CT, USA, cat. no. Q55-110), French press, or cell homogenizer.
- BioTek Synergy LX Multi-Mode Reader (Agilent, Santa Clara, CA, USA)
- NanoDrop One to check the OD (Thermofisher Scientific, cat. no. ND-ONE-W)
- Refrigerated Centrifuge and Centrifuge Bottles (500 mL and 50 mL) (Eppendorf, Enfield, CT, USA)
- AKTA Start for protein purification (Cytiva, cat. no. 29022094)
- Oakridge Tubes (Thermofisher Scientific)
- Both 50 and 15 mL falcon tubes (Genessee Scientific, cat. no. 21-106 & 21-103)
- Ultrafiltration Centrifugal Concentrating Devices (Centricon) with Appropriate Molecular-Weight Cut-Offs (Sigma Aldrich, St. Louis, MO, USA)
- ChemiDoc Imaging System (Biorad, cat. no. 12003154)
- Trans-Blot Turbo Transfer System (Biorad, cat. no. 1704150)
- Mini-Sub Cell GT (Biorad, cat. no. 1664000EDU)
- Mini Rocker (Benchmark Scientific, Sayreville, NJ, USA, cat. no. BR1000)
- Mini-Protean Tetra Vertical Electrophoresis Cell (Biorad, cat. no. 1658001FC)
2.5. Solutions
- Kanamycin solution (50 mg/mL)—Dissolve 0.5 g of Kanamycin sulfate in 10 mL of deionized water, vortex to ensure complete solubility, sterilize by filtering through a 0.2 µm filter, aliquot, and store at −20 °C for up to six months. Add 1 mL of kanamycin stock (at 50 mg/mL) per liter of LB media to obtain a final concentration of 50 µg/mL.
- Chloramphenicol solution (30 mg/mL)—Dissolve 0.34 g of chloramphenicol in 10 mL of 100% ethanol, vortex to ensure complete solubility, sterilize by filtering through a 0.2 µm filter, aliquot, and store at −20 °C for up to six months. Add 1 mL of chloramphenicol stock (at 30 mg/mL) per liter of LB media to obtain a final concentration of 30 µg/mL.
- IPTG solution (1 M)—Weigh 2.38 g IPTG, dissolve in 8 mL deionized water, adjust to a final volume of 10 mL, sterile filter using a 0.22 µm syringe filter, aliquot the stock, and store at −20 °C for up to one year.
- Staining solution (400 mL)—Prepare a staining solution by combining 180 mL of deionized water, 180 mL of methanol, 40 mL of acetic acid, and 7 g of Coomassie Blue, ensuring thorough mixing.
- Destaining solution (1 L)—Create a destaining solution by mixing 400 mL of methanol and 200 mL of acetic acid, and filling the container to 1 L with deionized water, followed by thorough mixing.
- Loading dye solution—Add 50 µL of BME to 950 µL of 2× Laemmli Sample Buffer
- LB agar—Combine 20 g of LB agar powder with 500 mL of deionized water, autoclave, cool, and pour into Petri dishes.
- LB media/broth—Mix 10 g of LB broth in 500 mL of deionized water, autoclave, and store at 4 °C.
- PMSF (Phenyl Methylsulfonylfluoride) solution: Concentration: 34.8 mg/mL in 100% ethanol (Storage: −20 °C)
- Glycerol stock—Mix an overnight bacterial culture with 50% glycerol at a 1:1 ratio, aliquot into cryovials, and freeze at −80 °C for long-term storage as glycerol stocks.
2.6. Buffers
- Lysis Buffer: Composition: 20 mM Tris-HCl pH 8.0, 250 mM KCl, 20 mM imidazole, 10% glycerol
- Wash Buffer: Composition: 20 mM Tris-HCl pH 8.0, 800 mM KCl, 30 mM imidazole, 10% glycerol
- Elution Buffer: Composition: 20 mM HEPES pH 8.0, 500 mM KCl, 250 mM imidazole, 10% glycerol
- Exchange Buffer: Composition: 20 mM HEPES pH 7.5, 500 mM KCl, 20% glycerol, 1 mM DTT
- Note: Add DTT just before dialysis. For concentrating, add DTT to the fresh Exchange Buffer the next day.
- SDS-PAGE Running Buffer: Prepare 1 X running buffer by diluting the 10 X buffer to 1 L. Add 100 mL of 10× commercially available running buffer to 900 mL of deionized water.
- 10 X TBS: Composition: 12 g Tris Base, 44 g NaCl, set to pH 7.6
- 1 X TBST: Composition: 100 mL of 10 X TBS, 500 μL Tween20, make volume up to 1 L
- Blocking Buffer: Composition: 2.5 g skim milk, 50 mL 1 X TBST
- 0.5 M EDTA (pH 8.0): Composition: Add 186.1 g of EDTA in 800 mL, adjust pH and then make up to 1 L.
