
Obesity, Metabolic Syndrome, and Osteoarthritis Require Integrative Understanding and Management
 , and
, and
Abstract
:1. Introduction
2. The Epidemic of WAT Pathology and Its Correlation with Joint Health and OA
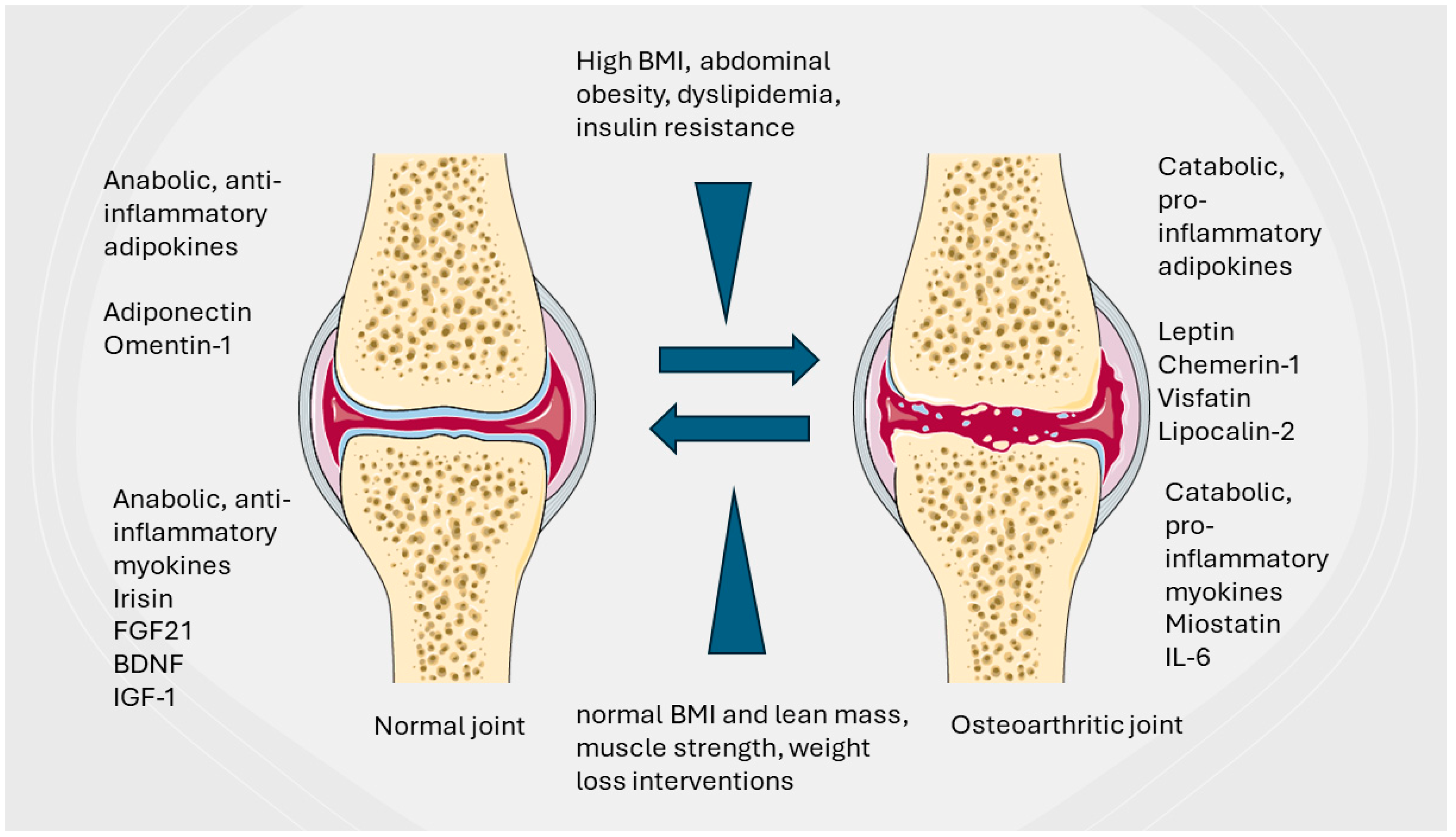
2.1. Metabolic Syndrome and Obesity in OA Pathogenesis
2.2. Epidemiologic Evidence Correlating Obesity and Metabolic Syndrome with OA
2.3. All Articular Joint Tissues Are Affected by Obesity and MetS
2.3.1. Synovial Tissue and Synovial Macrophages
2.3.2. Chondrocyte and Cartilage Layer
2.3.3. Subchondral Bone
2.3.4. Articular and Periarticular Fat Deposits
2.3.5. Articular and Periarticular Stabilization Structures—Menisci and Ligaments
3. Obesity and MetS Adipokine Disbalance Impact OA Occurrence and Progression
3.1. Leptin
3.2. Resistin
3.3. Visfatin
3.4. Lipocalin 2
3.5. Chemerin
3.6. Adiponectin
3.7. Progranulin
3.8. Vaspin
3.9. Omentin-1
4. White Adipose Tissue Distribution Disorders and Their Implications for OA Pathogenesis
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, D.; Shen, J.; Zhao, W.; Wang, T.; Han, L.; Hamilton, J.L.; Im, H.-J. Osteoarthritis: Toward a comprehensive understanding of pathological mechanism. Bone Res. 2017, 5, 16044. [Google Scholar] [CrossRef]
- Greene, M.; Loeser, R. Aging-related inflammation in osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1966–1971. [Google Scholar] [CrossRef]
- GBD 2021 Osteoarthritis Collaborators. Global, regional, and national burden of osteoarthritis, 1990–2020 and projections to 2050: A systematic analysis for the Global Burden of Disease Study 2021. Lancet Rheumatol. 2023, 5, e508–e522. [Google Scholar] [CrossRef]
- Wei, G.; Lu, K.; Umar, M.; Zhu, Z.; Lu, W.W.; Speakman, J.R.; Chen, Y.; Tong, L.; Chen, D. Risk of metabolic abnormalities in osteoarthritis: A new perspective to understand its pathological mechanisms. Bone Res. 2023, 11, 63. [Google Scholar] [CrossRef]
- Aderinto, N.; Abdulbasit, M.O.; Tangmi, A.D.E.; Okesanya, J.O.; Mubarak, J.M. Unveiling the growing significance of metabolism in modulating immune cell function: Exploring mechanisms and implications; a review. Ann. Med. Surg. 2023, 85, 5511–5522. [Google Scholar] [CrossRef]
- Terkawi, M.A.; Ebata, T.; Yokota, S.; Takahashi, D.; Endo, T.; Matsumae, G.; Shimizu, T.; Kadoya, K.; Iwasaki, N. Low-Grade Inflammation in the Pathogenesis of Osteoarthritis: Cellular and Molecular Mechanisms and Strategies for Future Therapeutic Intervention. Biomedicines 2022, 10, 1109. [Google Scholar] [CrossRef]
- Naumovs, V.; Groma, V.; Mednieks, J. From Low-Grade Inflammation in Osteoarthritis to Neuropsychiatric Sequelae: A Narrative Review. Int. J. Mol. Sci. 2022, 23, 16031. [Google Scholar] [CrossRef]
- Yunus, M.H.M.; Nordin, A.; Kamal, H. Pathophysiological Perspective of Osteoarthritis. Medicina 2020, 56, 614. [Google Scholar] [CrossRef]
- White, U. Adipose tissue expansion in obesity, health, and disease. Front. Cell Dev. Biol. 2023, 11, 1188844. [Google Scholar] [CrossRef]
- Wang, X.; Xu, M.; Li, Y. Adipose Tissue Aging and Metabolic Disorder, and the Impact of Nutritional Interventions. Nutrients 2022, 14, 3134. [Google Scholar] [CrossRef]
- Fantuzzi, G. Adipose tissue, adipokines, and inflammation. J. Allergy Clin. Immunol. 2005, 115, 911–919. [Google Scholar] [CrossRef]
- Bing, C.; Russell, S.; Becket, E.; Pope, M.; Tisdale, M.J.; Trayhurn, P.; Jenkins, J.R. Adipose atrophy in cancer cachexia: Morphologic and molecular analysis of adipose tissue in tumour-bearing mice. Br. J. Cancer 2006, 95, 1028–1037. [Google Scholar] [CrossRef]
- Schrover, I.M.; Van Der Graaf, Y.; Spiering, W.; Visseren, F.L.; SMART study group. The relation between body fat distribution, plasma concentrations of adipokines and the metabolic syndrome in patients with clinically manifest vascular disease. Eur. J. Prev. Cardiol. 2018, 25, 1548–1557. [Google Scholar] [CrossRef]
- Canale, M.P.; di Villahermosa, S.M.; Martino, G.; Rovella, V.; Noce, A.; De Lorenzo, A.; Di Daniele, N. Obesity-related metabolic syndrome: Mechanisms of sympathetic overactivity. Int. J. Endocrinol. 2013, 2013, 865965. [Google Scholar] [CrossRef]
- Czaja-Stolc, S.; Potrykus, M.; Stankiewicz, M.; Kaska, Ł.; Małgorzewicz, S. Pro-Inflammatory Profile of Adipokines in Obesity Contributes to Pathogenesis, Nutritional Disorders, and Cardiovascular Risk in Chronic Kidney Disease. Nutrients 2022, 14, 1457. [Google Scholar] [CrossRef]
- Mangion, D.; Pace, N.P.; Formosa, M.M. The relationship between adipokine levels and bone mass—A systematic review. Endocrinol. Diabetes Metab. 2023, 6, e408. [Google Scholar] [CrossRef]
- Scotece, M.; Conde, J.; Vuolteenaho, K.; Koskinen, A.; López, V.; Gómez-Reino, J.; Lago, F.; Moilanen, E.; Gualillo, O. Adipokines as drug targets in joint and bone disease. Drug Discov. Today 2014, 19, 241–258. [Google Scholar] [CrossRef]
- Giardullo, L.; Corrado, A.; Maruotti, N.; Cici, D.; Mansueto, N.; Cantatore, F.P. Adipokine role in physiopathology of inflammatory and degenerative musculoskeletal diseases. Int. J. Immunopathol. Pharmacol. 2021, 35, 20587384211015034. [Google Scholar] [CrossRef]
- Mansilla, E.; Diaz Aquino, V.; Zambón, D.; Marin, G.H.; Mártire, K.; Roque, G.; Ichim, T.; Riordan, N.H.; Patel, A.; Sturla, F.; et al. Could metabolic syndrome, lipodystrophy, and aging be mesenchymal stem cell exhaustion syndromes? Stem Cells Int. 2011, 2011, 943216. [Google Scholar] [CrossRef]
- Rohm, M.; Zeigerer, A.; Machado, J.; Herzig, S. Energy metabolism in cachexia. EMBO Rep. 2019, 20, e47258. [Google Scholar] [CrossRef]
