
The Role of Lipoprotein(a) in Peripheral Artery Disease



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Epidemiology
3. Structure/Genetics
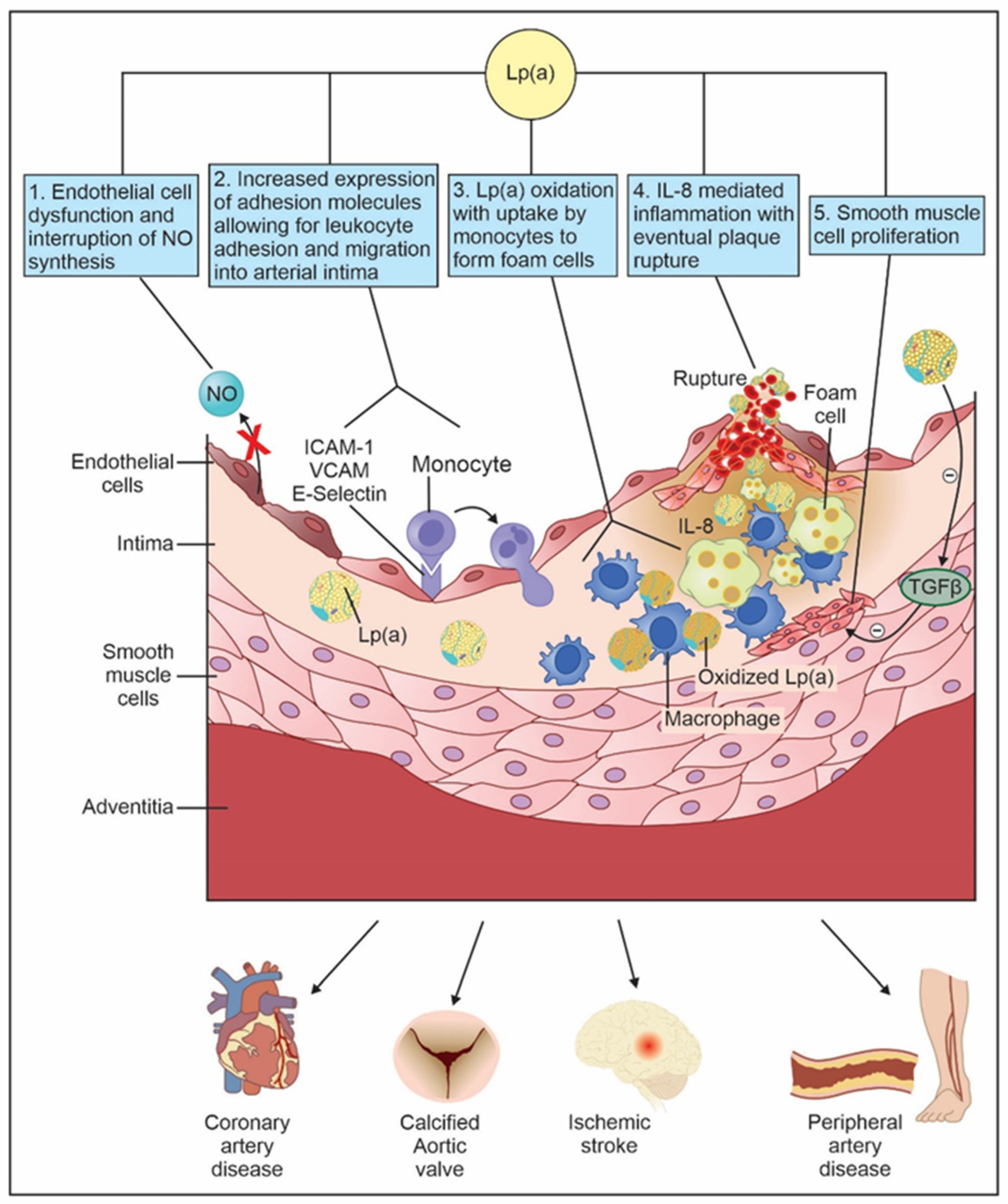
4. Pathogenesis
5. Diagnostic Testing
6. Screening
7. Lp(a) in PAD
8. Management
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berg, K. A New Serum Type System in Man—The Lp System. Acta Pathol. Microbiol. Scand. 1963, 59, 369–382. [Google Scholar] [CrossRef]
- Berg, K.; Dahlén, G.; Frick, M.H. Lp(a) lipoprotein and pre-beta1-lipoprotein in patients with coronary heart disease. Clin. Genet. 1974, 6, 230–235. [Google Scholar] [CrossRef]
- Maloberti, A.; Fabbri, S.; Colombo, V.; Gualini, E.; Monticelli, M.; Daus, F.; Busti, A.; Galasso, M.; De Censi, L.; Algeri, M.; et al. Lipoprotein(a): Cardiovascular Disease, Aortic Stenosis and New Therapeutic Option. Int. J. Mol. Sci. 2022, 24, 170. [Google Scholar] [CrossRef]
- Wilson, D.P.; Jacobson, T.A.; Jones, P.H.; Koschinsky, M.L.; McNeal, C.J.; Nordestgaard, B.G.; Orringer, C.E. Use of Lipoprotein(a) in clinical practice: A biomarker whose time has come: A scientific statement from the National Lipid Association. J. Clin. Lipidol. 2019, 13, 374–392. [Google Scholar] [CrossRef]
- Raitakari, O.; Kivelä, A.; Pahkala, K.; Rovio, S.; Mykkänen, J.; Ahola-Olli, A.; Loo, B.M.; Lyytikäinen, L.P.; Lehtimäki, T.; Kähönen, M.; et al. Long-term tracking and population characteristics of lipoprotein (a) in the Cardiovascular Risk in Young Finns Study. Atherosclerosis 2022, 356, 18–27. [Google Scholar] [CrossRef]
- Ward, N.C.; Kostner, K.M.; Sullivan, D.R.; Nestel, P.; Watts, G.F. Molecular, Population, and Clinical Aspects of Lipoprotein(a): A Bridge Too Far? J. Clin. Med. 2019, 8, 2073. [Google Scholar] [CrossRef]
- Khan, T.Z.; Bornstein, S.R.; Barbir, M. Lipoprotein(a): The underutilized risk factor for cardiovascular disease. Glob. Cardiol. Sci. Pract. 2019, 2019, e201911. [Google Scholar] [CrossRef]
- Shu, J.; Santulli, G. Update on peripheral artery disease: Epidemiology and evidence-based facts. Atherosclerosis 2018, 275, 379–381. [Google Scholar] [CrossRef]
- Fowkes, F.G.R.; Rudan, D.; Rudan, I.; Aboyans, V.; Denenberg, J.O.; McDermott, M.M.; Norman, P.E.; Sampson, U.K.; Williams, L.J.; Mensah, G.A.; et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: A systematic review and analysis. Lancet 2013, 382, 1329–1340. [Google Scholar] [CrossRef]
- Criqui, M.H.; Matsushita, K.; Aboyans, V.; Hess, C.N.; Hicks, C.W.; Kwan, T.W.; McDermott, M.M.; Misra, S.; Ujueta, F.; American Heart Association Council on Epidemiology and Prevention; et al. Lower Extremity Peripheral Artery Disease: Contemporary Epidemiology, Management Gaps, and Future Directions: A Scientific Statement from the American Heart Association. Circulation 2021, 144, e171–e191. [Google Scholar] [CrossRef]
- Sartipy, F.; Sigvant, B.; Lundin, F.; Wahlberg, E. Ten Year Mortality in Different Peripheral Arterial Disease Stages: A Population Based Observational Study on Outcome. Eur. J. Vasc. Endovasc. Surg. 2018, 55, 529–536. [Google Scholar] [CrossRef]
- Scully, R.E.; Arnaoutakis, D.J.; DeBord Smith, A.; Semel, M.; Nguyen, L.L. Estimated annual health care expenditures in individuals with peripheral arterial disease. J. Vasc. Surg. 2018, 67, 558–567. [Google Scholar] [CrossRef]
- Kosmas, C.E.; Silverio, D.; Sourlas, A.; Peralta, R.; Montan, P.D.; Guzman, E.; Garcia, M.J. Role of lipoprotein (a) in peripheral arterial disease. Ann. Transl. Med. 2019, 7 (Suppl. S6), S242. [Google Scholar] [CrossRef]