- 50 X TAE: Composition: 243.3 g Tris Base, 57.2 mL glacial acetic acid, 100 mL 0.5 M EDTA (pH 8.0), and make up volume to 1 L.
3. Procedure
3.1. Preparation of Competent Cells
- Autoclave the following materials: 500 mL LB media, 2500 mL centrifuge bottles, Eppendorf tubes, pipette tips (if needed), 0.1 M CaCl2, and 0.1 M CaCl2 in 30% glycerol.
- Inoculate 10 mL of LB media with BL21-pLysS (or any desired competent cell) and grow for 12–16 h on shaker at 250 rpm.
- Take 1% of previous culture (e.g., 100 μL of 10 mL) in 500 mL LB media and grow the competent cells on the shaker at 250 rpm until optical density (OD) is between 0.4–0.5. Transfer to the autoclaved centrifuge bottles and centrifuge at 4500 rpm for 15 min at 4 °C.
- Remove the supernatant and resuspend the pellet in 1 mL of 0.1 M CaCl2, mixing carefully on ice. Incubate on ice for 15 min. Then, centrifuge for 4500 rpm for 15 min at 4 °C and discard the supernatant.
- Prechill 0.1 M CaCl2 in 30% glycerol and Eppendorf tubes and then resuspend the pellet in 1 mL of 0.1 M CaCl2 in 30% glycerol and mix carefully on ice.
- Aliquot 50 μL in prechilled Eppendorf tubes and store all tubes at −80 °C.
3.2. Preparation of Plasmid from Addgene
- Spread a small amount of the agar stab from Addgene on an LB agar plate with kanamycin (1 μL per 1 mL). Leave for 12–16 h at 37 °C.
- Next day, isolate 1 colony and put in 10 mL of LB media and 10 μL of kanamycin and leave for 12–16 h at 37 °C.
- To create a glycerol stock, combine and gently mix 500 μL from the bacterial culture and 500 μL of 50% glycerol (1:1 ratio) and store at −80 °C. The SpCas9-His plasmid is isolated from the remaining bacterial culture using the QIAprep Spin Miniprep Kit protocol.
3.3. Bacterial Transformation
- For transformation, if competent cells are frozen at −80 °C, thaw on ice for ~15 min.
- Add 5 μL of His-Cas9 plasmid (concentration—5 ng/μL) and 50 μL of competent cells to a 1.5 mL microcentrifuge tube and incubate on ice for 30 min.Note—For transformation to E. coli competent cells, use 10–50 ng of the Cas9 plasmid, and the maximum volume that can be used in the transformation is about 10% of the total volume.
- Heat shock at 42 °C for 45 s and then immediately put it on ice for 3 min. Add 250 μL of SOC media and incubate on shaker for 1 h at 37 °C.
- Plate 50–100 μL on an agar plate with kanamycin (50 µg/mL) for BL21(DE3), BL21(DE3)-pLysS, and BL21(DE3)-Star, and Chloramphenicol (30 µg/mL) and kanamycin (50 µg/mL) for Rosetta2 as the antibiotic and incubate at 37 °C in an incubator for 12–16 h.
- After incubation time, take 1 colony from the plate and use it to inoculate in 10 mL of LB media and 10 μL of antibiotic (kanamycin for all the 4 competent cells and kanamycin and chloramphenicol for Rosetta2) (1 μL for every 1 mL of LB media) and incubate for 12–16 h.
- To create a glycerol stock, combine and gently mix 500 μL from each culture (BL21(DE3), BL21(DE3)-pLysS, BL21(DE3)Star, and Rosetta2) and 500 μL of 50% glycerol (1:1 ratio) and store at −80 °C.
3.4. Small-Scale Expression of Recombinant SpCas9-His Protein
3.4.1. Preparation of Starter Culture
- In a 50 mL centrifuge tube, add 10 mL of autoclaved LB broth and 10 µL of kanamycin (50 μg/mL) for BL21(DE3), BL21(DE3)-pLysS, and BL21(DE3)Star, and chloramphenicol (30 µg/mL) and kanamycin (50 µg/mL) for Rosetta2.
- Transfer 5 µL from the His-Cas9 glycerol stocks into the LB broth with the appropriate antibiotic.
- Incubate the bacterial culture on a shaker (250 rpm) at 37 °C overnight (12–15 h).
3.4.2. Preparation of Small-Scale Bacterial Culture for Protein Overexpression
- In a 250 mL flask, prepare 50 mL of sterile LB broth. Inoculate with 2.5 mL (5%) of the starter culture and the appropriate antibiotic concentration (50 µL).
- Grow the cells at 37 °C and 250 rpm until the optical density at 600 nm reaches ~0.4 to 0.6.
