Abstract
Amyotrophic lateral sclerosis is a severe neurodegenerative disease whose exact cause is still unclear. Currently, research attention is turning to the mitochondrion as a critical organelle of energy metabolism. Current knowledge is sufficient to confirm the involvement of the mitochondria in the pathophysiology of the disease, since the mitochondria are involved in many processes in the cell; however, the exact mechanism of involvement is still unclear. We used peripheral blood mononuclear cells isolated from whole fresh blood from patients with amyotrophic lateral sclerosis for measurement and matched an age- and sex-matched set of healthy subjects. The group of patients consisted of patients examined and diagnosed at the neurological clinic of the University Hospital Martin. The set of controls consisted of healthy individuals who were actively searched, and controls were selected on the basis of age and sex. The group consisted of 26 patients with sporadic forms of ALS (13 women, 13 men), diagnosed based on the definitive criteria of El Escorial. The average age of patients was 54 years, and the average age of healthy controls was 56 years. We used a high-resolution O2K respirometry method, Oxygraph-2k, to measure mitochondrial respiration. Basal respiration was lower in patients by 29.48%, pyruvate-stimulated respiration (respiratory chain complex I) was lower by 29.26%, and maximal respiratory capacity was lower by 28.15%. The decrease in succinate-stimulated respiration (respiratory chain complex II) was 26.91%. Our data confirm changes in mitochondrial respiration in ALS patients, manifested by the reduced function of complex I and complex II of the respiratory chain. These defects are severe enough to confirm this disease’s hypothesized mitochondrial damage. Therefore, research interest in the future should be directed towards a deeper understanding of the involvement of mitochondria and respiratory complexes in the pathophysiology of the disease. This understanding could develop new biomarkers in diagnostics and subsequent therapeutic interventions.
1. Introduction
Amyotrophic sclerosis is a fatal neurodegenerative disease with a median survival of 3 to 5 years [1]. The disease is characterized by the progressive neurodegeneration of motor neurons in the brain and spinal cord at all levels of the motor system. The prevalence of this disease is approximately 6/100,000 persons [2]. Even nowadays, there is no evidence to fully elucidate the pathophysiology of this severe disease. Possible theories deal with the influence of genetics and environmental factors, but the involvement of multiple factors in developing the disease is not excluded. The literature also describes possible mechanisms such as genetic mutations, oxidative stress, excitotoxicity, mitochondrial and proteasomal dysfunction, impaired axonal transport, and neuroinflammation [3]. Table 1 provides a more detailed overview. A thorough understanding of the pathophysiology of the disease is a crucial basis for the subsequent development of effective therapeutic interventions. The unidirectional focus on possible pathophysiology may account for the unsatisfactory outcomes of potential therapeutic approaches.

Table 1.
Overview of pathophysiological mechanisms underlying the development of ALS and potential use in therapy. ROS—reactive oxygen species, CNS—central nervous system, ER—endoplasmic reticulum, OPC—oligodendrocyte precursor cells, * drugs used in clinical practice.
The clinical symptoms of the disease can be divided according to the affected area/level into bulbar and cervical or lumbar forms (spinal form of the disease) [22]. The onset of the bulbar form of the disease is usually represented by slurred speech, difficulty chewing and swallowing, excessive choking, and weakness or twitching of the muscles of the face, jaw, throat, and vocal cords, especially the tongue. The spinal form of the disease is characterized by symptoms associated with focal muscle weakness and flaccidity, the onset of which may be distal or proximal in the upper and lower limbs. As the damage progresses, spasticity develops in the weakened atrophic limbs. There is also a known respiratory form of the disease, where respiratory symptoms are the first to appear without more severe damage to the limbs or bulbs. Common symptoms of this form of ALS include nocturnal hypoventilation, dyspnoea, orthopnoea, sleep disturbances, morning headaches, excessive daytime somnolence, anorexia, reduced concentration, and irritability or mood changes [23]. This form of the disease affects approximately 5% of patients [24]. The most common cause of death in patients is respiratory muscle paralysis and subsequent respiratory failure.
Diagnosis of this disease is also challenging, involving clinical assessment of the patient’s condition and exclusion of other potentially treatable diseases. The average diagnostic delay for this disease is 10 to 16 months [25]. Diagnosis of the disease is primarily clinical and relies on El Escorial criteria. The diagnostic procedure consists of obtaining a detailed history, conducting a clinical examination, and using electrodiagnostic, imaging, and laboratory methods. A thorough family history, especially the presence of motor neuron diseases, should be considered. As part of the examination, we observe symptoms in people with bulbar ALS related to speech and swallowing, while the first symptoms in people with limb or spinal ALS are related to the limbs and trunk [26]. The gold standard in assessing patient status and rate of progression is currently the ALS Functional Rating Scale (ALSFRS) and its revised form (ALSFRS-R). This questionnaire includes questions targeting bulbar function, fine and gross motor tasks, and respiratory function. However, more advanced staging systems are now available (King’s Staging System, Milano Torino Staging System, Rasch Overall ALS Disability Scale) [1]. Nerve conduction studies are required as part of electrodiagnostic methods to demonstrate motor nerve involvement, indicated by reduced action potential of the compound muscle, prolonged distal motor latency, and reduced conduction velocity without evidence of conduction blocks. Another method is needle EMG, which is necessary for assessing acute and chronic denervation in ALS patients. Muscle ultrasonography can increase diagnostic certainty in this disease due to its excellent sensitivity for detecting fasciculations. A relatively new approach is nerve ultrasonography, which has shown smaller nerves and nerve roots in ALS patients; it also has the potential to differentiate from multifocal motor neuropathy [27]. Classical MRI is used to rule out other diseases. Still, an interesting method is MRI sequence assessment of functional connectivity to determine impairment of the peripheral and central motor nervous system in ALS [28]. Another method is genetic testing, which can find confirmed genetic variants and rule out other genetic disorders. Within laboratory diagnostics, potentially promising biomarkers in diagnosing this disease are neurofilaments, inflammatory biomarkers, Chitinase, Tau Protein, TDP-43, and Creatine Kinase [1]. There are currently three drugs approved for the treatment of ALS (Riluzole and Edavarone, Sodium phenylbutyrate, and Taurursodiol PB/TURSO), which do not cure the disease but help slow the progression of this serious disease.