- Thomas, P.E.; Vedel-Krogh, S.; Nielsen, S.F.; Nordestgaard, B.G.; Kamstrup, P.R. Lipoprotein(a) and Risks of Peripheral Artery Disease, Abdominal Aortic Aneurysm, and Major Adverse Limb Events. J. Am. Coll. Cardiol. 2023, 82, 2265–2276. [Google Scholar] [CrossRef]
- Tsimikas, S.; Fazio, S.; Ferdinand, K.C.; Ginsberg, H.N.; Koschinsky, M.L.; Marcovina, S.M.; Moriarty, P.M.; Rader, D.J.; Remaley, A.T.; Reyes-Soffer, G.; et al. NHLBI Working Group Recommendations to Reduce Lipoprotein(a)-Mediated Risk of Cardiovascular Disease and Aortic Stenosis. J. Am. Coll. Cardiol. 2018, 71, 177–192. [Google Scholar] [CrossRef]
- Bilen, O.; Kamal, A.; Virani, S.S. Lipoprotein abnormalities in South Asians and its association with cardiovascular disease: Current state and future directions. World J. Cardiol. 2016, 8, 247–257. [Google Scholar] [CrossRef]
- Paré, G.; Çaku, A.; McQueen, M.; Anand, S.S.; Enas, E.; Clarke, R.; Boffa, M.B.; Koschinsky, M.; Wang, X.; Yusuf, S.; et al. Lipoprotein(a) Levels and the Risk of Myocardial Infarction Among 7 Ethnic Groups. Circulation 2019, 139, 1472–1482. [Google Scholar] [CrossRef]
- Lee, S.R.; Prasad, A.; Choi, Y.S.; Xing, C.; Clopton, P.; Witztum, J.L.; Tsimikas, S. LPA Gene, Ethnicity, and Cardiovascular Events. Circulation 2017, 135, 251–263. [Google Scholar] [CrossRef]
- Guan, W.; Cao, J.; Steffen, B.T.; Post, W.S.; Stein, J.H.; Tattersall, M.C.; Kaufman, J.D.; McConnell, J.P.; Hoefner, D.M.; Warnick, R.; et al. Race is a key variable in assigning lipoprotein(a) cutoff values for coronary heart disease risk assessment: The Multi-Ethnic Study of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 996–1001. [Google Scholar] [CrossRef]
- Makshood, M.; Joshi, P.H.; Kanaya, A.M.; Ayers, C.; Budoff, M.; Tsai, M.Y.; Blaha, M.; Michos, E.D.; Post, W.S. Lipoprotein (a) and aortic valve calcium in South Asians compared to other race/ethnic groups. Atherosclerosis 2020, 313, 14–19. [Google Scholar] [CrossRef]
- Bhatia, H.S.; Hurst, S.; Desai, P.; Zhu, W.; Yeang, C. Lipoprotein(a) Testing Trends in a Large Academic Health System in the United States. J. Am. Heart Assoc. 2023, 12, e031255. [Google Scholar] [CrossRef]
- Kelsey, M.D.; Mulder, H.; Chiswell, K.; Lampron, Z.M.; Nilles, E.; Kulinski, J.P.; Joshi, P.H.; Jones, W.S.; Chamberlain, A.M.; Leucker, T.M.; et al. Contemporary patterns of lipoprotein(a) testing and associated clinical care and outcomes. Am. J. Prev. Cardiol. 2023, 14, 100478. [Google Scholar] [CrossRef]
- Schmidt, K.; Noureen, A.; Kronenberg, F.; Utermann, G. Structure, function, and genetics of lipoprotein (a). J. Lipid Res. 2016, 57, 1339–1359. [Google Scholar] [CrossRef]
- Behbodikhah, J.; Ahmed, S.; Elyasi, A.; Kasselman, L.J.; De Leon, J.; Glass, A.D.; Reiss, A.B. Apolipoprotein B and Cardiovascular Disease: Biomarker and Potential Therapeutic Target. Metabolites 2021, 11, 690. [Google Scholar] [CrossRef]
- Banerjee, R.; Weideman, S.; Fernandez-Vazquez, D.; Banerjee, A.; Hasan, A.; Tsai, S. Role of Lipoprotein A in Lower Extremity Peripheral Artery Disease. Am. J. Cardiol. 2023, 198, 47–49. [Google Scholar] [CrossRef]
- Maranhão, R.C.; Carvalho, P.O.; Strunz, C.C.; Pileggi, F. Lipoprotein (a): Structure, pathophysiology and clinical implications. Arq. Bras. Cardiol. 2014, 103, 76–84. [Google Scholar] [CrossRef]