- Boutari, C.; Mantzoros, C.S. A 2022 update on the epidemiology of obesity and a call to action: As its twin COVID-19 pandemic appears to be receding, the obesity and dysmetabolism pandemic continues to rage on. Metabolism 2022, 133, 155217. [Google Scholar] [CrossRef]
- Poobalan, A.; Aucott, L. Obesity Among Young Adults in Developing Countries: A Systematic Overview. Curr. Obes. Rep. 2016, 5, 2–13. [Google Scholar] [CrossRef]
- González-Álvarez, M.A.; Lázaro-Alquézar, A.; Simón-Fernández, M.B. Global Trends in Child Obesity: Are Figures Converging? Int. J. Environ. Res. Public Health 2020, 17, 9252. [Google Scholar] [CrossRef]
- World Obesity Atlas 2023. Available online: https://data.worldobesity.org/publications/WOF-Obesity-Atlas-V5.pdf (accessed on 25 March 2024).
- Ackerman, I.N.; Kemp, J.L.; Crossley, K.M.; Culvenor, A.G.; Hinman, R.S. Hip and Knee Osteoarthritis Affects Younger People, Too. J. Orthop. Sports Phys. Ther. 2017, 47, 67–79. [Google Scholar] [CrossRef]
- Swarup, S.; Goyal, A.; Grigorova, Y.; Zeltser, R. Metabolic Syndrome. 2022 Oct 24. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Dickson, B.M.; Roelofs, A.J.; Rochford, J.J.; Wilson, H.M.; De Bari, C. The burden of metabolic syndrome on osteoarthritic joints. Arthritis Res. Ther. 2019, 21, 289. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Tian, W.; Wang, Y.; Rong, J.; Bao, C.; Liu, Y.; Zhao, Y.; Wang, C. Body mass index and susceptibility to knee osteoarthritis: A systematic review and meta-analysis. Jt. Bone Spine 2012, 79, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Park, Y.-M.; Ko, S.-H.; Hyun, K.-S.; Choi, Y.-H.; Min, D.-U.; Han, K.; Koh, H.-S. Association of general and central obesity, and their changes with risk of knee osteoarthritis: A nationwide population-based cohort study. Sci. Rep. 2023, 13, 3796. [Google Scholar] [CrossRef]
- Messier, S.P.; Gutekunst, D.J.; Davis, C.; DeVita, P. Weight loss reduces knee-joint loads in overweight and obese older adults with knee osteoarthritis. Arthritis Rheum. 2005, 52, 2026–2032. [Google Scholar] [CrossRef]
- Suh, D.; Han, K.; Hong, J.; Park, J.; Bae, J.; Moon, Y.; Kim, J. Body composition is more closely related to the development of knee osteoarthritis in women than men: A cross-sectional study using the Fifth Korea National Health and Nutrition Examination Survey (KNHANES V-1, 2). Osteoarthr. Cartil. 2016, 24, 605–611. [Google Scholar] [CrossRef]
- Felson, D.T. The epidemiology of knee osteoarthritis: Results from the Framingham Osteoarthritis study. Semin. Arthritis Rheum. 1990, 20 (Suppl. S1), 42–50. [Google Scholar] [CrossRef]
- Nelson, A.E.; Hu, D.; Arbeeva, L.; Alvarez, C.; Cleveland, R.J.; Schwartz, T.A.; Murphy, L.B.; Helmick, C.G.; Callahan, L.F.; Renner, J.B.; et al. The Prevalence of Knee Symptoms, Radiographic, and Symptomatic Osteoarthritis at Four Time Points: The Johnston County Osteoarthritis Project, 1999–2018. ACR Open Rheumatol. 2021, 3, 558–565. [Google Scholar] [CrossRef]
- Ikram, M.A.; Brusselle, G.G.O.; Murad, S.D.; van Duijn, C.M.; Franco, O.H.; Goedegebure, A.; Klaver, C.C.W.; Nijsten, T.E.C.; Peeters, R.P.; Stricker, B.H.; et al. The Rotterdam Study: 2018 update on objectives, design and main results. Eur. J. Epidemiol. 2017, 32, 807–850. [Google Scholar] [CrossRef]
- Reijman, M.; Pols, H.A.P.; Bergink, A.P.; Hazes, J.M.W.; Belo, J.N.; Lievense, A.M.; A Bierma-Zeinstra, S.M. Body mass index associated with onset and progression of osteoarthritis of the knee but not of the hip: The Rotterdam Study. Ann. Rheum. Dis. 2007, 66, 158–162. [Google Scholar] [CrossRef]
- Raud, B.; Gay, C.; Guiguet-Auclair, C.; Bonnin, A.; Gerbaud, L.; Pereira, B.; Duclos, M.; Boirie, Y.; Coudeyre, E. Level of obesity is directly associated with the clinical and functional consequences of knee osteoarthritis. Sci. Rep. 2020, 10, 3601. [Google Scholar] [CrossRef]
- Messier, S.P.; Mihalko, S.L.; Legault, C.; Miller, G.D.; Nicklas, B.J.; DeVita, P.; Beavers, D.P.; Hunter, D.J.; Lyles, M.F.; Eckstein, F.; et al. Effects of intensive diet and exercise on knee joint loads, inflammation, and clinical outcomes among overweight and obese adults with knee osteoarthritis: The IDEA randomized clinical trial. JAMA 2013, 310, 1263–1273. [Google Scholar] [CrossRef]
- Törmälehto, S.; Aarnio, E.; Mononen, M.E.; Arokoski, J.P.A.; Korhonen, R.K.; Martikainen, J.A. Eight-year trajectories of changes in health-related quality of life in knee osteoarthritis: Data from the Osteoarthritis Initiative (OAI). PLoS ONE 2019, 14, e0219902. [Google Scholar] [CrossRef]
- Katz, J.N.; Arant, K.R.; Loeser, R.F. Diagnosis and Treatment of Hip and Knee Osteoarthritis: A Review. JAMA 2021, 325, 568–578. [Google Scholar] [CrossRef]
- Heuts, E.A.; de Jong, L.D.; Hazebroek, E.J.; Wagener, M.; Somford, M.P. The influence of bariatric surgery on hip and knee joint pain: A systematic review. Surg. Obes. Relat. Dis. 2021, 17, 1637–1653. [Google Scholar] [CrossRef]
- Xiong, J.; Long, J.; Chen, X.; Li, Y.; Song, H. Dyslipidemia Might Be Associated with an Increased Risk of Osteoarthritis. BioMed Res. Int. 2020, 2020, 3105248. [Google Scholar] [CrossRef]
- Lo, K.; Au, M.; Ni, J.; Wen, C. Association between hypertension and osteoarthritis: A systematic review and meta-analysis of observational studies. J. Orthop. Transl. 2021, 32, 12–20. [Google Scholar] [CrossRef]
- Yang, Z.-J.; Liu, Y.; Liu, Y.-L.; Qi, B.; Yuan, X.; Shi, W.-X.; Miao, L. Osteoarthritis and hypertension: Observational and Mendelian randomization analyses. Arthritis Res. Ther. 2024, 26, 88. [Google Scholar] [CrossRef] [PubMed]