Mitochondrial dysfunction in ALS patients has been known for more than 50 years, and the involvement of mitochondria in the pathophysiology of this disease is now widely accepted [29]. Motor neurons are particularly vulnerable to mitochondrial malfunctions due to high energy demands. However, it is not known whether mitochondria have a role as a primary trigger in the pathophysiology of the disease or if this is a consequence of other cellular processes. Mitochondrial dysfunction has been demonstrated in ALS patients as well as in experimental models in vivo and in vitro. Abnormal mitochondrial morphology, increased reactive oxygen species production, defects in mitochondrial dynamics, altered respiratory chain enzyme activities, and impaired Ca2 homeostasis have been described [30]. When mitochondrial homeostasis is disrupted, not only oxidative phosphorylation is reduced but the mitochondrial respiratory chain is disrupted. This can further damage mitochondria and lead to the release of mitochondrial proapoptotic proteins such as cytochrome c and apoptosis-inducing factor, ultimately leading to programmed cell death [31]. A comprehensive view of the role of mitochondria and its involvement in the pathomechanisms of this disease may contribute to the development of new diagnostic and therapeutic strategies.
Our study aimed to determine mitochondrial respiration in 26 patients with amyotrophic lateral sclerosis (13 males and 13 females) and subsequent comparison with healthy subjects, correlated based on age and sex.
2. Materials and Methods
2.1. Patient Cohort
The study population consisted of patients examined and diagnosed at the Department of Neurology, University Hospital Martin, Martin, Slovakia, between June 2021 and February 2023. The study population included 26 subjects (13 males and 13 females, the mean age was 54 years, and the mean ALSFR-S score at the time of diagnosis was 35.11 points. The mean diagnostic delay was 12.26 months. Twenty-one patients developed the spinal form of the disease, and five patients developed the bulbar form of the disease. The oldest patient was 80 years old, and the youngest was 28 years old. All the patients were Caucasian. Patients were diagnosed according to El Escorial criteria, which were satisfied by all patients. EMG findings in all patients met the criteria for diagnosing ALS (according to Awai Shima criteria) [32,33]. Inclusion and exclusion criteria for patients are summarized in Table 2.
Table 2.
Inclusion and exclusion criteria of ALS patients.
Some patients also had comorbidities: (1) cardiovascular comorbidities, the most common of which were arterial hypertension (12 patients), ischemic heart disease (2 patients), myocardial infarction (1 patient), and cardiac autonomic neuropathy (1 patient); (2) respiratory comorbidities: bronchial asthma (3 patients), chronic obstructive pulmonary disease (1 patient), and spastic bronchitis (1 patient); (3) liver comorbidities: hepatic steatosis (3 patients), hepatopathy (2 patients), and mild hepatomegaly (1 patient); (4) thyroid comorbidities: Graves–Basedow disease (1 patient), Sjogren’s syndrome (1 patient), thyroiditis (1 patient), and thyroidopathy (1 patient); (5) metabolic comorbidities: hyperlipidemia (5 patients), diabetes mellitus (3 patients), hyperuricemia (3 patients), and hyperbilirubinemia (1 patient); (6) autoimmune comorbidities: rheumatoid arthritis (1 patient); (7) neurologic diseases: epilepsy (2 patients); and (8) other comorbidities: lyme disease (2 patients), anemia (1 patient), hypovitaminosis vit. D (1 patient). Detailed characteristics of the patient cohort are available in Table 3.
Table 3.
Characteristics of patient cohort: diagnostic delay is the time from the appearance of the first symptoms to a definitive diagnosis; disease duration is the time from the first symptoms of the disease to the measurement of mitochondrial respiration; ΔFS ratio (rate of disease progression) is the rate of disease progression, ΔFS = (ALSFR-S total − ALSFR-S actual)/the duration of the disease from the onset of symptoms. Milgamma N-benfotiamine (40 mg), pyridoxinium chloride (90 mg), cyanocobalamin (0.25 g).
The control group was matched based on a correlation of age and sex with no symptoms or history of neurological disease. The control group consisted of 13 males and 13 females; the mean age was 56. The inclusion and exclusion criteria for the control group are summarized in Table 4.
Table 4.
Inclusion and exclusion criteria of a control group.
Written informed consent was obtained from all patients before the examination. The informed consent process followed ethical guidelines and ensured participants were well-informed and voluntarily participated in the research protocol. The research project was approved by the ethics committee under the number EC 43/2021. A 10 mL quantity of venous blood was collected through a single venipuncture. EDTA tubes were used for the collection. The blood was transported to the laboratory in the shortest possible time and then immediately processed and analyzed. Isolation of PBMCs was performed within a maximum of 1 hour after collection.
2.2. Peripheral Blood Mononuclear Cell Isolation
PBMCs (peripheral blood mononuclear cells) isolated from fresh venous blood were used for measurement. Fresh peripheral venous blood samples were collected into EDTA-containing tubes. A commercially available Leucosep kit (Greiner bio-one GmbH, Kremsmünster, Austria) was used to isolate PBMCs followed by post-treatment according to the instructions for use. Histopaque-1077 (Sigma-Aldrich, St. Louis, MO, USA) warmed at room temperature was used as the separation medium. Subsequently, the buffy coat was gently aspirated and washed twice with DPBS.
Further details of the methodology adopted can be found in Sumbalova et al. [34]. Subsequently, the washed PBMC pellet was dispersed in 250 µL of MiR05 respiration medium (Oroboros Instruments GmbH, Innsbruck, Austria), and 100 µL of cell suspension was injected into each 2 mL glass chamber.