- Rehberger Likozar, A.; Zavrtanik, M.; Šebeštjen, M. Lipoprotein(a) in atherosclerosis: From pathophysiology to clinical relevance and treatment options. Ann. Med. 2020, 52, 162–177. [Google Scholar] [CrossRef]
- Kronenberg, F. Human Genetics and the Causal Role of Lipoprotein(a) for Various Diseases. Cardiovasc. Drugs Ther. 2016, 30, 87–100. [Google Scholar] [CrossRef]
- Coassin, S.; Kronenberg, F. Lipoprotein(a) beyond the kringle IV repeat polymorphism: The complexity of genetic variation in the LPA gene. Atherosclerosis 2022, 349, 17–35. [Google Scholar] [CrossRef]
- Hopewell, J.C.; Clarke, R.; Parish, S.; Armitage, J.; Lathrop, M.; Hager, J.; Collins, R. Lipoprotein(a) genetic variants associated with coronary and peripheral vascular disease but not with stroke risk in the Heart Protection Study. Circ. Cardiovasc. Genet. 2011, 4, 68–73. [Google Scholar] [CrossRef]
- Clarke, R.; Peden, J.F.; Hopewell, J.C.; Kyriakou, T.; Goel, A.; Heath, S.C.; Parish, S.; Barlera, S.; Franzosi, M.G.; Rust, S.; et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N. Engl. J. Med. 2009, 361, 2518–2528. [Google Scholar] [CrossRef]
- Klarin, D.; Lynch, J.; Aragam, K.; Chaffin, M.; Assimes, T.L.; Huang, J.; Lee, K.M.; Shao, Q.; Huffman, J.E.; Natarajan, P.; et al. Genome-wide association study of peripheral artery disease in the Million Veteran Program. Nat. Med. 2019, 25, 1274–1279. [Google Scholar] [CrossRef]
- Takami, S.; Yamashita, S.; Kihara, S.; Ishigami, M.; Takemura, K.; Kume, N.; Kita, T.; Matsuzawa, Y. Lipoprotein(a) enhances the expression of intercellular adhesion molecule-1 in cultured human umbilical vein endothelial cells. Circulation 1998, 97, 721–728. [Google Scholar] [CrossRef]
- Poon, M.; Zhang, X.; Dunsky, K.G.; Taubman, M.B.; Harpel, P.C. Apolipoprotein(a) induces monocyte chemotactic activity in human vascular endothelial cells. Circulation 1997, 96, 2514–2519. [Google Scholar] [CrossRef]
- Rath, M.; Niendorf, A.; Reblin, T.; Dietel, M.; Krebber, H.J.; Beisiegel, U. Detection and quantification of lipoprotein(a) in the arterial wall of 107 coronary bypass patients. Arteriosclerosis 1989, 9, 579–592. [Google Scholar] [CrossRef]
- Cho, T.; Jung, Y.; Koschinsky, M.L. Apolipoprotein(a), through its strong lysine-binding site in KIV(10’), mediates increased endothelial cell contraction and permeability via a Rho/Rho kinase/MYPT1-dependent pathway. J. Biol. Chem. 2008, 283, 30503–30512. [Google Scholar] [CrossRef]
- Deb, A.; Caplice, N.M. Lipoprotein(a): New insights into mechanisms of atherogenesis and thrombosis. Clin. Cardiol. 2004, 27, 258–264. [Google Scholar] [CrossRef]
- Javadifar, A.; Rastgoo, S.; Banach, M.; Jamialahmadi, T.; Johnston, T.P.; Sahebkar, A. Foam Cells as Therapeutic Targets in Atherosclerosis with a Focus on the Regulatory Roles of Non-Coding RNAs. Int. J. Mol. Sci. 2021, 22, 2529. [Google Scholar] [CrossRef]
- Grainger, D.J.; Kemp, P.R.; Liu, A.C.; Lawn, R.M.; Metcalfe, J.C. Activation of transforming growth factor-beta is inhibited in transgenic apolipoprotein(a) mice. Nature 1994, 370, 460–462. [Google Scholar] [CrossRef]
- Lampsas, S.; Xenou, M.; Oikonomou, E.; Pantelidis, P.; Lysandrou, A.; Sarantos, S.; Goliopoulou, A.; Kalogeras, K.; Tsigkou, V.; Kalpis, A.; et al. Lipoprotein(a) in Atherosclerotic Diseases: From Pathophysiology to Diagnosis and Treatment. Molecules 2023, 28, 969. [Google Scholar] [CrossRef]