- Tchetina, E.V.; A Markova, G.; Sharapova, E.P. Insulin Resistance in Osteoarthritis: Similar Mechanisms to Type 2 Diabetes Mellitus. J. Nutr. Metab. 2020, 2020, 4143802. [Google Scholar] [CrossRef] [PubMed]
- Zaharia, O.P.; Pesta, D.H.; Bobrov, P.; Kupriyanova, Y.; Herder, C.; Karusheva, Y.; Bódis, K.; Bönhof, G.J.; Knitza, J.; Simon, D.; et al. Reduced Muscle Strength Is Associated with Insulin Resistance in Type 2 Diabetes Patients with Osteoarthritis. J. Clin. Endocrinol. Metab. 2021, 106, e1062–e1073. [Google Scholar] [CrossRef]
- Culemann, S.; Grüneboom, A.; Krönke, G. Origin and function of synovial macrophage subsets during inflammatory joint disease. Adv Immunol. 2019, 143, 75–98. [Google Scholar] [CrossRef] [PubMed]
- Haubruck, P.; Pinto, M.M.; Moradi, B.; Little, C.B.; Gentek, R. Monocytes, Macrophages, and Their Potential Niches in Synovial Joints-Therapeutic Targets in Post-Traumatic Osteoarthritis? Front. Immunol. 2021, 12, 763702. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, M.; Zhao, J.; Zheng, M.; Yang, H. Imbalance of M1/M2 macrophages is linked to severity level of knee osteoarthritis. Exp. Ther. Med. 2018, 16, 5009–5014. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.-J.; Szczesny, B.; Wan, X.; Putluri, N.; Garg, N.J. Pentose Phosphate Shunt Modulates Reactive Oxygen Species and Nitric Oxide Production Controlling Trypanosoma cruzi in Macrophages. Front. Immunol. 2018, 9, 202. [Google Scholar] [CrossRef] [PubMed]
- Lepetsos, P.; Papavassiliou, A.G. ROS/oxidative stress signaling in osteoarthritis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2016, 1862, 576–591. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lin, C.; Zeng, C.; Wang, Z.; Wang, H.; Lu, J.; Liu, X.; Shao, Y.; Zhao, C.; Pan, J.; et al. Synovial macrophage M1 polarisation exacerbates experimental osteoarthritis partially through R-spondin-2. Ann. Rheum. Dis. 2018, 77, 1524–1534. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, E.Y.; Liu, J.; Yu, J.J. Inhibition of the mechanistic target of rapamycin induces cell survival via MAPK in tuberous sclerosis complex. Orphanet J. Rare Dis. 2020, 15, 209. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [PubMed]
- Steenvoorden, M.M.C.; Huizinga, T.W.J.; Verzijl, N.; Bank, R.A.; Ronday, H.K.; Luning, H.A.F.; Lafeber, F.P.J.G.; Toes, R.E.M.; DeGroot, J. Activation of receptor for advanced glycation end products in osteoarthritis leads to increased stimulation of chondrocytes and synoviocytes. Arthritis Rheum. 2006, 54, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Lin, C.; Lu, Y.; Guan, H.; Qi, W.; Zhang, H.; Shao, Y.; Zeng, C.; Zhang, R.; Zhang, H.; et al. FABP4 secreted by M1-polarized macrophages promotes synovitis and angiogenesis to exacerbate rheumatoid arthritis. Bone Res. 2022, 10, 45, Erratum in Bone Res. 2023, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Ayele, T.M.; Muche, Z.T.; Teklemariam, A.B.; Bogale, A.; Abebe, E.C. Role of JAK2/STAT3 Signaling Pathway in the Tumorigenesis, Chemotherapy Resistance, and Treatment of Solid Tumors: A Systemic Review. J. Inflamm. Res. 2022, 15, 1349–1364. [Google Scholar] [CrossRef]
- Jiang, H.; Pu, Y.; Li, Z.H.; Liu, W.; Deng, Y.; Liang, R.; Zhang, X.M.; Zuo, H.D. Adiponectin, May Be a Potential Protective Factor for Obesity-Related Osteoarthritis. Diabetes Metab. Syndr. Obes. 2022, 15, 1305–1319. [Google Scholar] [CrossRef]
- Thapa, B.; Lee, K. Metabolic influence on macrophage polarization and pathogenesis. BMB Rep. 2019, 52, 360–372. [Google Scholar] [CrossRef]
- Zhou, S.; Lu, W.; Chen, L.; Ge, Q.; Chen, D.; Xu, Z.; Shi, D.; Dai, J.; Li, J.; Ju, H.; et al. AMPK deficiency in chondrocytes accelerated the progression of instability-induced and ageing-associated osteoarthritis in adult mice. Sci. Rep. 2017, 7, 43245. [Google Scholar] [CrossRef]
- De Luna-Preitschopf, A.; Zwickl, H.; Nehrer, S.; Hengstschläger, M.; Mikula, M. Rapamycin Maintains the Chondrocytic Phenotype and Interferes with Inflammatory Cytokine Induced Processes. Int. J. Mol. Sci. 2017, 18, 1494. [Google Scholar] [CrossRef]
- Dhanabalan, K.M.; Dravid, A.A.; Agarwal, S.; Sharath, R.K.; Padmanabhan, A.K.; Agarwal, R. Intra-articular injection of rapamycin microparticles prevent senescence and effectively treat osteoarthritis. Bioeng. Transl. Med. 2022, 8, e10298. [Google Scholar] [CrossRef]
- Medina-Luna, D.; Santamaría-Olmedo, M.G.; Zamudio-Cuevas, Y.; Martínez-Flores, K.; Fernández-Torres, J.; Martínez-Nava, G.A.; Clavijo-Cornejo, D.; Hernández-Díaz, C.; Olivos-Meza, A.; Gomez-Quiroz, L.E.; et al. Hyperlipidemic microenvironment conditionates damage mechanisms in human chondrocytes by oxidative stress. Lipids Health Dis. 2017, 16, 114. [Google Scholar] [CrossRef]
- Haywood, J.; Yammani, R. Free fatty acid palmitate activates unfolded protein response pathway and promotes apoptosis in meniscus cells. Osteoarthr. Cartil. 2016, 24, 942–945. [Google Scholar] [CrossRef] [PubMed]
- Felson, D.; Misra, D.; LaValley, M.; Clancy, M.; Chen, X.; Lichtenstein, A.; Matthan, N.; Torner, J.; Lewis, C.; Nevitt, M. Fatty acids and osteoarthritis: The MOST study. Osteoarthr. Cartil. 2021, 29, 973–978. [Google Scholar] [CrossRef]
- Singh, V.; Oliashirazi, A.; Tan, T.; Fayyad, A.; Shahi, A. Clinical and Pathophysiologic Significance of MRI Identified Bone Marrow Lesions Associated with Knee Osteoarthritis. Arch. Bone Jt. Surg. 2019, 7, 211–219. [Google Scholar]