- Extract 1 mL of bacterial cells from the flask as a pre-induction (un-induced) sample for SDS-PAGE analysis.
- Induce the culture for SpCas9-His overexpression by adding 50 μL of 1 M IPTG stock solution, achieving a final concentration of 1 mM. Incubate the culture at 30 °C on the shaker.
- Induction typically takes 5–6 h. Following induction, centrifuge the remaining bacterial cell culture in 50 mL centrifuge bottles at 6000 rpm (10,620× g) for 20 min at 4 °C. Discard the supernatant without disturbing the bacterial cell pellet.
- Resuspend the cells (in 1 mL of freshly prepared lysis buffer) and centrifuge again at 6000 rpm (10,620× g) for 15 min at 4 °C.
- Discard the clear supernatant, resuspend the cells in lysis buffer and load it as a post-induction (induced) sample for subsequent SDS-PAGE analysis, looking for a distinct band indicating the molecular size of SpCas9-His (~160 kDa).
3.5. Large-Scale Expression of Recombinant SpCas9-His Protein
3.5.1. Preparation of Starter Culture
- Sterilize a flask containing 50–100 mL of LB broth by autoclaving. Then, add kanamycin (50 μg/mL) to the sterile LB media.
- Add 100 µL of the glycerol stock of SpCas9-His into the LB broth with kanamycin.
- Incubate the bacterial culture on a shaker (250 rpm) at 37 °C for 12–15 h.
3.5.2. Preparation of Large-Scale Bacterial Culture for Protein Overexpression
- In a 1 L Erlenmeyer flask, prepare 400 mL of sterile LB broth. Inoculate with 20 mL (5%) of the starter culture and the appropriate antibiotic concentration.
- Grow the cells under consistent conditions at 37 °C and 250 rpm until the optical density at 600 nm reaches ~0.4 to 0.6.
- Induce the culture to overexpress SpCas9-His by adding 40 μL of 1 M IPTG stock solution, achieving a final concentration of 0.5 mM. Incubate the culture at 30 °C.
- Induction typically takes 5–6 h. Following induction, centrifuge the bacterial cell culture in 500 mL centrifuge bottles at 6000 rpm for 30 min at 4 °C. Discard the supernatant without disturbing the bacterial cell pellet.
- Transfer resuspended cells (in 50 mL of freshly prepared lysis buffer) to a new falcon tube (50 mL) and centrifuge again at 6000 rpm (10,620× g) for 20 min at 4 °C.
- Discard the clear supernatant, freeze the cell pellet at −80 °C for future use, and thaw when needed at room temperature before placing it on ice for further application.
3.6. Purification Procedure for Recombinant SpCas9-His Protein
3.6.1. Bacterial Cell Lysis
- Begin by thawing the cell pellet obtained from the freezer. Add 15 mL of lysis buffer and allow it to thaw for 10 min at room temperature. Subsequently, suspend the cell pellet until the solution becomes turbid. Keep the cell suspension on an ice bath.