- Klezovitch, O.; Edelstein, C.; Scanu, A.M. Stimulation of interleukin-8 production in human THP-1 macrophages by apolipoprotein(a). Evidence for a critical involvement of elements in its C-terminal domain. J. Biol. Chem. 2001, 276, 46864–46869. [Google Scholar] [CrossRef]
- Scipione, C.A.; Sayegh, S.E.; Romagnuolo, R.; Tsimikas, S.; Marcovina, S.M.; Boffa, M.B.; Koschinsky, M.L. Mechanistic insights into Lp(a)-induced IL-8 expression: A role for oxidized phospholipid modification of apo(a). J. Lipid Res. 2015, 56, 2273–2285. [Google Scholar] [CrossRef]
- Loscalzo, J.; Weinfeld, M.; Fless, G.M.; Scanu, A.M. Lipoprotein(a), fibrin binding, and plasminogen activation. Arteriosclerosis 1990, 10, 240–245. [Google Scholar] [CrossRef]
- Ugovšek, S.; Šebeštjen, M. Lipoprotein(a)-The Crossroads of Atherosclerosis, Atherothrombosis and Inflammation. Biomolecules 2021, 12, 26. [Google Scholar] [CrossRef]
- Etingin, O.R.; Hajjar, D.P.; Hajjar, K.A.; Harpel, P.C.; Nachman, R.L. Lipoprotein (a) regulates plasminogen activator inhibitor-1 expression in endothelial cells. A potential mechanism in thrombogenesis. J. Biol. Chem. 1991, 266, 2459–2465. [Google Scholar] [CrossRef]
- Cegla, J.; France, M.; Marcovina, S.M.; Neely, R.D.G. Lp(a): When and how to measure it. Ann. Clin. Biochem. 2021, 58, 16–21. [Google Scholar] [CrossRef]
- Kamstrup, P.R. Lipoprotein(a) and Cardiovascular Disease. Clin. Chem. 2021, 67, 154–166. [Google Scholar] [CrossRef]
- Reyes-Soffer, G.; Ginsberg, H.N.; Berglund, L.; Duell, P.B.; Heffron, S.P.; Kamstrup, P.R.; Lloyd-Jones, D.M.; Marcovina, S.M.; Yeang, C.; Koschinsky, M.L. Lipoprotein(a): A Genetically Determined, Causal, and Prevalent Risk Factor for Atherosclerotic Cardiovascular Disease: A Scientific Statement from the American Heart Association. Arterioscler. Thromb. Vasc. Biol. 2022, 42, e48–e60. [Google Scholar] [CrossRef]
- Kronenberg, F.; Mora, S.; Stroes, E.S.G.; Ference, B.A.; Arsenault, B.J.; Berglund, L.; Dweck, M.R.; Koschinsky, M.; Lambert, G.; Mach, F.; et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: A European Atherosclerosis Society consensus statement. Eur. Heart J. 2022, 43, 3925–3946. [Google Scholar] [CrossRef]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2019, 73, 3168–3209. [Google Scholar] [CrossRef]
- Enkhmaa, B.; Berglund, L. Non-genetic influences on lipoprotein(a) concentrations. Atherosclerosis 2022, 349, 53–62. [Google Scholar] [CrossRef]
- US Preventive Services Task Force; Curry, S.J.; Krist, A.H.; Owens, D.K.; Barry, M.J.; Caughey, A.B.; Davidson, K.W.; Doubeni, C.A.; Epling, J.W.; Kemper, A.R.; et al. Screening for Peripheral Artery Disease and Cardiovascular Disease Risk Assessment with the Ankle-Brachial Index: US Preventive Services Task Force Recommendation Statement. JAMA 2018, 320, 177–183. [Google Scholar] [CrossRef]
- Aboyans, V.; Ricco, J.B.; Bartelink, M.L.E.L.; Björck, M.; Brodmann, M.; Cohnert, T.; Collet, J.P.; Czerny, M.; De Carlo, M.; Debus, S.; et al. 2017 ESC Guidelines on the Diagnosis and Treatment of Peripheral Arterial Diseases, in collaboration with the European Society for Vascular Surgery (ESVS). Eur. Heart J. 2018, 39, 763–816. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Pearson, G.J.; Thanassoulis, G.; Anderson, T.J.; Barry, A.R.; Couture, P.; Dayan, N.; Francis, G.A.; Genest, J.; Grégoire, J.; Grover, S.A.; et al. 2021 Canadian Cardiovascular Society Guidelines for the Management of Dyslipidemia for the Prevention of Cardiovascular Disease in Adults. Can. J. Cardiol. 2021, 37, 1129–1150. [Google Scholar] [CrossRef]