- Azzini, G.O.M.; Santos, G.S.; Visoni, S.B.C.; Azzini, V.O.M.; dos Santos, R.G.; Huber, S.C.; Lana, J.F. Metabolic syndrome and subchondral bone alterations: The rise of osteoarthritis–A review. J. Clin. Orthop. Trauma 2020, 11 (Suppl. S5), S849–S855. [Google Scholar] [CrossRef] [PubMed]
- Findlay, D.M. Vascular pathology and osteoarthritis. Rheumatology 2007, 46, 1763–1768. [Google Scholar] [CrossRef] [PubMed]
- Triantaphyllidou, I.-E.; Kalyvioti, E.; Karavia, E.; Lilis, I.; Kypreos, K.; Papachristou, D. Perturbations in the HDL metabolic pathway predispose to the development of osteoarthritis in mice following long-term exposure to western-type diet. Osteoarthr. Cartil. 2013, 21, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Davies-Tuck, M.L.; Hanna, F.; Davis, S.R.; Bell, R.J.; Davison, S.L.; E Wluka, A.; Adams, J.; Cicuttini, F.M. Total cholesterol and triglycerides are associated with the development of new bone marrow lesions in asymptomatic middle-aged women-a prospective cohort study. Arthritis Res. Ther. 2009, 11, R181. [Google Scholar] [CrossRef] [PubMed]
- Doré, D.; de Hoog, J.; Giles, G.; Ding, C.; Cicuttini, F.; Jones, G. A longitudinal study of the association between dietary factors, serum lipids, and bone marrow lesions of the knee. Arthritis Res. Ther. 2012, 14, R13. [Google Scholar] [CrossRef] [PubMed]
- Veronese, N.; Cooper, C.; Reginster, J.-Y.; Hochberg, M.; Branco, J.; Bruyère, O.; Chapurlat, R.; Al-Daghri, N.; Dennison, E.; Herrero-Beaumont, G.; et al. Type 2 diabetes mellitus and osteoarthritis. Semin. Arthritis Rheum. 2019, 49, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Hamada, D.; Maynard, R.; Schott, E.; Drinkwater, C.J.; Ketz, J.P.; Kates, S.L.; Jonason, J.H.; Hilton, M.J.; Zuscik, M.J.; Mooney, R.A. Suppressive Effects of Insulin on Tumor Necrosis Factor–Dependent Early Osteoarthritic Changes Associated With Obesity and Type 2 Diabetes Mellitus. Arthritis Rheumatol. 2016, 68, 1392–1402. [Google Scholar] [CrossRef]
- Ioan-Facsinay, A.; Kloppenburg, M. An emerging player in knee osteoarthritis: The infrapatellar fat pad. Arthritis Res. Ther. 2013, 15, 225–229. [Google Scholar] [CrossRef]
- Paduszyński, W.; Jeśkiewicz, M.; Uchański, P.; Gackowski, S.; Radkowski, M.; Demkow, U. Hoffa’s Fat Pad Abnormality in the De-velopment of Knee Osteoarthritis. Adv. Exp. Med. Biol. 2018, 1039, 95–102. [Google Scholar] [CrossRef]
- Labusca, L.; Zugun-Eloae, F. The Unexplored Role of Intra-articular Adipose Tissue in the Homeostasis and Pathology of Articular Joints. Front. Vet. Sci. 2018, 5, 35. [Google Scholar] [CrossRef]
- Steidle-Kloc, E.; Dannhauer, T.; Wirth, W.; Eckstein, F. Responsiveness of Infrapatellar Fat Pad Volume Change to Body Weight Loss or Gain: Data from the Osteoarthritis Initiative. Cells Tissues Organs 2018, 205, 53–62. [Google Scholar] [CrossRef]
- Duan, L.; Ma, Y.; Wang, Y.; Liu, J.; Tan, Z.; Wu, Q.; Wu, Y.; Yu, X. Infrapatellar fat pads participate in the development of knee osteoarthritis in obese patients via the activation of the NF-κB signaling pathway. Int. J. Mol. Med. 2020, 46, 2260–2270. [Google Scholar] [CrossRef]
- Greif, D.N.; Kouroupis, D.; Murdock, C.J.; Griswold, A.J.; Kaplan, L.D.; Best, T.M.; Correa, D. Infrapatellar Fat Pad/Synovium Complex in Early-Stage Knee Osteoarthritis: Potential New Target and Source of Therapeutic Mesenchymal Stem/Stromal Cells. Front. Bioeng. Biotechnol. 2020, 8, 860. [Google Scholar] [CrossRef]
- Jayasekera, N.; Aprato, A.; Villar, R.N. Fat pad entrapment at the hip: A new diagnosis. PLoS ONE 2014, 9, e83503. [Google Scholar] [CrossRef]
- Bodden, J.; Ok, A.H.; Joseph, G.B.; Nevitt, M.C.; McCulloch, C.E.; Lane, N.E.; Link, T.M. Joint-adjacent Adipose Tissue by MRI is Associated With Prevalence and Progression of Knee Degenerative Changes: Data from the Osteoarthritis Initiative. J. Magn. Reson. Imaging 2021, 54, 155–165. [Google Scholar] [CrossRef]
- Englund, M.; Roemer, F.W.; Hayashi, D.; Crema, M.D.; Guermazi, A. Meniscus pathology, osteoarthritis and the treatment con-troversy. Nat. Rev. Rheumatol. 2012, 8, 412–419. [Google Scholar] [CrossRef]
- Rai, M.F.; Sandell, L.J.; Cheverud, J.M.; Brophy, R.H. Relationship of age and body mass index to the expression of obesity and osteoarthritis-related genes in human meniscus. Int. J. Obes. 2013, 37, 1238–1246. [Google Scholar] [CrossRef]
- Chen, L.; Zheng, J.J.Y.; Li, G.; Yuan, J.; Ebert, J.R.; Li, H.; Papadimitriou, J.; Wang, Q.; Wood, D.; Jones, C.W.; et al. Pathogenesis and clinical management of obesity-related knee osteoarthritis: Impact of mechanical loading. J. Orthop. Transl. 2020, 24, 66–75. [Google Scholar] [CrossRef]
- Melrose, J.; Fuller, E.S.; Little, C.B. The biology of meniscal pathology in osteoarthritis and its contribution to joint disease: Beyond simple mechanics. Connect. Tissue Res. 2017, 58, 282–294. [Google Scholar] [CrossRef]
- Alsayed, H.N.; Alkhateeb, M.A.; Aldossary, A.A.; Houbani, K.M.; Aljamaan, Y.M. Risk of anterior cruciate ligament injury in population with elevated body mass index. Med. Glas. 2023, 20, 83–87. [Google Scholar] [CrossRef]
- Chalmers, P.N.; Mall, N.A.; Moric, M.; Sherman, S.L.; Paletta, G.P.; Cole, B.J.; Bach, B.R. Does ACL reconstruction alter natural history?: A systematic literature review of long-term outcomes. J. Bone Jt. Surg. Am. 2014, 96, 292–300. [Google Scholar] [CrossRef]
- DiSilvestro, K.J.; Jauregui, J.J.; Glazier, E.; Cherkalin, D.; Bennett, C.H.; Packer, J.D.; Henn, R.F., III. Outcomes of anterior cruciate ligament re-construction in obese and overweight patients: A systematic review. Clin. J. Sport Med. 2019, 29, 257–261. [Google Scholar] [CrossRef]