- Employ ultrasonication with an amplitude of 20 W output, applying alternate cycles of 20 s ON and OFF for efficient cell lysis.
- To further enhance cell lysis, subject the post-sonicated sample to a high-pressure French press homogenizer, applying pressure at 30,000 psi (pounds per square inch) for 3 cycles to extract intracellular components efficiently.
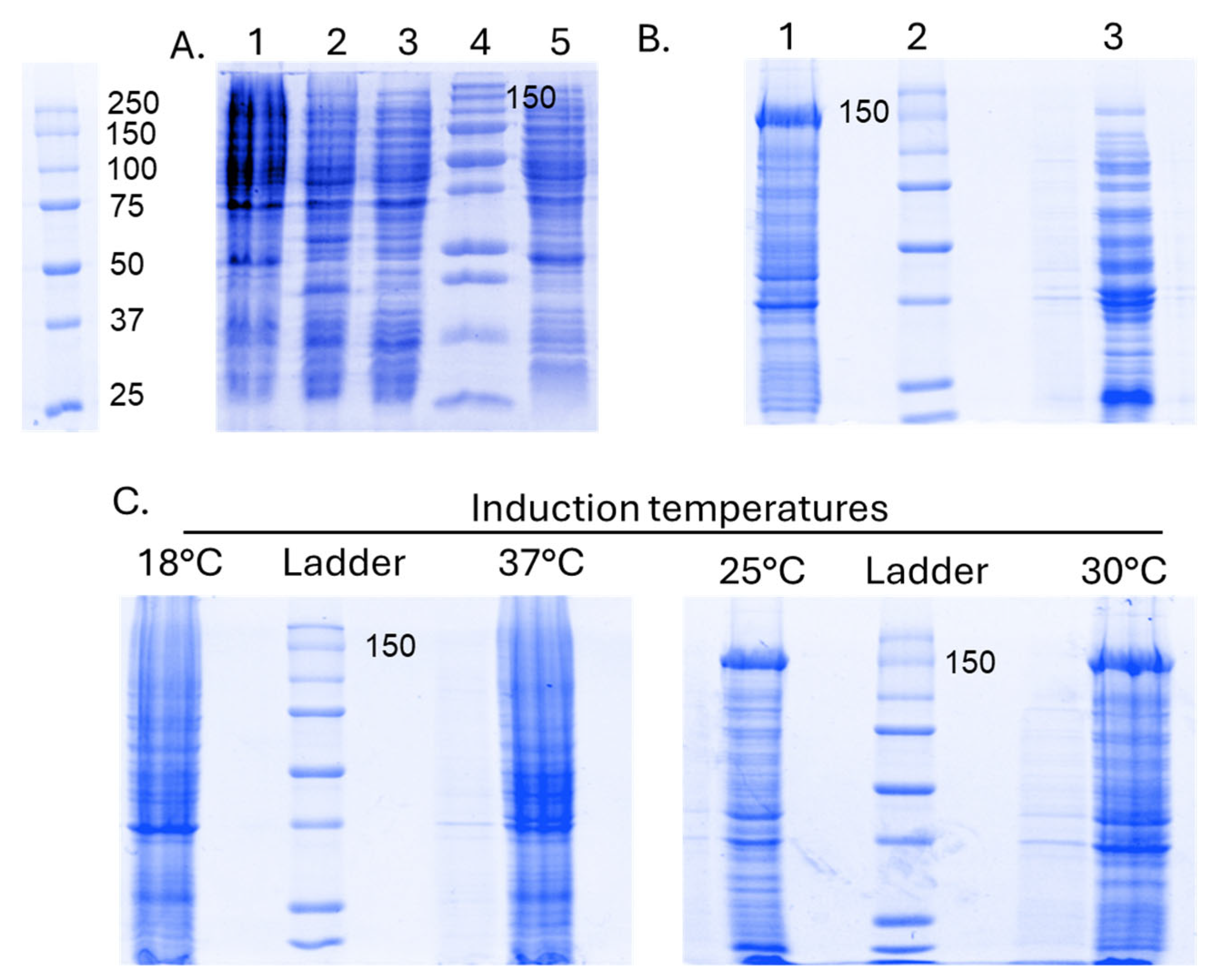
3.6.2. Purification Using Nickel-Sepharose Affinity Chromatography (Figure 3)
- Centrifuge disrupted cell suspension at 12,100 rpm for 30 min at 4 °C. Before loading onto the column, pass the supernatant through a 0.45 mm polyethersulfone membrane to eliminate any residual particulates.
- Extract a small aliquot from the supernatant for SDS-PAGE analysis.
- Apply the supernatant onto a Ni-Sepharose resin column pre-equilibrated with lysis buffer. Maintain a consistent flow rate of 1–2 mL/min. Wash the column with the lysis buffer until a stable baseline is achieved.
- Wash the column with 100 mL of wash buffer or until the A280 readings approach background levels. Save the wash-through.
- Elute the SpCas9-His protein with 50 mL of elution buffer or until the A280 is close to background. Ensure to zero the NanoDrop/spectrophotometer with the appropriate buffer if the latter method is employed.
- Identify fractions containing the SpCas9-His protein through SDS-PAGE.
- Concentrate the protein as desired and perform buffer exchange, using exchange buffer to eliminate residual imidazole through Vivaspin 20–50 kDa MWCO spin filters. Aliquot samples and store at −80 °C.Note—Aim to reduce the imidazole concentration below 3 mM from the original 250 mM concentration in the Elution Buffer. This process may take a significant amount of time, so wait until the concentrate is around 1 to 2 mL before diluting the remaining imidazole with a much smaller volume of Exchange Buffer. Otherwise, the entire concentration process may take more than two cycles.
- Measure the protein concentration by Bradford assay.
- Freeze 0.5 mL aliquots for long-term storage at −80 °C.

3.7. SDS Page
3.7.1. Preparing the Gel
- Press together two glass plates using the casting frame so the indention of the back plate is in between, leaving a slight opening in the plates. Put the casting frame in the casting stand and load deionized water onto the plate opening to the top. Check for leakage.
- To run SpCas9-His, make 10.5% resolving gel in a 15 mL conical tube. The following volumes coordinate with 1 gel. (Note: 10% APS and TEMED need to be added last because they initiate polymerization)
- a.
- The 10.5% resolving gel is composed of 1.3 mL of deionized water, 1.75 mL of 30% acrylamide, 1.875 mL Tris (Base) at pH = 8.8, 50 μL of 10% SDS, 50 μL of 10% APS, and 10 μL of TEMED.