- Koschinsky, M.L.; Bajaj, A.; Boffa, M.B.; Dixon, D.L.; Ferdinand, K.C.; Gidding, S.S.; Gill, E.A.; Jacobson, T.A.; Michos, E.D.; Safarova, M.S.; et al. A focused update to the 2019 NLA scientific statement on use of lipoprotein(a) in clinical practice. J. Clin. Lipidol. 2024, in press. [CrossRef]
- Guédon, A.F.; De Freminville, J.B.; Mirault, T.; Mohamedi, N.; Rance, B.; Fournier, N.; Paul, J.L.; Messas, E.; Goudot, G. Association of Lipoprotein(a) Levels with Incidence of Major Adverse Limb Events. JAMA Netw. Open 2022, 5, e2245720. [Google Scholar] [CrossRef]
- Gurdasani, D.; Sjouke, B.; Tsimikas, S.; Hovingh, G.K.; Luben, R.N.; Wainwright, N.W.; Pomilla, C.; Wareham, N.J.; Khaw, K.T.; Boekholdt, S.M.; et al. Lipoprotein(a) and risk of coronary, cerebrovascular, and peripheral artery disease: The EPIC-Norfolk prospective population study. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 3058–3065. [Google Scholar] [CrossRef]
- Dieplinger, B.; Lingenhel, A.; Baumgartner, N.; Poelz, W.; Dieplinger, H.; Haltmayer, M.; Kronenberg, F.; Mueller, T. Increased serum lipoprotein(a) concentrations and low molecular weight phenotypes of apolipoprotein(a) are associated with symptomatic peripheral arterial disease. Clin. Chem. 2007, 53, 1298–1305. [Google Scholar] [CrossRef]
- Golledge, J.; Rowbotham, S.; Velu, R.; Quigley, F.; Jenkins, J.; Bourke, M.; Bourke, B.; Thanigaimani, S.; Chan, D.C.; Watts, G.F. Association of Serum Lipoprotein (a) With the Requirement for a Peripheral Artery Operation and the Incidence of Major Adverse Cardiovascular Events in People with Peripheral Artery Disease. J. Am. Heart Assoc. 2020, 9, e015355. [Google Scholar] [CrossRef]
- Tzoulaki, I.; Murray, G.D.; Lee, A.J.; Rumley, A.; Lowe, G.D.O.; Fowkes, F.G.R. Inflammatory, haemostatic, and rheological markers for incident peripheral arterial disease: Edinburgh Artery Study. Eur. Heart J. 2007, 28, 354–362. [Google Scholar] [CrossRef]
- Volpato, S.; Vigna, G.B.; McDermott, M.M.; Cavalieri, M.; Maraldi, C.; Lauretani, F.; Bandinelli, S.; Zuliani, G.; Guralnik, J.M.; Fellin, R.; et al. Lipoprotein(a), inflammation, and peripheral arterial disease in a community-based sample of older men and women (the InCHIANTI study). Am. J. Cardiol. 2010, 105, 1825–1830. [Google Scholar] [CrossRef]
- Sanchez Muñoz-Torrero, J.F.; Rico-Martín, S.; Álvarez, L.R.; Aguilar, E.; Alcalá, J.N.; Monreal, M.; FRENA Investigators. Lipoprotein (a) levels and outcomes in stable outpatients with symptomatic artery disease. Atherosclerosis 2018, 276, 10–14. [Google Scholar] [CrossRef]
- Yi, C.; Junyi, G.; Fengju, L.; Qing, Z.; Jie, C. Association between lipoprotein(a) and peripheral arterial disease in coronary artery bypass grafting patients. Clin. Cardiol. 2023, 46, 512–520. [Google Scholar] [CrossRef]
- Ridker, P.M.; Stampfer, M.J.; Rifai, N. Novel risk factors for systemic atherosclerosis: A comparison of C-reactive protein, fibrinogen, homocysteine, lipoprotein(a), and standard cholesterol screening as predictors of peripheral arterial disease. JAMA 2001, 285, 2481–2485. [Google Scholar] [CrossRef]
- Pradhan, A.D.; Shrivastava, S.; Cook, N.R.; Rifai, N.; Creager, M.A.; Ridker, P.M. Symptomatic peripheral arterial disease in women: Nontraditional biomarkers of elevated risk. Circulation 2008, 117, 823–831. [Google Scholar] [CrossRef]