2.3. High-Resolution Respirometry
Mitochondrial function was determined by high-resolution O2K respirometry, Oxygraph-2k (Oroboros Instruments GmbH, Innsbruck, AT, Austria). Measurements were performed in a two-chamber system at 37 °C with a stirring speed of 750 rpm. The amplified signal from the oxygen sensor was recorded by computer with scanning intervals of 2 s using DatLab 7 software (Oroboros Instruments, Innsbruck, AT). Before starting all experiments, the respirometer was calibrated at air saturation and 37 °C. Manual titration of inhibitors and uncouplers was performed using Hamilton syringes. A standard coupling control protocol (CCP) was used to determine mitochondrial function. All aliquots of substrates and inhibitors were stored at −20 °C. Pyruvate was prepared fresh daily. Following the Oroboros guidelines [35], the resulting O2 flux values were used to calculate respiration in different states of coupling control, including basal respiration (ROUTINE), LEAK, maximal electron transport (ET) capacity, respiratory reserve, ATP-coupled respiration, and succinate-driven stimulation. The measurements allowed analysis of the main parameters of mitochondrial respiration: ROUTINE (R) respiration, which represents physiological cell respiration without being affected and is measured after titration of the cell suspension into chambers containing mitochondrial respiration medium; pyruvate-stimulated respiration (P, 10 mM), which is measured after addition of P as substrate for I. in intact cells; oligomycin (Omy, 10 nM), which is used to induce the LEAK respiratory state; maximal ET capacity, which represents the attainment of maximal respiration by sequential titration of Uncoupler (U*-CCCP, 0.5 µM sequential titration); Rotenone (Rot, 500 nM) as a complex I (CI) inhibitor, which is used for ROX; Succinate (S, 10 mM) stimulation of mitochondrial respiration, which is only observed in nonliving/permeabilized cells; digitonin (Dig, 10 mg/mL sequential addition), which is used to permeabilize the entire population of cells, providing a reference state for succinate-stimulated ETS capacity; cytochrome C titration assay (c, 10 µM), which provides information on the integrity of the outer mitochondrial membrane; and antimycin A (Ama, 2.5 µM), which is a complex III inhibitor. Residual oxygen consumption (ROX) is assessed to correct for the flow of intact cellular respiration, and ROX is respiration that is associated with other oxygen consumption processes in the cell in addition to the respiratory system.
2.4. Determination of Protein Concentration
Protein concentration determinations were performed to determine the exact amount of mitochondria. Protein concentration was determined using the DC Protein assay (BioRad, Hercules, CA, USA). The first step was centrifugation at 1000× g for 10 min, followed by aspiration without disturbing the visible protein pellet. The pellet was then dispersed and adequately mixed in 200 microliters of RIPA analysis and extraction buffer (Thermo Scientific, Waltham, MA, USA). This was followed by a 30-min incubation at 4 °C, during which protein lysis occurred. This was followed by centrifugation at 14,000× g for 15 min at 4 °C. A 96-well plate was used to measure the proteins, and 5 microliters of analyte and 5 microliters of standards were pipetted with increasing concentrations from 0.125 mg/mL to 2 mg/mL. Pre-diluted protein assay standards were used for standard preparation: BSA (bovine serum albumin) Set (Thermo Scientific). Subsequently, 25 microliters of reagent A (basic copper tartrate solution) and 200 microliters of reagent B (dilute folin reagent) were added to each well. The plate was incubated for 15 min in the dark. The absorbance was measured on a Synergy H4 instrument (BioRad, Hercules, CA, USA) at 750 nm. The protein concentration in milligrams was calculated based on the calibration curve of individual absorbance.
2.5. Statistical Analysis
Statistical analysis was performed using PRISM GraphPad version 9.1.2 (La Jolla, CA, USA). All figures were also created in this program. The unpaired (Mann–Whitney) t-test was used in the statistical analysis. Significance values were set at p > 0.05, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, and **** p ≤ 0.0001. Spearman’s test of correlation was also used.
3. Results
We focused on measuring mitochondrial respiration in ALS patients against sex- and age-matched controls. Our cohort consisted of 26 patients. We used a high-resolution respirometry method, Oroboros (AT), for the measurement. After adding intact cells, we observed basal respiration, i.e., energy demand under steady-state conditions. Subsequently, we added pyruvate, which is the substrate for complex I. After the addition of oligomycin, ATPase was inhibited. Subsequently, we measured the maximal respiratory capacity using sequential uncoupler titration. With the addition of rotenone, complex I was inhibited. Subsequently, we added succinate, the substrate for the second complex. With the gradual addition of digitonin, permeabilization of the plasma membrane occurs, and mitochondrial respiration (dependent on complex II) increases until complete permeabilization, without disruption of the inner cell membrane, where succinate and ADP enter the cells. Cytochrome C was added to test the integrity of the outer mitochondrial membrane. The final step was the addition of Antimicin A, which serves to inhibit complex III. The mean respiratory values of ALS patients and healthy controls are shown in Table 5 and Figure 1 and Figure 2.
Table 5.
Mean values with standard deviations of mitochondrial respiration in patients with amyotrophic lateral sclerosis and healthy controls.
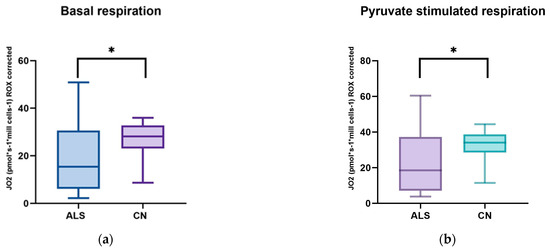
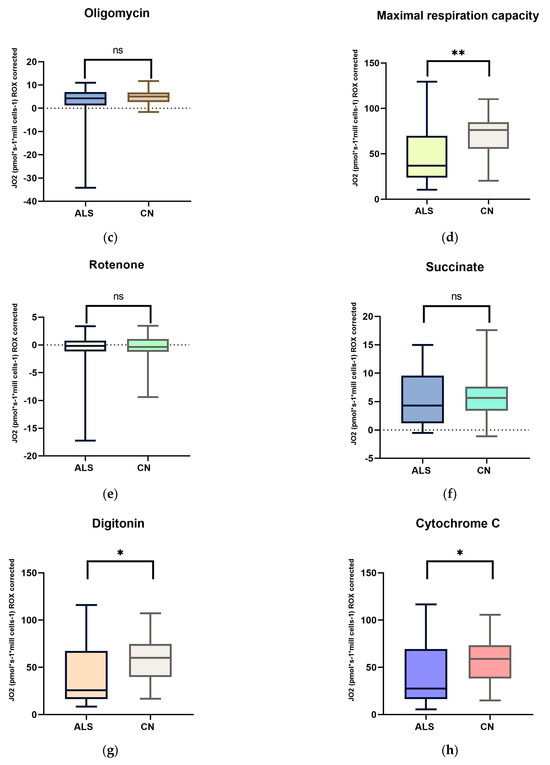
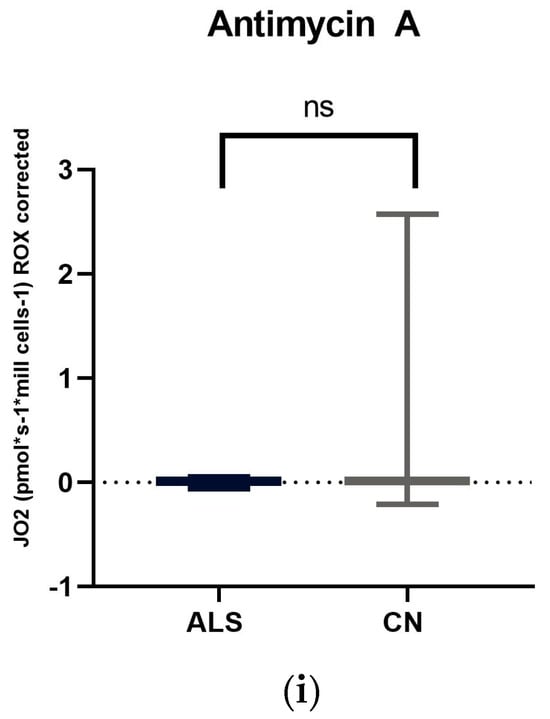
Figure 1.