- Pour 10.5% resolving gel into plates and add 1 mL of isopropyl alcohol to eliminate free radicals from the gel.
- After solidification make a 5% stacking gel solution in a 15 mL conical tube. The following volumes coordinate with 1 gel. (See Note above)
- a.
- The 5% stacking gel is composed of 1.875 mL of deionized water, 0.415 mL of 30% acrylamide, 0.62 mL Tris (Base) at pH = 6.8, 25 μL of 10% SDS, 25 μL of 10% APS, and 10 μL of TEMED.
- Pour 5% stacking gel into plates after removing isopropyl alcohol and insert the comb. If gel is not immediately used, wrap gels including the comb in a wet paper towel and foil and store at 4 °C.
3.7.2. Running Gel
- Put the gel into the Mini-PROTEAN Tetra Companion Running Module and put into the buffer tank.
- Load the buffer tank with SDS-PAGE running buffer until the designated lines. Ensure the module is full regardless of the level of buffer in the buffer tank.
- Load samples into wells along with ladder and run for approximately 60 min at 200 V and 100 A.
3.8. Protein Storage
- After the gel is run and pure protein is observed, quantify the protein concentration using a Bradford assay.
- Store the protein at a final concentration~1 mg/mL (usually the protein concentration is higher than 1 mg/mL after buffer exchange). Prepare single use aliquots and store at −80 °C. The protein remains active for over a year.
3.9. Western Blot
- During SDS-PAGE, prepare 10 X TBS and 1 X TBST buffers (See Section 2.6 for recipe).
- After SDS-PAGE is completed, remove the back plate and cut off the stacking gel.
- Using the Trans-Blot Turbo System, put the bottom blot of the Midi 0.2 μm Nitrocellulose Transfer Packs in one of the compartments. (Note: Be very careful not to touch the blot’s surface while taking out of the package and handling. Additionally, make sure hands and all instruments are coated with deionized water to reduce possible contamination).
- Add your gel and then the top blot, ensuring each layer is flush through rolling. Run Trans-Blot Turbo on High Molecular Weight.
- Prepare 50 mL of blocking buffer (See Section 2.6 for recipe) in a 50 mL conical tube.
- Transfer the bottom blot into box and pour in approximately 15 mL of blocking buffer and incubate on a rocker for 1 h at RT.
- Dilute the primary antibody (1:1000) in blocking buffer to a final volume of 15 mL and incubate on the rocker for 1 h at RT or overnight at 4 °C.
- Rinse the blot for 5 min 5 times with 1 X TBST, not directly pouring on the plot.
- Prepare an additional 15 mL of blocking buffer. Then dilute the secondary antibody (1:10,000) in blocking buffer to a final volume of 15 mL and incubate on the rocker for 1 h at RT. [Note: secondary antibody is conjugated to horseradish peroxidase (HRP)]
- Rinse the blot for 5 min 6 times with 1 X TBST, not directly pouring on the plot.
- Prepare the Clarity Max Western ECL Substrate Mixture by combining 6 mL of Peroxide Reagent and 6 mL of Luminol/Enhancer Reagent in a 15 mL conical tube. Add it to the blot and incubate on the rocker for approximately 2 min.
- To visualize luminescence, place blot in the ChemiDoc on the platform and roll out to make the blot flush with the platform. If marks are saturated in the image, rinse blot with deionized water to remove ECL mixture.
3.10. In Vitro Digestion
3.10.1. The Construction of Guide RNA and Amplification of Target Gene
- In this experiment, a previously validated guide RNA (gRNA) targeting the human CKM gene is utilized. The detailed protocol for constructing in vitro transcribed gRNA is referenced in Padmaswari, M. H. et al. [25].
- To begin, the human genome is extracted from the HEK293 cell line. The CKM fragment is then amplified using appropriate primers with Q5® High-Fidelity DNA Polymerase (Figure 4).
- Following amplification, the PCR product is purified using Qiaquick PCR purification. This purified CKM fragment serves as the template for subsequent steps in the experiment.
3.10.2. The Incubation of CRISPR Ribonucleoprotein Complex
- Prepare a reaction mixture of 27 μL at room temperature in the following sequential order:
Component Volume Final Concentration 10 X NEBufferTM r3.1 (NEB# B6003) 3 μL 1 X 300 nM gRNA 3 μL 30 nM 1 μM SpCas9-His 1 μL 30 nM Nuclease free water 20 μL
- 2.
- Pre-incubate the mixture at 25 °C for 10 min.
- 3.
- Gently add 3 μL of a 30 nM purified DNA fragment into the mixture, achieving a final concentration of 3 nM.
- 4.
- Thoroughly mix the components using pipette mixing.
- 5.
- Incubate the mixture at 37 °C for 15–60 min.