- Evers, B.J.; Bosch, M.H.J.V.D.; Blom, A.B.; van der Kraan, P.M.; Koeter, S.; Thurlings, R.M. Post-traumatic knee osteoarthritis; the role of inflammation and hemarthrosis on disease progression. Front. Med. 2022, 9, 973870. [Google Scholar] [CrossRef] [PubMed]
- Azamar-Llamas, D.; Hernández-Molina, G.; Ramos-Ávalos, B.; Furuzawa-Carballeda, J. Adipokine Contribution to the Pathogenesis of Osteoarthritis. Mediat. Inflamm. 2017, 2017, 5468023. [Google Scholar] [CrossRef] [PubMed]
- Poonpet, T.; Honsawek, S. Adipokines: Biomarkers for osteoarthritis? World J. Orthop. 2014, 5, 319–327. [Google Scholar] [CrossRef]
- Xie, C.; Chen, Q. Adipokines: New Therapeutic Target for Osteoarthritis? Curr. Rheumatol. Rep. 2019, 21, 71. [Google Scholar] [CrossRef]
- Conde, J.; Scotece, M.; Gómez, R.; Lopez, V.; Gómez-Reino, J.J.; Gualillo, O. Adipokines and osteoarthritis: Novel molecules involved in the pathogenesis and progression of disease. Arthritis 2011, 2011, 203901. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lin, Y.; Yan, C.H.; Zhang, W. Adipokine Signaling Pathways in Osteoarthritis. Front. Bioeng. Biotechnol. 2022, 10, 865370. [Google Scholar] [CrossRef]
- Hu, P.-F.; Bao, J.-P.; Wu, L.-D. The emerging role of adipokines in osteoarthritis: A narrative review. Mol. Biol. Rep. 2011, 38, 873–878. [Google Scholar] [CrossRef]
- Sobieh, B.H.; El-Mesallamy, H.O.; Kassem, D.H. Beyond mechanical loading: The metabolic contribution of obesity in osteoarthritis unveils novel therapeutic targets. Heliyon 2023, 9, e15700. [Google Scholar] [CrossRef]
- Karvonen-Gutierrez, C.A.; Harlow, S.D.; Mancuso, P.; Jacobson, J.; de Leon, C.F.M.; Nan, B. Association of leptin levels with radiographic knee osteoarthritis among a cohort of midlife women. Arthritis Care Res. 2013, 65, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Zhang, J.; Yang, H.; Sun, Y. The role of leptin in osteoarthritis. Medicine 2018, 97, e0257. [Google Scholar] [CrossRef] [PubMed]
- Koskinen, A.; Vuolteenaho, K.; Nieminen, R.; Moilanen, T.; Moilanen, E. Leptin enhances MMP-1, MMP-3 and MMP-13 production in human osteoarthritic cartilage and correlates with MMP-1 and MMP-3 in synovial fluid from OA patients. Clin. Exp. Rheumatol. 2011, 29, 57–64. [Google Scholar]
- Conde, J.; Scotece, M.; López, V.; Gómez, R.; Lago, F.; Pino, J.; Gómez-Reino, J.J.; Gualillo, O. Adiponectin and leptin induce vcam-1 expression in human and murine chondrocytes. PLoS ONE 2012, 7, e52533. [Google Scholar] [CrossRef]
- Yang, W.-H.; Liu, S.-C.; Tsai, C.-H.; Fong, Y.-C.; Wang, S.-J.; Chang, Y.-S.; Tang, C.-H. Leptin induces IL-6 expression through OBRL receptor signaling pathway in human synovial fibroblasts. PLoS ONE 2013, 8, e75551. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Liu, Z.; Shen, C.; Li, H.; Ding, J.; Jin, F.; Sha, L.; Zhang, Z. Microarray study of gene expression profile to identify new candidate genes involved in the molecular mechanism of leptin-induced knee joint osteoarthritis in rat. Hereditas 2017, 155, 4. [Google Scholar] [CrossRef]
- Lambova, S.N.; Batsalova, T.; Moten, D.; Stoyanova, S.; Georgieva, E.; Belenska-Todorova, L.; Kolchakova, D.; Dzhambazov, B. Serum Leptin and Resistin Levels in Knee Osteoarthritis—Clinical and Radiologic Links: Towards Precise Definition of Metabolic Type Knee Osteoarthritis. Biomedicines 2021, 9, 1019. [Google Scholar] [CrossRef]
- Jamaluddin, S.; Weakley, S.M.; Yao, Q.; Chen, C. Resistin: Functional roles and therapeutic considerations for cardiovascular disease. Br. J. Pharmacol. 2012, 165, 622–632. [Google Scholar] [CrossRef]
- Naqvi, S.K.B.; Murtaza, I.; Javed, Q. Role of resistin genetic variations in knee osteoarthritis pathogenesis, a cross sectional study. Mol. Biol. Rep. 2019, 46, 2657–2663. [Google Scholar] [CrossRef]
- Zhang, J.; Qin, Y.; Zheng, X.; Qiu, J.; Gong, L.; Mao, H.; Jia, W.; Guo, J. The relationship between human serum resistin level and body fat content, plasma glucose as well as blood pressure. Zhonghua Yi Xue Za Zhi 2002, 82, 1609–1612. [Google Scholar]
- Perruccio, A.V.; Mahomed, N.N.; Chandran, V.; Gandhi, R. Plasma adipokine levels and their association with overall burden of painful joints among individuals with hip and knee osteoarthritis. J. Rheumatol. 2014, 41, 334–337. [Google Scholar] [CrossRef]
- Zhao, C.-W.; Gao, Y.-H.; Song, W.-X.; Liu, B.; Ding, L.; Dong, N.; Qi, X. An Update on the Emerging Role of Resistin on the Pathogenesis of Osteoarthritis. Mediat. Inflamm. 2019, 2019, 1532164. [Google Scholar] [CrossRef]
- Zhang, Z.; Xing, X.; Hensley, G.; Chang, L.-W.; Liao, W.; Abu-Amer, Y.; Sandell, L.J. Resistin induces expression of proinflammatory cytokines and chemokines in human articular chondrocytes via transcription and messenger RNA stabilization. Arthritis Rheum. 2010, 62, 1993–2003. [Google Scholar] [CrossRef]
- Haider, D.G.; Pleiner, J.; Francesconi, M.; Wiesinger, G.F.; Müller, M.; Wolzt, M. Exercise training lowers plasma visfatin concentrations in patients with type 1 diabetes. J. Clin. Endocrinol. Metab. 2006, 91, 4702–4704. [Google Scholar] [CrossRef]
- Duan, Y.; Hao, D.; Li, M.; Wu, Z.; Li, D.; Yang, X.; Qiu, G. Increased synovial fluid visfatin is positively linked to cartilage degradation biomarkers in osteoarthritis. Rheumatol. Int. 2012, 32, 985–990. [Google Scholar] [CrossRef]
- Chen, W.-P.; Bao, J.-P.; Feng, J.; Hu, P.-F.; Shi, Z.-L.; Wu, L.-D. Increased serum concentrations of visfatin and its production by different joint tissues in patients with osteoarthritis. Clin. Chem. Lab. Med. 2010, 48, 1141–1145. [Google Scholar] [CrossRef] [PubMed]