Comparison of mitochondrial respiration in patients with amyotrophic lateral sclerosis and healthy subjects: (a) basal respiration significant difference p = 0.0123; (b) pyruvate −stimulated respiration significant difference p = 0.011; (c) respiration after addition of oligomycin, no significant difference p = 0.4843; (d) maximal respiratory capacity, significant difference p = 0.0059; (e) respiration after addition of rotenone, no significant difference p = 0.817; (f) respiration after stimulation with succinate without significant differences p = 0.5673; (g) respiration after addition of digitonin, significant difference p = 0.0144; (h) respiration after addition of cytochrome C significant difference p = 0.0253; and (i) respiration after addition of Antimycin A without significant differences p = 0.1604. ns: p > 0.05; * p ≤ 0.05; ** p ≤ 0.01.
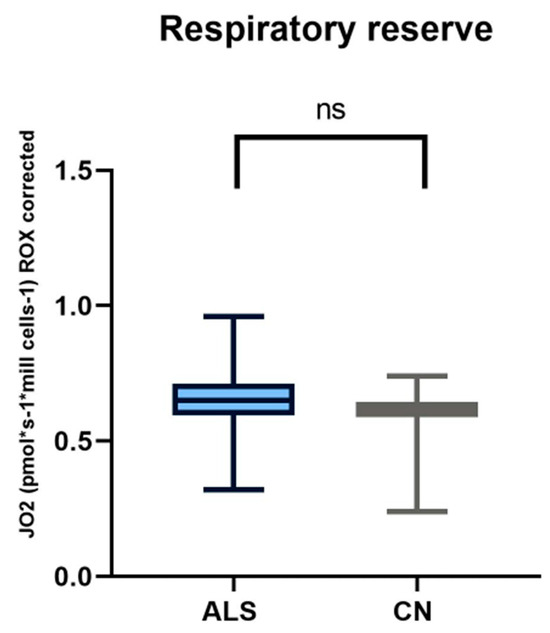
Figure 2.
In comparing respiratory reserve in patients with amyotrophic lateral sclerosis and healthy controls, we observed no significant differences.
We observed significant changes between ALS patients and healthy controls in basal respiration (p = 0.0123), pyruvate-stimulated respiration (p = 0.011), and maximal respiratory capacity (p = 0.0059), changes were also observed in succinate-stimulated respiration, after the addition of digitonin (p = 0.0144) and Cytochrome C (p = 0.0253).
To assess significant differences in respiration after the addition of digitonin and cytochrome C, Spearman’s correlation test was performed. Since respiration after adding cytochrome C should not increase complex II-dependent respiration, this test demonstrated no loss of cytochrome C from the outer mitochondrial membrane, and mitochondrial integrity is preserved. This test showed a significantly high correlation (r = 0.9925; p < 0.0001). Spearman’s correlation is shown in Figure 3.
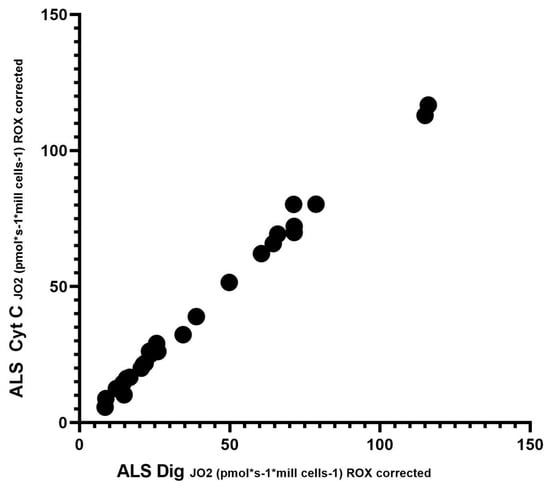
Figure 3.
Correlation between digitonin and cytochrome C; Spearman’s correlation test.
In our cohort of patients, we observed significant differences in basal respiration pyruvate-stimulated respiration, which is indicative of malfunctioning of complex I. Furthermore, maximal respiratory capacity was significantly reduced in patients. We also observed significant differences after adding digitonin and Cytochrome C (succinate stimulated respiration), indicating malfunctioning of complex II. We then used Spearman’s correlation test, which showed a linear relationship between respiration after adding digitonin and Cytochrome C.
4. Discussion
Amyotrophic lateral sclerosis is a severe and incurable neurodegenerative disease. Despite considerable research efforts, its pathophysiology is unclear. We aimed to determine mitochondrial respiration in ALS patients and sex- and age-correlated controls. We observed a decrease in basal respiration and respiration after pyruvate stimulation, indicating reduced complex I function. Maximal respiratory capacity was also significantly lower in patients. Respiration after adding digitonin (succinate-stimulated respiration) was also significantly lower in patients, indicating decreased complex II function.