- 6.
- Add 1 μL of Proteinase K to the reaction and ensure thorough mixing.
- 7.
- Allow the reaction to incubate at room temperature for 10 min.
3.10.3. Fragment Analysis
- Prepare a 2% TAE agarose gel.
- Adjust the DNA ladder concentration into 100 ng/μL and load 1 μL of the DNA ladder.
- Load 30 μL of the in vitro digestion reaction.
- Run the gel for 30 min at 100 V.
- Visualize the fragment in gel imager.
4. Anticipated Results
5. Notes/Troubleshooting
- No Protein Expression: If no protein is observed, it indicates plasmid loss. Retransform BL21 Rosetta2 or other competent cells or use another aliquot from the bacterial stock. Verify the plasmid’s presence in the bacterial stock.
- Expression Host Choice and Yield Parameters: Selecting an appropriate expression host is critical for cloning. Prokaryotes, favored for their ease of handling, cost-effectiveness, and rapid cell growth, may pose challenges in upscaling for large-scale production. Key parameters influencing expression yields include growth temperature, shaker aeration speed, bacterial generation time, induction time, and IPTG concentration. Each variable requires individual testing and optimization. Determining the strain with the highest SpCas9-His expression rate among E. coli competent cells is the initial step, followed by testing various IPTG concentrations and post-induction times and temperatures. This process, evaluated at various volumes, should be compared against existing literature methods for continuous improvement.
- Histidine-Binding Affinity: The recombinant protein’s binding affinity to heparin is contingent on net charge, influenced by pH. Optimal elution under salt gradient conditions and incubation of the heparin-Sepharose column at 4 °C are recommended for efficient binding.
- Unexpected Protein Size and Proteolysis: Protease contamination or instability of the target protein can lead to proteolysis. Adding protease inhibitors during purification and adjusting temperature conditions is advised. Ni-Sepharose matrix efficiency relies on proper maintenance and storage in 20% ethanol/lysis buffer with 1 mM sodium azide at 4 °C.
- Detection Challenges: In cases of low protein expression, Western blotting with anti-SpCas9 or anti-His antibodies are recommended for specific detection, even at low concentrations.
- Time Considerations: The entire procedure, from transformation to the final purification of the SpCas9 protein, takes approximately 126 h. These optimization steps are crucial for making large-scale production more readily achievable.
- Poor Solubility of Purified Protein: Incompatibility of the lysis buffer or purification conditions with the protein could lead to inefficient lysis or purification. Optimizing lysis and purification buffers to enhance solubility, and including additives like urea or guanidine hydrochloride, if necessary, is recommended.
- Loss of Protein Stability: Inadequate storage conditions could result in denaturation during storage. The protein should be stored in a suitable buffer at recommended temperatures.Contaminants in Purified Protein: Incomplete washing or elution steps may lead to cross-contamination during purification. Improving washing steps and ensuring proper elution are essential. Implementing additional purification steps may be needed.
6. Patent
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Assenberg, R.; Wan, P.T.; Geisse, S.; Mayr, L.M. Advances in Recombinant Protein Expression for Use in Pharmaceutical Research. Curr. Opin. Struct. Biol. 2013, 23, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Wingfield, P.T. Overview of the Purification of Recombinant Proteins. Curr. Protoc. Protein Sci. 2015, 80, 6.1.1–6.1.35. [Google Scholar] [CrossRef] [PubMed]
- Maity, S.; Al-Ameer, M.; Gundampati, R.K.; Agrawal, S.; Kumar, T.K.S. Heparin-Binding Affinity Tag: A Novel Affinity Tag for Simple and Efficient Purification of Recombinant Proteins. In Protein Downstream Processing: Design, Development, and Application of High and Low-Resolution Methods; Labrou, N.E., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2021; pp. 311–328. [Google Scholar] [CrossRef]