- Junker, S.; Frommer, K.W.; Krumbholz, G.; Tsiklauri, L.; Gerstberger, R.; Rehart, S.; Steinmeyer, J.; Rickert, M.; Wenisch, S.; Schett, G.; et al. Expression of adipokines in osteoarthritis osteophytes and their effect on osteoblasts. Matrix Biol. 2017, 62, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.-H.; Yun, H.S.; Kim, J.; Um, H.-D.; Lee, K.-H.; Kang, C.-M.; Lee, S.-J.; Chun, J.-S.; Hwang, S.-G. Nicotinamide phosphoribosyltransferase is essential for interleukin-1β-mediated dedifferentiation of articular chondrocytes via sirt1 and extracellular signal-regulated kinase (ERK) complex signaling. J. Biol. Chem. 2011, 286, 28619–28631. [Google Scholar] [CrossRef] [PubMed]
- Dvir-Ginzberg, M.; Steinmeyer, J. Towards elucidating the role of SirT1 in osteoarthritis. Front. Biosci. 2013, 18, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Al Jaberi, S.; Cohen, A.; D’souza, C.; Abdulrazzaq, Y.M.; Ojha, S.; Bastaki, S.; Adeghate, E.A. Lipocalin-2: Structure, function, distribution and role in metabolic disorders. Biomed. Pharmacother. 2021, 142, 112002. [Google Scholar] [CrossRef] [PubMed]
- Villalvilla, A.; García-Martín, A.; Largo, R.; Gualillo, O.; Herrero-Beaumont, G.; Gómez, R. The adipokine lipocalin-2 in the context of the osteoarthritic osteochondral junction. Sci. Rep. 2016, 6, 29243, Erratum in Sci Rep. 2016, 6, 30666. [Google Scholar] [CrossRef] [PubMed]
- Zayed, N.; Li, X.; Chabane, N.; Benderdour, M.; Martel-Pelletier, J.; Pelletier, J.-P.; Duval, N.; Fahmi, H. Increased expression of lipocalin-type prostaglandin D2 synthase in osteoarthritic cartilage. Arthritis Res. Ther. 2008, 10, R146. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.-S.; Chun, J.-S. Upregulation of lipocalin-2 (LCN2) in osteoarthritic cartilage is not necessary for cartilage destruction in mice. Osteoarthr. Cartil. 2017, 25, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Gómez, R.; Scotece, M.; Conde, J.; Lopez, V.; Pino, J.; Lago, F.; Gómez-Reino, J.J.; Gualillo, O. Nitric oxide boosts TLR-4 mediated lipocalin 2 expression in chondrocytes. J. Orthop. Res. 2013, 31, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Gulkesen, A.; Akgol, G.; Poyraz, A.K.; Aydin, S.; Denk, A.; Yildirim, T.; Kaya, A. Lipocalin 2 as a clinical significance in rheumatoid arthritis. Central Eur. J. Immunol. 2017, 42, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Vergara, A.; Fernández-Pittol, M.J.; Muñoz-Mahamud, E.; Morata, L.; Bosch, J.; Vila, J.; Soriano, A.; Casals-Pascual, C. Evaluation of Lipocalin-2 as a Biomarker of Periprosthetic Joint Infection. J. Arthroplast. 2019, 34, 123–125. [Google Scholar] [CrossRef]
- Helfer, G.; Wu, Q.-F. Chemerin: A multifaceted adipokine involved in metabolic disorders. J. Endocrinol. 2018, 238, R79–R94. [Google Scholar] [CrossRef]
- Santana, L.J.C.; Herrera, F.R.; Rojas, A.P.; Lozano, D.J.M.; Prieto, N.; Castañeda, M.B. Serum chemerin in a cohort of Colombian patients with primary osteoarthritis. Reumatol. Clin. 2021, 17, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Du, G.; Li, L.; Liang, H.; Zhang, B. Association of chemerin levels in synovial fluid with the severity of knee osteoarthritis. Biomarkers 2012, 17, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Ren, L.; Guo, C.J.; Wan, N.J.; Niu, D.S. Chemerin affects the metabolic and proliferative capabilities of chondrocytes by in-creasing the phosphorylation of AKT/ERK. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3656–3662. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Shao, Z.; Zhang, C.; Chen, L.; Al Mamun, A.; Zhao, N.; Cai, J.; Lou, Z.; Wang, X.; Chen, J. Chemerin facilitates intervertebral disc degeneration via TLR4 and CMKLR1 and activation of NF-kB signaling pathway. Aging 2020, 12, 11732–11753. [Google Scholar] [CrossRef] [PubMed]
- Eisinger, K.; Bauer, S.; Schäffler, A.; Walter, R.; Neumann, E.; Buechler, C.; Müller-Ladner, U.; Frommer, K.W. Chemerin induces CCL2 and TLR4 in synovial fibroblasts of patients with rheumatoid arthritis and osteoarthritis. Exp. Mol. Pathol. 2012, 92, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Khoramipour, K.; Chamari, K.; Hekmatikar, A.A.; Ziyaiyan, A.; Taherkhani, S.; Elguindy, N.M.; Bragazzi, N.L. Adiponectin: Structure, Physiological Functions, Role in Diseases, and Effects of Nutrition. Nutrients 2021, 13, 1180. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-H.; Chen, L.; Hsieh, M.-S.; Chang, C.-P.; Chou, D.-T.; Tsai, S.-H. Evidence for a protective role for adiponectin in osteoarthritis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2006, 1762, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Hu, Z.-C.; Shen, L.-Y.; Shang, P.; Xu, H.-Z.; Liu, H.-X. Association of osteoarthritis and circulating adiponectin levels: A systematic review and meta-analysis. Lipids Health Dis. 2018, 17, 189. [Google Scholar] [CrossRef]
- Francin, P.-J.; Abot, A.; Guillaume, C.; Moulin, D.; Bianchi, A.; Gegout-Pottie, P.; Jouzeau, J.-Y.; Mainard, D.; Presle, N. Association between adiponectin and cartilage degradation in human osteoarthritis. Osteoarthr. Cartil. 2014, 22, 519–526. [Google Scholar] [CrossRef]
- Townley, R.A.; Boeve, B.F.; Benarroch, E.E. Progranulin: Functions and neurologic correlations. Neurology 2018, 90, 118–125, Erratum in Neurology 2018, 90, 1127. [Google Scholar] [CrossRef]
- Zhao, Y.-P.; Liu, B.; Tian, Q.-Y.; Wei, J.-L.; Richbourgh, B.; Liu, C.-J. Progranulin protects against osteoarthritis through interacting with TNF-α and β-Catenin signalling. Ann. Rheum. Dis. 2015, 74, 2244–2253. [Google Scholar] [CrossRef] [PubMed]