The mitochondria is a highly dynamic organelle crucial in cell survival and for maintaining normal metabolism, energy homeostasis, calcium homeostasis, cell death mechanisms, redox balance, cell growth, and differentiation [36]. It is also involved in antioxidant metabolism and axonal transport [37]. Mitochondria are abundantly represented and actively transported in regions of neurons with intense demands for energy in the form of ATP, such as axon hillocks, nodes of Ranvier, and synaptic regions. Their subcellular composition can change dynamically according to the physiological needs of the cell [38]. However, the damage and subsequent malfunction of mitochondria results in a decrease in ATP production, leading to neuronal damage.
Mitochondrial damage also mediates intraneuronal damage and death of motor neurons through calcium-mediated excitotoxicity, activation of the intrinsic apoptotic pathway, and increased ROS production [39,40,41,42]. The progressive changes in mitochondrial morphology, bioenergetics, and calcium homeostasis have been linked to pathological changes in ALS. Furthermore, many genes (SOD1, FUS, VAPB, TARDBP, OPTN, VCP, C9orf72) shown to be involved in the pathophysiology of the disease are closely linked to mitochondria and their function [43]. In studies of disease models in vitro [44,45] and animal models [46,47,48,49], mitochondrial damage was also observed. Last but not least, this damage has also been demonstrated by studies on patients or in muscles [50,51,52,53], peripheral blood [54,55,56,57], and spinal cord [58].
In our work, we used PBMCs to investigate the changes; the main advantage is their easy accessibility, as they are isolated from blood. Therefore, monitoring their abnormalities could be a useful prognostic and diagnostic marker of ALS. Araujo et al. also used PBMCs to look at mitochondrial damage in ALS. Their experiments led to the following conclusions: lower mitochondrial calcium uptake/retention, mitochondrial depolarization, and impaired redox homeostasis. They also showed a decrease in biogenesis and mitochondrial number and a decrease in energy-producing metabolic compounds such as ATP and pyruvate production in the PBMC cells studied [59].
We used a high-resolution respirometry method to analyze mitochondrial respiration. This method is probably the most rigorous procedure to assess the state of mitochondrial respiration, by adding specific electron-providing substrates or inhibitors of individual complexes [60]. In ALS patients, we observed significantly lower basal respiration, pyruvate-stimulated respiration, maximal respiratory capacity, and also a decrease in respiration after the addition of digitonin compared to healthy controls. Respiratory reserve did not show significant changes compared to healthy controls. Basal respiration represents the oxygen consumption rate in ATP production and proton leak. Changes in this basic parameter in patients may indicate altered or malfunctioning mitochondria [61]. Consistent with our results is another work where the authors observed a lower basal respiration rate and, consequently, reduced production in the mitochondria of ALS patients [62]. Ehinger et al., however, came to contradictory results in ALS patients. In their work, they observed a 36% increase in basal respiration and a 23% increase in maximal respiratory capacity compared to the control group, and they did not observe any significant differences in escape respiration. However, the authors described a major limitation of their study, the small sample size of patients [63].
Previous work has shown that complex I dysfunction significantly contributes to mitochondrial dysfunction in ALS. This dysfunction was also observed in our patient cohort. A decrease in complex I activity has also been demonstrated by the work of other authors [64,65,66,67]. The mitochondrial respiratory chain is responsible for producing energy in the form of ATP, and complex I is the first and largest enzyme in this chain, contributing to most of the proton motive force that drives ATP synthesis [68]. Inhibition of mitochondrial complex I in ALS has been known about since 1998 [49,69]. The role of complex I is to oxidize NADH and reduce ubiquinone to create part of the proton gradient required for ATP synthesis. Mutations in genes encoding complex I subunits have been found in ALS patients, and postmortem examinations have revealed reduced complex I activity in the motor neurons of ALS patients. In addition, animal models of ALS have shown that increasing complex I activity can improve motor function and prolong lifespan in these animals, suggesting that targeting complex I could be a potential treatment option for ALS [70]. Hor et al., in their study, demonstrated decreased mitochondrial respiration and increased glycolysis in ALS patients. Specifically, they observed reduced basal respiration, lower ATP production, and maximal respiratory capacity in patients [71].
The differences in maximal respiratory capacity that we also observed in our patients may signal dysfunction in respiratory complexes or cellular or mitochondrial substrate uptake/loading [72]. They then continued their study by creating isogenic cell lines; by activating SIRT3 and treating with nicotinamide, they observed a reversal of the defect in mitochondrial respiration [71]. Lastes et al., in their study, observed higher basal cell respiration in ALS patients than in the control group. However, reserve respiratory capacity, a parameter important in coping with increased energy demand, was significantly lower. If the reserve respiratory capacity is insufficient to provide enough ATP, it may indicate cell death and damage [73]. They also observed higher lactate production in ALS patients. An interesting observation was increased glycolytic capacity and decreased glycolytic reserve. The authors conclude that such mitochondrial damage would lead to increased ROS production and ATP production via the glycolysis pathway [74]. An increase in the production of reactive oxygen species was also observed in another study, which also showed depolarization of the mitochondria, impaired oxidative phosphorylation, decreased ATP production, and defective import of mitochondrial proteins [62].
In our work, we observed a decrease in mitochondrial respiration after adding digitonin when respiration is dependent on complex II. Complex II is an essential component of the Krebs cycle as well as the mitochondrial respiratory chain, both of which play an important role in ATP production [75]. As complex II is located at the junction of two key pathways, the Krebs cycle and oxidative phosphorylation, there is a suggestion that the function, regulation, and response of complex II to pathophysiological stimuli are key to the bioenergetics of the cell and the possible development of disease [76]. Previously, authors have shown that damaged complex II is capable of producing oxygen radicals [77]. Since most of the work focuses on complex I, we could not find any work to compare the results. An alternative interpretation of the digitonin effect is the hypersensitivity of the cells to the addition of digitonin, as the mitochondrial membrane underwent the first changes under apoptosis [78].