- Mishra, V. Affinity Tags for Protein Purification. Curr. Protein Pept. Sci. 2020, 21, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Kosobokova, E.N.; Skrypnik, K.A.; Kosorukov, V.S. Overview of Fusion Tags for Recombinant Proteins. Biochemistry 2016, 81, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.; Domingues, L. Guidelines to Reach High-Quality Purified Recombinant Proteins. Appl. Microbiol. Biotechnol. 2018, 102, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Einhauer, A.; Jungbauer, A. The FLAG Peptide, a Versatile Fusion Tag for the Purification of Recombinant Proteins. J. Biochem. Biophys. Methods 2001, 49, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Méresse, S. A Method to Introduce an Internal Tag Sequence into a Salmonella Chromosomal Gene. Methods Mol. Biol. 2015, 1225, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Raran-Kurussi, S.; Waugh, D.S. Expression and Purification of Recombinant Proteins in Escherichia Coli with a His6 or Dual His6-MBP Tag. Methods Mol. Biol. 2017, 1607, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Vologzhannikova, A.A.; Khorn, P.A.; Kazakov, A.S.; Permyakov, E.A.; Uversky, V.N.; Permyakov, S.E. Effects of His-Tags on Physical Properties of Parvalbumins. Cell Calcium 2019, 77, 1–7. [Google Scholar] [CrossRef]
- Le, N.T.P.; Phan, T.T.P.; Phan, H.T.T.; Truong, T.T.T.; Schumann, W.; Nguyen, H.D. Influence of N-Terminal His-Tags on the Production of Recombinant Proteins in the Cytoplasm of Bacillus Subtilis. Biotechnol. Rep. 2022, 35, e00754. [Google Scholar] [CrossRef]
- Raran-Kurussi, S.; Cherry, S.; Zhang, D.; Waugh, D.S. Removal of Affinity Tags with TEV Protease. Methods Mol. Biol. 2017, 1586, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Kerr, R.; Agrawal, S.; Maity, S.; Koppolu, B.; Jayanthi, S.; Suresh Kumar, G.; Gundampati, R.K.; McNabb, D.S.; Zaharoff, D.A.; Kumar, T.K.S. Design of a Thrombin Resistant Human Acidic Fibroblast Growth Factor (hFGF1) Variant That Exhibits Enhanced Cell Proliferation Activity. Biochem. Biophys. Res. Commun. 2019, 518, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Spriestersbach, A.; Kubicek, J.; Schäfer, F.; Block, H.; Maertens, B. Purification of His-Tagged Proteins. Methods Enzym. Enzymol. 2015, 559, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. The New Frontier of Genome Engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Doudna, J.A. CRISPR Technology: A Decade of Genome Editing Is Only the Beginning. Science 2023, 379, eadd8643. [Google Scholar] [CrossRef]
- Morisaka, H.; Yoshimi, K.; Okuzaki, Y.; Gee, P.; Kunihiro, Y.; Sonpho, E.; Xu, H.; Sasakawa, N.; Naito, Y.; Nakada, S.; et al. CRISPR-Cas3 Induces Broad and Unidirectional Genome Editing in Human Cells. Nat. Commun. 2019, 10, 5302. [Google Scholar] [CrossRef] [PubMed]
- Teng, A.C.T.; Tavassoli, M.; Shrestha, S.; Marks, R.M.; McFadden, M.J.; Evagelou, S.L.; Lindsay, K.; Vandenbelt, A.; Li, W.; Ivakine, E.; et al. An Efficient and Cost-Effective Purification Protocol for Staphylococcus Aureus Cas9 Nuclease. STAR Protoc. 2023, 4, 101933. [Google Scholar] [CrossRef] [PubMed]
- Carmignotto, G.P.; Azzoni, A.R. On the Expression of Recombinant Cas9 Protein in E. Coli BL21(DE3) and BL21(DE3) Rosetta Strains. J. Biotechnol. 2019, 306, 62–70. [Google Scholar] [CrossRef]
- Singpant, P.; Tubsuwan, A.; Sakdee, S.; Ketterman, A.J.; Jearawiriyapaisarn, N.; Kurita, R.; Nakamura, Y.; Songdej, D.; Tangprasittipap, A.; Bhukhai, K.; et al. Recombinant Cas9 Protein Production in an Endotoxin-Free System and Evaluation with Editing the BCL11A Gene in Human Cells. Protein Expr. Purif. 2023, 210, 106313. [Google Scholar] [CrossRef]
- Evmenov, K.; Pustogarov, N.; Panteleev, D.; Safin, A.; Alkalaeva, E. An Efficient Expression and Purification Protocol for SpCas9 Nuclease and Evaluation of Different Delivery Methods of Ribonucleoprotein. Int. J. Mol. Sci. 2024, 25, 1622. [Google Scholar] [CrossRef]
- Flottmann, F.; Pohl, G.M.; Gummert, J.; Milting, H.; Brodehl, A. A Detailed Protocol for Expression, Purification, and Activity Determination of Recombinant SaCas9. STAR Protoc. 2022, 3, 101276. [Google Scholar] [CrossRef] [PubMed]