- Zierfuss, B.; Höbaus, C.; Feldscher, A.; Hannes, A.; Mrak, D.; Koppensteiner, R.; Stangl, H.; Schernthaner, G.H. Lipoprotein (a) and long-term outcome in patients with peripheral artery disease undergoing revascularization. Atherosclerosis 2022, 363, 94–101. [Google Scholar] [CrossRef]
- Gerhard-Herman, M.D.; Gornik, H.L.; Barrett, C.; Barshes, N.R.; Corriere, M.A.; Drachman, D.E.; Fleisher, L.A.; Fowkes, F.G.; Hamburg, N.M.; Kinlay, S.; et al. 2016 AHA/ACC Guideline on the Management of Patients with Lower Extremity Peripheral Artery Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2017, 135, e686–e725. [Google Scholar] [CrossRef]
- Westin, G.G.; Armstrong, E.J.; Bang, H.; Yeo, K.K.; Anderson, D.; Dawson, D.L.; Pevec, W.C.; Amsterdam, E.A.; Laird, J.R. Association between statin medications and mortality, major adverse cardiovascular event, and amputation-free survival in patients with critical limb ischemia. J. Am. Coll. Cardiol. 2014, 63, 682–690. [Google Scholar] [CrossRef]
- Kumbhani, D.J.; Steg, P.G.; Cannon, C.P.; Eagle, K.A.; Smith, S.C.; Goto, S.; Ohman, E.M.; Elbez, Y.; Sritara, P.; Baumgartner, I.; et al. Statin therapy and long-term adverse limb outcomes in patients with peripheral artery disease: Insights from the REACH registry. Eur. Heart J. 2014, 35, 2864–2872. [Google Scholar] [CrossRef]
- Ramos, R.; García-Gil, M.; Comas-Cufí, M.; Quesada, M.; Marrugat, J.; Elosua, R.; Sala, J.; Grau, M.; Martí, R.; Ponjoan, A.; et al. Statins for Prevention of Cardiovascular Events in a Low-Risk Population with Low Ankle Brachial Index. J. Am. Coll. Cardiol. 2016, 67, 630–640. [Google Scholar] [CrossRef]
- Heart Protection Study Collaborative Group. Randomized trial of the effects of cholesterol-lowering with simvastatin on peripheral vascular and other major vascular outcomes in 20,536 people with peripheral arterial disease and other high-risk conditions. J. Vasc. Surg. 2007, 45, 645–654; discussion 653–654. [Google Scholar] [CrossRef]
- Arsenault, B.J.; Barter, P.; DeMicco, D.A.; Bao, W.; Preston, G.M.; LaRosa, J.C.; Grundy, S.M.; Deedwania, P.; Greten, H.; Wenger, N.K.; et al. Prediction of cardiovascular events in statin-treated stable coronary patients of the treating to new targets randomized controlled trial by lipid and non-lipid biomarkers. PLoS ONE 2014, 9, e114519. [Google Scholar] [CrossRef]
- Khera, A.V.; Everett, B.M.; Caulfield, M.P.; Hantash, F.M.; Wohlgemuth, J.; Ridker, P.M.; Mora, S. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: An analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin). Circulation 2014, 129, 635–642. [Google Scholar] [CrossRef]
- Fraley, A.E.; Schwartz, G.G.; Olsson, A.G.; Kinlay, S.; Szarek, M.; Rifai, N.; Libby, P.; Ganz, P.; Witztum, J.L.; Tsimikas, S.; et al. Relationship between biomarkers of oxidized low-density lipoprotein, statin therapy, quantitative coronary angiography, and atheroma: Volume observations from the REVERSAL trial. J. Am. Coll. Cardiol. 2009, 53, 2186–2196. [Google Scholar] [CrossRef]
- Choi, S.H.; Chae, A.; Miller, E.; Messig, M.; Ntanios, F.; DeMaria, A.N.; Nissen, S.E.; Witztum, J.L.; Tsimikas, S. Relationship between biomarkers of oxidized low-density lipoprotein, statin therapy, quantitative coronary angiography, and atheroma: Volume observations from the REVERSAL (Reversal of Atherosclerosis with Aggressive Lipid Lowering) study. J. Am. Coll. Cardiol. 2008, 52, 24–32. [Google Scholar] [CrossRef]