Defects in mitochondrial respiration have been implicated in various neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease. Studies focusing on Parkinson’s disease have observed unaltered basal respiration [79] and increased maximal respiratory capacity [79,80]. Shirinzi et al. also observed an increase in respiratory reserve [79]. Other studies have shown low activity of complex I [81,82] and low activities of complexes II and IV [82]. Work focused on Alzheimer’s disease has shown reductions in basal respiration and maximal respiratory capacity [83]. Other work has observed increased complex II and IV activity [84]. Frontotemporal lobar degeneration (FTLD) is a clinically and pathologically heterogeneous syndrome characterized by a progressive decline in behavior or language associated with degeneration of the frontal and anterior temporal lobes [85]. We were unable to find studies describing mitochondrial respiration in this disorder. Although some papers have focused on defects in mitochondrial respiration in individual diseases, there is no available evidence that examines differences between neurodegenerative diseases.
Available evidence suggests that at the intracellular level, mitochondria are the earliest targets in the pathophysiology of ALS. One expected consequence of impaired mitochondrial respiration is reduced ATP production and subsequent bioenergetic failure [86]. Another consequence of impaired mitochondrial respiration is increased reactive oxygen species (ROS) production. Under normal circumstances, the small amount of molecular oxygen in the mitochondria is reduced to ROSs (such as superoxide radicals) instead of being converted to water. However, in the normal state, these levels are minimal due to the antioxidants present in the mitochondria. According to the available knowledge, a disturbance of complex I will disrupt this homeostasis, and a dramatic increase in reactive oxygen species will occur [87]. One consequence is mitochondrial instability, which will trigger a negative cycle of increasing ROS production, damage, and mitochondrial malfunction. This can further damage mitochondria and lead to the release of mitochondrial proapoptotic proteins (cytochrome C, apoptosis-inducing factor) and ultimately to programmed cell death [88]. The result of this chain of events is neurodegeneration. Potential strategies to improve mitochondrial function include the use of dichloroacetate (the pyruvate dehydrogenase inhibitor DCA stimulates the conversion of pyruvate to acetyl coenzyme A, thereby supplying additional energy substrates to the TCA cycle) [89], ketogenic and high-fat diets, acetylcarnitine (plays a key role in the transport of long-chain fatty acids across mitochondrial membranes and limits the rate of β-oxidation [90]), and mitochondria-targeted antioxidants. In addition, antiapoptotic agents such as the mPTP-targeting agents minocycline and rasagiline are discussed [91]. In the future, it can be anticipated that novel ways of pharmacologically or genetically modulating mitochondrial turnover, movement, and dynamics in ALS, as well as restoring bioenergetic balance in the body, may become promising therapeutic strategies.
In summary, our data demonstrated impaired respiratory function in our group of patients with amyotrophic lateral sclerosis. We observed differences compared to healthy controls in basal respiration and impairment of complex I and complex II of the respiratory chain.
High-resolution respirometry is an excellent means of assessing the control of mitochondrial respiration or studying cell oxygen kinetics in response to the delivery of specific electron-providing substrates, proton ionophores, and/or inhibitors of mitochondrial complexes. However, it should be mentioned that the study of amyotrophic sclerosis had limitations. Since this disease is rare, a significant limitation is the low number of patients. An overview of the limitations and ideas for further research are presented in Table 6.
Table 6.
Study limitations and suggestions for further research.
5. Conclusions
Amyotrophic lateral sclerosis is a devastating neurodegenerative disease that is currently incurable. Although it is a rare disease, its incidence is expected to increase in the future. Data from preclinical, epidemiological, histopathological, and clinical studies indicate that mitochondrial dysfunction is a significant factor in the development of this disease. Current experimental evidence suggests that several mitochondrial pathways contribute to the pathogenesis of ALS. Our work demonstrates significant alterations in mitochondrial respiration in patients with amyotrophic lateral sclerosis. We observed decreased activity of complex I. An interesting finding was the reduced activity of complex II, as most studies focused their activity only on complex I. However, despite the overwhelming evidence suggesting that mitochondrial function is essential in the pathogenesis of ALS, the ability to identify the initial event in the cascade of changes that lead to neurodegeneration remains poorly understood. Therefore, future research should focus on investigating the mechanisms that lead to this damage. In addition, studying the interactions between mitochondrial respiration and other cellular processes involved in ALS, such as protein aggregation and oxidative stress, may also provide valuable insights into the mechanism of the disease. A better understanding of the involvement of mitochondria in the causes of this disease could lead to the discovery of appropriate diagnostic tools and, consequently, the development of new approaches to the therapy of this serious disease.
Author Contributions
Conceptualization, P.P., A.E. and M.T.-K.; methodology and validation, P.P. and A.E.; formal analysis, P.P.; writing—preparation of original draft, P.P.; writing—review and editing, P.P., P.H. and Z.T.; resources, M.T.-K. and M.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the guidelines of the Declaration of Helsinki and approved by the ethics committee of the Jessenius Medical Faculty in Martin (no. EC43/2021, 29 June 2021).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Acknowledgments