- Hayat, S.M.G.; Farahani, N.; Golichenari, B.; Sahebkar, A. Recombinant Protein Expression in Escherichia coli (E. coli): What We Need Know. Curr. Pharm. Des. 2018, 24, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Studier, F.W. Mechanism of Inhibition of Bacteriophage T7 RNA Polymerase by T7 Lysozyme. J. Mol. Biol. 1997, 269, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Padmaswari, M.H.; Bulliard, G.; Agrawal, S.; Jia, M.; Nelson, C. Precision and Efficacy of RNA-Guided DNA Integration in High-Expressing Muscle Loci. bioRxiv 2024. [Google Scholar] [CrossRef] [PubMed]
- Kane, J.F. Effects of Rare Codon Clusters on High-Level Expression of Heterologous Proteins in Escherichia Coli. Curr. Opin. Biotechnol. 1995, 6, 494–500. [Google Scholar] [CrossRef]
- Wang, Q.; Cobine, P.A.; Coleman, J.J. Efficient Genome Editing in Fusarium Oxysporum Based on CRISPR/Cas9 Ribonucleoprotein Complexes. Fungal Genet. Biol. 2018, 117, 21–29. [Google Scholar] [CrossRef]
- Fleitas, A.L.; Señorale, M.; Vidal, S. A Robust Expression and Purification Method for Production of SpCas9-GFP-MBP Fusion Protein for In vitro Applications. Methods Protoc. 2022, 5, 44. [Google Scholar] [CrossRef]
- Rajagopalan, N.; Kagale, S.; Bhowmik, P.; Song, H. A Two-Step Method for Obtaining Highly Pure Cas9 Nuclease for Genome Editing, Biophysical, and Structural Studies. Methods Protoc. 2018, 1, 17. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Strain | Features | Benefits | Growth Condition | Relative Ranking in Terms of the Protein Yield in This Study |
|---|---|---|---|---|
| BL21(DE3) | Inclusion of T7 polymerase. | Expression of nontoxic genes. | Kanamycin (50 μg/mL) | 3 |
| BL21(DE3)-Star | Includes mutated RNase for increased mRNA stability. | Tight control expression. Expression of both soluble and insoluble proteins. | Kanamycin (50 μg/mL) | 2 |
| BL21(DE3)-pLysS | Includes pLysS plasmid expressing T7 lysosome to hinder pre-induction expression. | Expression of toxic genes. Mitigates leaky expression. | Kanamycin (50 μg/mL) | 1 |
| Rosetta2 | Inclusion of rare codons not generally found in E. coli | Expression of heterogenous proteins containing rare codons. | Kanamycin (50 μg/mL) and Chloramphenicol (30 μg/mL) | 4 |
| Process | Hands on Time (h) | Approximate Time Required (h) |
|---|---|---|
| Preparation of plasmid from Addgene (bacterial growth + colony pick-up + plasmid isolation) | 1 | 14 + 14 + 1 |
| Bacterial transformation (transformation + colony pick-up + glycerol stock prep) | 1.5 | 2 + 14 + 0.5 |
| Small-scale expression (bacterial growth + induction + harvesting + SDS PAGE) | 3 | 18 + 6 + 1 + 2 |
| Large-scale expression (bacterial growth + induction + harvesting) | 4 | 18 + 6 + 1 |
| Protein purification (bacterial cell lysis + purification + SDS PAGE) | 6 | 0.5 + 5 + 2 |
| Buffer exchange and protein storage | 1 | 6 + 0.5 |
| Western Blot (SDS PAGE + protein transfer and detection) | 4 | 2 + 6 |
| In vitro digestion | 2 | 6 |
| Total | 22.5 | 126 h/6 days |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agrawal, S.; Padmaswari, M.H.; Stokes, A.L.; Maxenberger, D.; Reese, M.; Khalil, A.; Nelson, C.E. Optimizing Recombinant Cas9 Expression: Insights from E. coli BL21(DE3) Strains for Enhanced Protein Purification and Genome Editing. Biomedicines 2024, 12, 1226. https://doi.org/10.3390/biomedicines12061226
Agrawal S, Padmaswari MH, Stokes AL, Maxenberger D, Reese M, Khalil A, Nelson CE. Optimizing Recombinant Cas9 Expression: Insights from E. coli BL21(DE3) Strains for Enhanced Protein Purification and Genome Editing. Biomedicines. 2024; 12(6):1226. https://doi.org/10.3390/biomedicines12061226
Chicago/Turabian StyleAgrawal, Shilpi, Made Harumi Padmaswari, Abbey L. Stokes, Daniel Maxenberger, Morgan Reese, Adila Khalil, and Christopher E. Nelson. 2024. "Optimizing Recombinant Cas9 Expression: Insights from E. coli BL21(DE3) Strains for Enhanced Protein Purification and Genome Editing" Biomedicines 12, no. 6: 1226. https://doi.org/10.3390/biomedicines12061226
APA StyleAgrawal, S., Padmaswari, M. H., Stokes, A. L., Maxenberger, D., Reese, M., Khalil, A., & Nelson, C. E. (2024). Optimizing Recombinant Cas9 Expression: Insights from E. coli BL21(DE3) Strains for Enhanced Protein Purification and Genome Editing. Biomedicines, 12(6), 1226. https://doi.org/10.3390/biomedicines12061226