- Capoulade, R.; Chan, K.L.; Yeang, C.; Mathieu, P.; Bossé, Y.; Dumesnil, J.G.; Tam, J.W.; Teo, K.K.; Mahmut, A.; Yang, X.; et al. Oxidized Phospholipids, Lipoprotein(a), and Progression of Calcific Aortic Valve Stenosis. J. Am. Coll. Cardiol. 2015, 66, 1236–1246. [Google Scholar] [CrossRef]
- Willeit, P.; Ridker, P.M.; Nestel, P.J.; Simes, J.; Tonkin, A.M.; Pedersen, T.R.; Schwartz, G.G.; Olsson, A.G.; Colhoun, H.M.; Kronenberg, F.; et al. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: Individual patient-data meta-analysis of statin outcome trials. Lancet 2018, 392, 1311–1320. [Google Scholar] [CrossRef]
- van Capelleveen, J.C.; van der Valk, F.M.; Stroes, E.S.G. Current therapies for lowering lipoprotein (a). J. Lipid Res. 2016, 57, 1612–1618. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bittner, V.A.; Diaz, R.; Goodman, S.G.; Kim, Y.U.; Jukema, J.W.; Pordy, R.; Roe, M.T.; et al. Peripheral Artery Disease and Venous Thromboembolic Events After Acute Coronary Syndrome: Role of Lipoprotein(a) and Modification by Alirocumab: Prespecified Analysis of the ODYSSEY OUTCOMES Randomized Clinical Trial. Circulation 2020, 141, 1608–1617. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Bonaca, M.P.; Nault, P.; Giugliano, R.P.; Keech, A.C.; Pineda, A.L.; Kanevsky, E.; Kuder, J.; Murphy, S.A.; Jukema, J.W.; Lewis, B.S.; et al. Low-Density Lipoprotein Cholesterol Lowering with Evolocumab and Outcomes in Patients with Peripheral Artery Disease: Insights from the FOURIER Trial (Further Cardiovascular Outcomes Research With PCSK9 Inhibition in Subjects With Elevated Risk). Circulation 2018, 137, 338–350. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Fazio, S.; Giugliano, R.P.; Stroes, E.S.G.; Kanevsky, E.; Gouni-Berthold, I.; Im, K.; Pineda, A.L.; Wasserman, A.M.; Češka, R.; et al. Lipoprotein(a), PCSK9 Inhibition, and Cardiovascular Risk. Circulation 2019, 139, 1483–1492. [Google Scholar] [CrossRef]
- Chennamsetty, I.; Kostner, K.M.; Claudel, T.; Vinod, M.; Frank, S.; Weiss, T.S.; Trauner, M.; Kostner, G.M. Nicotinic acid inhibits hepatic APOA gene expression: Studies in humans and in transgenic mice. J. Lipid Res. 2012, 53, 2405–2412. [Google Scholar] [CrossRef]
- Kamanna, V.S.; Kashyap, M.L. Mechanism of action of niacin. Am. J. Cardiol. 2008, 101, 20B–26B. [Google Scholar] [CrossRef]
- AIM-HIGH Investigators; Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef]
- Albers, J.J.; Slee, A.; O’Brien, K.D.; Robinson, J.G.; Kashyap, M.L.; Kwiterovich, P.O.; Xu, P.; Marcovina, S.M. Relationship of apolipoproteins A-1 and B, and lipoprotein(a) to cardiovascular outcomes: The AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglyceride and Impact on Global Health Outcomes). J. Am. Coll. Cardiol. 2013, 62, 1575–1579. [Google Scholar] [CrossRef]
- HPS2-THRIVE Collaborative Group; Landray, M.J.; Haynes, R.; Hopewell, J.C.; Parish, S.; Aung, T.; Tomson, J.; Wallendszus, K.; Craig, M.; Jiang, L.; et al. Effects of extended-release niacin with laropiprant in high-risk patients. N. Engl. J. Med. 2014, 371, 203–212. [Google Scholar] [CrossRef]
- Waldmann, E.; Parhofer, K.G. Lipoprotein apheresis to treat elevated lipoprotein (a). J. Lipid Res. 2016, 57, 1751–1757. [Google Scholar] [CrossRef]
- Leebmann, J.; Roeseler, E.; Julius, U.; Heigl, F.; Spitthoever, R.; Heutling, D.; Breitenberger, P.; Maerz, W.; Lehmacher, W.; Heibges, A.; et al. Lipoprotein apheresis in patients with maximally tolerated lipid-lowering therapy, lipoprotein(a)-hyperlipoproteinemia, and progressive cardiovascular disease: Prospective observational multicenter study. Circulation 2013, 128, 2567–2576. [Google Scholar] [CrossRef]