We would like to thank all the patients who were in this study. Thanks also to Martin Kolisek for his valuable advice during this study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Vidovic, M.; Müschen, L.H.; Brakemeier, S.; Machetanz, G.; Naumann, M.; Castro-Gomez, S. Current State and Future Directions in the Diagnosis of Amyotrophic Lateral Sclerosis. Cells 2023, 12, 736. [Google Scholar] [CrossRef] [PubMed]
- Hanhisuanto, M.; Solje, E.; Jokela, M.; Sipilä, J.O.T. Amyotrophic Lateral Sclerosis in Southwestern and Eastern Finland. Neuroepidemiology 2023, 57, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Brotman, R.G.; Moreno-Escobar, M.C.; Joseph, J.; Munakomi, S.; Pawar, G. Amyotrophic Lateral Sclerosis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Cerillo, J.L.; Parmar, M. Tofersen. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Mead, R.J.; Shan, N.; Reiser, H.J.; Marshall, F.; Shaw, P.J. Amyotrophic Lateral Sclerosis: A Neurodegenerative Disorder Poised for Successful Therapeutic Translation. Nat. Rev. Drug. Discov. 2023, 22, 185–212. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Andreucci, A.; Iwamoto, N.; Yin, Y.; Yang, H.; Liu, F.; Bulychev, A.; Hu, X.S.; Lin, X.; Lamore, S.; et al. Preclinical Evaluation of WVE-004, an Investigational Stereopure Oligonucleotide for the Treatment of C9orf72-Associated ALS or FTD. Mol. Ther. Nucleic Acids 2022, 28, 558–570. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, Y.; Deng, M. New Developments and Opportunities in Drugs Being Trialed for Amyotrophic Lateral Sclerosis from 2020 to 2022. Front. Pharmacol. 2022, 13, 1054006. [Google Scholar] [CrossRef] [PubMed]
- Yerton, M.; Winter, A.; Kostov, A.; Lieberman, C.; Gelevski, D.; Weber, H.; Doyle, M.; Kane, G.; Parikh, N.; Burke, K.M.; et al. An Expanded Access Protocol of RT001 in Amyotrophic Lateral Sclerosis-Initial Experience with a Lipid Peroxidation Inhibitor. Muscle Nerve 2022, 66, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Shukla, S. Role of Edaravone as a Treatment Option for Patients with Amyotrophic Lateral Sclerosis. Pharmaceuticals 2020, 14, 29. [Google Scholar] [CrossRef] [PubMed]
- Paganoni, S.; Quintana, M.; Sherman, A.V.; Vestrucci, M.; Wu, Y.; Timmons, J.; Cudkowicz, M. Pooled Resource Open-Access ALS Clinical Trials Consortium Analysis of Sodium Phenylbutyrate and Taurursodiol Survival Effect in ALS Using External Controls. Ann. Clin. Transl. Neurol. 2023, 10, 2297–2304. [Google Scholar] [CrossRef] [PubMed]
- Gautam, M.; Genç, B.; Helmold, B.; Ahrens, A.; Kuka, J.; Makrecka-Kuka, M.; Günay, A.; Koçak, N.; Aguilar-Wickings, I.R.; Keefe, D.; et al. SBT-272 Improves TDP-43 Pathology in ALS Upper Motor Neurons by Modulating Mitochondrial Integrity, Motility, and Function. Neurobiol. Dis. 2023, 178, 106022. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Bracci, P.M.; Azhir, A.; Forrest, B.D.; McGrath, M.S. Macrophage-Targeted Sodium Chlorite (NP001) Slows Progression of Amyotrophic Lateral Sclerosis (ALS) through Regulation of Microbial Translocation. Biomedicines 2022, 10, 2907. [Google Scholar] [CrossRef] [PubMed]
- Mora, J.S.; Bradley, W.G.; Chaverri, D.; Hernández-Barral, M.; Mascias, J.; Gamez, J.; Gargiulo-Monachelli, G.M.; Moussy, A.; Mansfield, C.D.; Hermine, O.; et al. Long-Term Survival Analysis of Masitinib in Amyotrophic Lateral Sclerosis. Ther. Adv. Neurol. Disord. 2021, 14, 17562864211030365. [Google Scholar] [CrossRef] [PubMed]
- Camu, W.; Mickunas, M.; Veyrune, J.-L.; Payan, C.; Garlanda, C.; Locati, M.; Juntas-Morales, R.; Pageot, N.; Malaspina, A.; Andreasson, U.; et al. Repeated 5-Day Cycles of Low Dose Aldesleukin in Amyotrophic Lateral Sclerosis (IMODALS): A Phase 2a Randomised, Double-Blind, Placebo-Controlled Trial. eBioMedicine 2020, 59, 102844. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Stoklund Dittlau, K.; Van Den Bosch, L. Axonal Transport Defects and Neurodegeneration: Molecular Mechanisms and Therapeutic Implications. Semin. Cell Dev. Biol. 2020, 99, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Raffaele, S.; Nguyen, N.; Milanese, M.; Mannella, F.C.; Boccazzi, M.; Frumento, G.; Bonanno, G.; Abbracchio, M.P.; Bonifacino, T.; Fumagalli, M. Montelukast Improves Disease Outcome in SOD1G93A Female Mice by Counteracting Oligodendrocyte Dysfunction and Aberrant Glial Reactivity. Br. J. Pharmacol. 2024; early view. [Google Scholar] [CrossRef]
- Raffaele, S.; Boccazzi, M.; Fumagalli, M. Oligodendrocyte Dysfunction in Amyotrophic Lateral Sclerosis: Mechanisms and Therapeutic Perspectives. Cells 2021, 10, 565. [Google Scholar] [CrossRef] [PubMed]
- Vanneste, J.; Van Den Bosch, L. The Role of Nucleocytoplasmic Transport Defects in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 12175. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.G.; Mitchell, J.D.; Lyon, M.; Moore, D.H. Riluzole for Amyotrophic Lateral Sclerosis (ALS)/Motor Neuron Disease (MND). Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2003, 4, 191–206. [Google Scholar] [CrossRef]
- Castillo, K.; Nassif, M.; Valenzuela, V.; Rojas, F.; Matus, S.; Mercado, G.; Court, F.A.; van Zundert, B.; Hetz, C. Trehalose Delays the Progression of Amyotrophic Lateral Sclerosis by Enhancing Autophagy in Motoneurons. Autophagy 2013, 9, 1308–1320. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Gonzalez, L.; Martinez, A. Emerging Clinical Investigational Drugs for the Treatment of Amyotrophic Lateral Sclerosis. Expert Opin. Investig. Drugs 2023, 32, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Štětkářová, I.; Ehler, E. Diagnostics of Amyotrophic Lateral Sclerosis: Up to Date. Diagnostics 2021, 11, 231. [Google Scholar] [CrossRef] [PubMed]