- Waluga-Kozlowska, E.; Kuznik-Trocha, K.; Komosinska-Vassev, K.; Olczyk, P.; Jura-Poltorak, A.; Winsz-Szczotka, K.; Telega, A.; Ivanova, D.; Strzoda, W.; Zimmermann, A.; et al. Progranulin and chemerin plasma level in obese patients with type 2 diabetes treated with a long-acting insulin analogue and premixed insulin analogue. J. Physiol. Pharmacol. 2021, 72, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Yang, Y.; Fan, M.; Chen, C.; Jiang, R.; Liang, L.; Xian, M.; Kuang, B.; Geng, N.; Feng, N.; et al. Progranulin regulation of autophagy contributes to its chondroprotective effect in osteoarthritis. Genes Dis. 2022, 10, 1582–1595. [Google Scholar] [CrossRef] [PubMed]
- González-Rodríguez, M.; Edjoudi, D.A.; Barreal, A.C.; Ruiz-Fernández, C.; Farrag, M.; González-Rodríguez, B.; Lago, F.; Capuozzo, M.; Gonzalez-Gay, M.A.; Varela, A.M.; et al. Progranulin in Musculoskeletal Inflammatory and Degenerative Disorders, Focus on Rheumatoid Arthritis, Lupus and Intervertebral Disc Disease: A Systematic Review. Pharmaceuticals 2022, 15, 1544. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.-L.; Fu, W.; Ding, Y.-J.; Hettinghouse, A.; Lendhey, M.; Schwarzkopf, R.; Kennedy, O.D.; Liu, C.-J. Progranulin derivative Atsttrin protects against early osteoarthritis in mouse and rat models. Arthritis Res. Ther. 2017, 19, 280. [Google Scholar] [CrossRef] [PubMed]
- Wada, J. Vaspin: A novel serpin with insulin-sensitizing effects. Expert Opin. Investig. Drugs 2008, 17, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Blüher, M. Vaspin in obesity and diabetes: Pathophysiological and clinical significance. Endocrine 2012, 41, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, K.; Zhang, S.; Guan, Z. Vaspin promotes chondrogenic differentiation of BMSCs via Akt activation in osteoarthritis. BMC Musculoskelet. Disord. 2022, 23, 344. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.-P.; Jiang, L.-F.; Chen, W.-P.; Hu, P.-F.; Wu, L.-D. Expression of vaspin in the joint and the levels in the serum and synovial fluid of patients with osteoarthritis. Int. J. Clin. Exp. Med. 2014, 7, 3447–3453. [Google Scholar] [PubMed]
- Respekta, N.; Pich, K.; Mlyczyńska, E.; Dobrzyń, K.; Ramé, C.; Kamiński, T.; Smolińska, N.; Dupont, J.; Rak, A. Plasma level of omentin-1, its expression, and its regulation by gonadotropin-releasing hormone and gonadotropins in porcine anterior pituitary cells. Sci. Rep. 2023, 13, 19325. [Google Scholar] [CrossRef]
- Zhao, A.; Xiao, H.; Zhu, Y.; Liu, S.; Zhang, S.; Yang, Z.; Du, L.; Li, X.; Niu, X.; Wang, C.; et al. Omentin-1: A newly discovered warrior against metabolic related diseases. Expert Opin. Ther. Targets 2022, 26, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.-Y.; Lin, Y.-Y.; Achudhan, D.; Chang, J.-W.; Liu, S.-C.; Lai, C.-Y.; Huang, Y.-L.; Tsai, C.-H.; Fong, Y.-C.; Chen, H.-T.; et al. Omentin-1 ameliorates the progress of osteoarthritis by promoting IL-4-dependent anti-inflammatory responses and M2 macrophage polarization. Int. J. Biol. Sci. 2023, 19, 5275–5289. [Google Scholar] [CrossRef] [PubMed]
- Chai, B.; Zheng, Z.-H.; Liao, X.; Li, K.-Y.; Liang, J.-S.; Huang, Y.-X.; Tong, C.-J.; Ou, D.-J.; Lu, J. The protective role of omentin-1 in IL-1β-induced chondrocyte senescence. Artif. Cells Nanomed. Biotechnol. 2020, 48, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Lana, J.F.S.D.; Lana, A.V.S.D.; da Fonseca, L.F.; Coelho, M.A.; Marques, G.G.; Mosaner, T.; Ribeiro, L.L.; Azzini, G.O.M.; Santos, G.S.; Fonseca, E.; et al. Stromal Vascular Fraction for Knee Osteoarthritis–An Update. J. Stem Cells Regen. Med. 2022, 18, 11–20. [Google Scholar] [CrossRef]
- Benedini, S.; Dozio, E.; Invernizzi, P.L.; Vianello, E.; Banfi, G.; Terruzzi, I.; Luzi, L.; Corsi Romanelli, M.M. Irisin: A Potential Link between Physical Exercise and Metabolism—An Observational Study in Differently Trained Subjects, from Elite Athletes to Sedentary People. J. Diabetes Res. 2017, 2017, 1039161. [Google Scholar] [CrossRef]
- Leustean, L.; Preda, C.; Teodoriu, L.; Mihalache, L.; Arhire, L.; Ungureanu, M.-C. Role of Irisin in Endocrine and Metabolic Disorders—Possible New Therapeutic Agent? Appl. Sci. 2021, 11, 5579. [Google Scholar] [CrossRef]
- Ma, C.; Ding, H.; Deng, Y.; Liu, H.; Xiong, X.; Yang, Y. Irisin: A New Code Uncover the Relationship of Skeletal Muscle and Cardiovascular Health During Exercise. Front. Physiol. 2021, 12, 620608. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Geng, T.; Samara, A.; Olstad, O.K.; He, J.; Agger, A.E.; Skallerud, B.H.; Landin, M.A.; Heyward, C.A.; Pullisaar, H.; et al. Recombinant irisin enhances the extracellular matrix formation, remodeling potential, and differentiation of human periodontal ligament cells cultured in 3D. J. Periodontal Res. 2023, 58, 336–349. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Peng, Y.; Hu, W.; Shi, H.; Li, P.; Que, Y.; Qiu, J.; Qiu, X.; Gao, B.; Zhou, H.; et al. Irisin enhances chondrogenic differentiation of human mesenchymal stem cells via Rap1/PI3K/AKT axis. Stem Cell Res. Ther. 2022, 13, 392. [Google Scholar] [CrossRef]
- Vadalà, G.; Di Giacomo, G.; Ambrosio, L.; Cannata, F.; Cicione, C.; Papalia, R.; Denaro, V. Irisin Recovers Osteoarthritic Chondrocytes In Vitro. Cells 2020, 9, 1478. [Google Scholar] [CrossRef]
- Wang, F.-S.; Kuo, C.-W.; Ko, J.-Y.; Chen, Y.-S.; Wang, S.-Y.; Ke, H.-J.; Kuo, P.-C.; Lee, C.-H.; Wu, J.-C.; Lu, W.-B.; et al. Irisin Mitigates Oxidative Stress, Chondrocyte Dysfunction and Osteoarthritis Development through Regulating Mitochondrial Integrity and Autophagy. Antioxidants 2020, 9, 810. [Google Scholar] [CrossRef]