- Poller, W.C.; Dreger, H.; Morgera, S.; Berger, A.; Flessenkämper, I.; Enke-Melzer, K. Lipoprotein apheresis in patients with peripheral artery disease and hyperlipoproteinemia(a). Atheroscler. Suppl. 2015, 18, 187–193. [Google Scholar] [CrossRef]
- Koutsogianni, A.D.; Liamis, G.; Liberopoulos, E.; Adamidis, P.S.; Florentin, M. Effects of Lipid-Modifying and Other Drugs on Lipoprotein(a) Levels-Potent Clinical Implications. Pharmaceuticals 2023, 16, 750. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Ditmarsch, M.; Kastelein, J.J.; Rigby, S.P.; Kling, D.; Curcio, D.L.; Alp, N.J.; Davidson, M.H. Lipid lowering effects of the CETP inhibitor obicetrapib in combination with high-intensity statins: A randomized phase 2 trial. Nat. Med. 2022, 28, 1672–1678. [Google Scholar] [CrossRef]
- Cuchel, M.; Meagher, E.A.; du Toit Theron, H.; Blom, D.J.; Marais, A.D.; Hegele, R.A.; Averna, M.R.; Sirotri, C.R.; Shah, P.K.; Gaudet, D.; et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: A single-arm, open-label, phase 3 study. Lancet 2013, 381, 40–46. [Google Scholar] [CrossRef]
- Tsimikas, S.; Karwatowska-Prokopczuk, E.; Gouni-Berthold, I.; Tardif, J.C.; Baum, S.J.; Steinhagen-Thiessen, E.; Shapiro, M.D.; Stroes, E.S.; Moriarty, P.M.; Nordestgaard, B.G.; et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N. Engl. J. Med. 2020, 382, 244–255. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Rosenson, R.S.; Gencer, B.; López, J.A.G.; Lepor, N.A.; Baum, S.J.; Stout, E.; Gaudet, D.; Knusel, B.; Kuder, J.F.; et al. Small Interfering RNA to Reduce Lipoprotein(a) in Cardiovascular Disease. N. Engl. J. Med. 2022, 387, 1855–1864. [Google Scholar] [CrossRef]
- Nissen, S.E.; Linnebjerg, H.; Shen, X.; Wolski, K.; Ma, X.; Lim, S.; Michael, L.; Ruotolo, G.; Gribble, G.; Navar, A.M.; et al. Lepodisiran, an Extended-Duration Short Interfering RNA Targeting Lipoprotein(a): A Randomized Dose-Ascending Clinical Trial. JAMA 2023, 330, 2075–2083. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K.; Watts, G.F.; Koren, M.J.; Fok, H.; Nicholls, S.J.; Rider, D.A.; Cho, L.; Romano, S.; Melgaard, C.; et al. Single Ascending and Multiple-Dose Trial of Zerlasiran, a Short Interfering RNA Targeting Lipoprotein(a): A Randomized Clinical Trial. JAMA 2024, 331, 1534–1543. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Nissen, S.E.; Fleming, C.; Urva, S.; Suico, J.; Berg, P.H.; Linnebjerg, H.; Ruotolo, G.; Turner, P.K.; Michael, L.F. Muvalaplin, an Oral Small Molecule Inhibitor of Lipoprotein(a) Formation: A Randomized Clinical Trial. JAMA 2023, 330, 1042–1053. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavlatos, N.; Kalra, D.K. The Role of Lipoprotein(a) in Peripheral Artery Disease. Biomedicines 2024, 12, 1229. https://doi.org/10.3390/biomedicines12061229
Pavlatos N, Kalra DK. The Role of Lipoprotein(a) in Peripheral Artery Disease. Biomedicines. 2024; 12(6):1229. https://doi.org/10.3390/biomedicines12061229
Chicago/Turabian StylePavlatos, Nicholas, and Dinesh K. Kalra. 2024. "The Role of Lipoprotein(a) in Peripheral Artery Disease" Biomedicines 12, no. 6: 1229. https://doi.org/10.3390/biomedicines12061229
APA StylePavlatos, N., & Kalra, D. K. (2024). The Role of Lipoprotein(a) in Peripheral Artery Disease. Biomedicines, 12(6), 1229. https://doi.org/10.3390/biomedicines12061229