- Lynskey, S.J.; Macaluso, M.J.; Gill, S.D.; McGee, S.L.; Page, R.S. Biomarkers of Osteoarthritis—A Narrative Review on Causal Links with Metabolic Syndrome. Life 2023, 13, 730. [Google Scholar] [CrossRef]
- Jia, S.; Yu, Z.; Bai, L. Exerkines and osteoarthritis. Front. Physiol. 2023, 14, 1302769. [Google Scholar] [CrossRef]
- Fiorenza, C.G.; Chou, S.H.; Mantzoros, C.S. Lipodystrophy: Pathophysiology and advances in treatment. Nat. Rev. Endocrinol. 2011, 7, 137–150. [Google Scholar] [CrossRef]
- Nagy, G.S.; Tsiodras, S.; Martin, L.D.; Avihingsanon, A.; Gavrila, A.; Hsu, W.C.; Karchmer, A.W.; Mantzoros, C.S. Human immunodeficiency virus type 1–related lipoatrophy and lipohypertrophy are associated with serum concentrations of leptin. Clin. Infect. Dis. 2003, 36, 795–802. [Google Scholar] [CrossRef]
- Villarroya, F.; Domingo, P.; Giralt, M. Drug-induced lipotoxicity: Lipodystrophy associated with HIV-1 infection and antiretroviral treatment. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2010, 1801, 392–399. [Google Scholar] [CrossRef]
- Collins, K.H.; Lenz, K.L.; Pollitt, E.N.; Ferguson, D.; Hutson, I.; Springer, L.E.; Oestreich, A.K.; Tang, R.; Choi, Y.-R.; Meyer, G.A.; et al. Adipose tissue is a critical regulator of osteoarthritis. Proc. Natl. Acad. Sci. USA 2021, 118, e2021096118. [Google Scholar] [CrossRef]
- Larrañaga-Vera, A.; Lamuedra, A.; Pérez-Baos, S.; Prieto-Potin, I.; Peña, L.; Herrero-Beaumont, G.; Largo, R. Increased synovial lipodystrophy induced by high fat diet aggravates synovitis in experimental osteoarthritis. Arthritis Res. Ther. 2017, 19, 264. [Google Scholar] [CrossRef]

{kind=link}
| Altering Factor | Impact on Joint Components | Pathological Mechanisms Leading to OA | Potential Therapeutic Targets |
|---|---|---|---|
| Macrophage Polarization | Synovium, local immune environment | Blood-borne macrophage infiltrates. Shift from anti-inflammatory M2 to pro-inflammatory M1 phenotype | Modulators of macrophage activity, cytokine inhibitors |
| Metabolic Pathways | Cartilage, synovial fluid, bone turnover, Hoffa fat pad | Activation of AMPK-mTORC1 pathway increased ROS production | mTOR inhibitors, antioxidants |
| Adipokines (e.g., Leptin, Adiponectin) | Cartilage, synovium, Hoffa fat pad, menisci in the knee | Influence inflammation and cartilage degradation | Drugs targeting adipokine pathways, biologic agents |
| Chondrocyte Metabolism | Cartilage | Impaired by impaired nutrient sensing and energy metabolism | Metabolic modulators, drugs improving cartilage repair |
| Subchondral Bone Changes | Bone | Reduced blood supply and mechanical support, increased propensity of bone marrow lesions | Vasoactive drugs, osteoporosis treatments (pharmacological and non-pharmacological) |
| Adipokine | Released by | Role in OA | Impact on Joint Components | Therapeutic Potential in OA | Reference |
|---|---|---|---|---|---|
| Leptin | Adipose tissue | Pro-inflammatory, stimulates MMP production | Increases cartilage degradation, higher synovial levels correlate with OA severity | Potential target for reducing inflammatory response | [96,97,98,99,100,101,102] |
| Resistin | Adipose tissue, synovium | Pro-inflammatory, involved in insulin resistance | Induces expression of matrix degradative proteins via NF-κB and cAMP/PKA | Gene polymorphisms and serum levels could serve as biomarkers or therapeutic targets | [103,104,105,106,107,108] |
| Chemerin | Adipose tissue | Pro-inflammatory, pro-angiogenic, pro-adipogenic | Induce release of inflammatory mediators (IL-1, IL-8, TNF-α) and MMP by chondrocytes and macrophages | Proposed as serum biomarker for OA detection | [122,123,124,125,126,127] |
| Adiponectin | Adipose tissue | Anti-inflammatory, increases insulin sensitivity | Inversely correlates with OA severity, affects cartilage and synovium | Protective role suggests potential for therapeutic enhancement | [128,129,130,131] |
| Visfatin | Adipose tissue, joint tissues | Pro-inflammatory, binds to insulin receptors | Associated with cartilage degradation markers, affects osteoblasts and chondrocytes | Targeting visfatin pathways could mitigate joint degradation | [95,96,97,98,99,100] |
| Lipocalin-2 (LCN2) | Adipose tissue, chondrocytes | Transport of molecules, involved in MMP release | Affects osteochondral junction, increased in OA cartilage | Investigated as biomarker and therapeutic target for early OA detection | [116,117,118,119,120,121] |
| Omentin-1 | Adipose tissue | Anti-inflammatory, affects macrophage polarization | Promotes M2 macrophage polarization, reduces cartilage degradation | Could be leveraged in therapies aimed at reducing inflammation | [137,138,139] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mocanu, V.; Timofte, D.V.; Zară-Dănceanu, C.-M.; Labusca, L. Obesity, Metabolic Syndrome, and Osteoarthritis Require Integrative Understanding and Management. Biomedicines 2024, 12, 1262. https://doi.org/10.3390/biomedicines12061262
Mocanu V, Timofte DV, Zară-Dănceanu C-M, Labusca L. Obesity, Metabolic Syndrome, and Osteoarthritis Require Integrative Understanding and Management. Biomedicines. 2024; 12(6):1262. https://doi.org/10.3390/biomedicines12061262
Chicago/Turabian StyleMocanu, Veronica, Daniel Vasile Timofte, Camelia-Mihaela Zară-Dănceanu, and Luminita Labusca. 2024. "Obesity, Metabolic Syndrome, and Osteoarthritis Require Integrative Understanding and Management" Biomedicines 12, no. 6: 1262. https://doi.org/10.3390/biomedicines12061262