- Zarei, S.; Carr, K.; Reiley, L.; Diaz, K.; Guerra, O.; Altamirano, P.F.; Pagani, W.; Lodin, D.; Orozco, G.; Chinea, A. A Comprehensive Review of Amyotrophic Lateral Sclerosis. Surg. Neurol. Int. 2015, 6, 171. [Google Scholar] [CrossRef] [PubMed]
- Wijesekera, L.C.; Leigh, P.N. Amyotrophic Lateral Sclerosis. Orphanet J. Rare Dis. 2009, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.; Morren, J.A.; Pioro, E.P. Time to Diagnosis and Factors Affecting Diagnostic Delay in Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 2020, 417, 117054. [Google Scholar] [CrossRef] [PubMed]
- Shellikeri, S.; Karthikeyan, V.; Martino, R.; Black, S.E.; Zinman, L.; Keith, J.; Yunusova, Y. The Neuropathological Signature of Bulbar-Onset ALS: A Systematic Review. Neurosci. Biobehav. Rev. 2017, 75, 378–392. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.; Vielhaber, S.; Schreiber, F.; Cartwright, M.S. Peripheral Nerve Imaging in Amyotrophic Lateral Sclerosis. Clin. Neurophysiol. 2020, 131, 2315–2326. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, M.; Bede, P.; Turner, M.R. Imaging Cerebral Activity in Amyotrophic Lateral Sclerosis. Front. Neurol. 2019, 9, 1148. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, X.; Huo, Z.; Chen, Y.; Liu, J.; Zhao, Z.; Meng, F.; Su, Q.; Bao, W.; Zhang, L.; et al. The Impact of Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis. Cells 2022, 11, 2049. [Google Scholar] [CrossRef] [PubMed]
- Genin, E.C.; Abou-Ali, M.; Paquis-Flucklinger, V. Mitochondria, a Key Target in Amyotrophic Lateral Sclerosis Pathogenesis. Genes 2023, 14, 1981. [Google Scholar] [CrossRef] [PubMed]
- Belosludtseva, N.V.; Matveeva, L.A.; Belosludtsev, K.N. Mitochondrial Dyshomeostasis as an Early Hallmark and a Therapeutic Target in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2023, 24, 16833. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. World Federation of Neurology Research Group on Motor Neuron Diseases El Escorial Revisited: Revised Criteria for the Diagnosis of Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, M.; Dengler, R.; Eisen, A.; England, J.D.; Kaji, R.; Kimura, J.; Mills, K.; Mitsumoto, H.; Nodera, H.; Shefner, J.; et al. Electrodiagnostic Criteria for Diagnosis of ALS. Clin. Neurophysiol. 2008, 119, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Sumbalova, Z.; Kucharska, J.; Palacka, P.; Rausova, Z.; Langsjoen, P.H.; Langsjoen, A.M.; Gvozdjakova, A. Platelet Mitochondrial Function and Endogenous Coenzyme Q10 Levels Are Reduced in Patients after COVID-19. Bratisl. Lek. Listy 2022, 123, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Gnaiger, E. Mitochondrial Pathways and Respiratory Control: An Introduction to OXPHOS Analysis, 5th ed.; Bioenergetics Communications: Innsbruck, Austria, 2020. [Google Scholar]
- Javadov, S.; Kozlov, A.V.; Camara, A.K.S. Mitochondria in Health and Diseases. Cells 2020, 9, 1177. [Google Scholar] [CrossRef] [PubMed]
- Kausar, S.; Wang, F.; Cui, H. The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Gal, J.; Kwinter, D.M.; Liu, X.; Zhu, H. Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis. Biochim. Biophys. Acta 2010, 1802, 45–51. [Google Scholar] [CrossRef]
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, Calcium and Mitochondria: A Triad in Synaptic Neurodegeneration. Transl. Neurodegener. 2022, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Kovács, R. Mitochondria and Neuronal Activity. Am. J. Physiol. Cell Physiol. 2007, 292, C641–C657. [Google Scholar] [CrossRef] [PubMed]
- Jadiya, P.; Garbincius, J.F.; Elrod, J.W. Reappraisal of Metabolic Dysfunction in Neurodegeneration: Focus on Mitochondrial Function and Calcium Signaling. Acta Neuropathol. Commun. 2021, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Norat, P.; Soldozy, S.; Sokolowski, J.D.; Gorick, C.M.; Kumar, J.S.; Chae, Y.; Yağmurlu, K.; Prada, F.; Walker, M.; Levitt, M.R.; et al. Mitochondrial Dysfunction in Neurological Disorders: Exploring Mitochondrial Transplantation. npj Regen. Med. 2020, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The Role of Mitochondria in Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef] [PubMed]
- Kaal, E.C.; Vlug, A.S.; Versleijen, M.W.; Kuilman, M.; Joosten, E.A.; Bär, P.R. Chronic Mitochondrial Inhibition Induces Selective Motoneuron Death in Vitro: A New Model for Amyotrophic Lateral Sclerosis. J. Neurochem. 2000, 74, 1158–1165. [Google Scholar] [CrossRef][Green Version]
- Grossini, E.; Garhwal, D.; Venkatesan, S.; Ferrante, D.; Mele, A.; Saraceno, M.; Scognamiglio, A.; Mandrioli, J.; Amedei, A.; De Marchi, F.; et al. The Potential Role of Peripheral Oxidative Stress on the Neurovascular Unit in Amyotrophic Lateral Sclerosis Pathogenesis: A Preliminary Report from Human and In Vitro Evaluations. Biomedicines 2022, 10, 691. [Google Scholar] [CrossRef] [PubMed]
- Miquel, E.; Cassina, A.; Martínez-Palma, L.; Souza, J.M.; Bolatto, C.; Rodríguez-Bottero, S.; Logan, A.; Smith, R.A.J.; Murphy, M.P.; Barbeito, L.; et al. Neuroprotective Effects of the Mitochondria-Targeted Antioxidant MitoQ in a Model of Inherited Amyotrophic Lateral Sclerosis. Free Radic. Biol. Med. 2014, 70, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Higgins, C.M.J.; Xu, Z. Mitochondrial Electron Transport Chain Complex Dysfunction in a Transgenic Mouse Model for Amyotrophic Lateral Sclerosis. J. Neurochem. 2002, 83, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, N.; Beal, M.F.; Matson, W.R.; Bogdanov, M.B. Increased Oxidative Damage to DNA in an Animal Model of Amyotrophic Lateral Sclerosis. Free Radic. Res. 2005, 39, 383–388. [Google Scholar] [CrossRef]
- Magrì, A.; Lipari, C.L.R.; Risiglione, P.; Zimbone, S.; Guarino, F.; Caccamo, A.; Messina, A. ERK1/2-Dependent TSPO Overactivation Associates with the Loss of Mitophagy and Mitochondrial Respiration in ALS. Cell Death Dis. 2023, 14, 122. [Google Scholar] [CrossRef] [PubMed]
- Crugnola, V.; Lamperti, C.; Lucchini, V.; Ronchi, D.; Peverelli, L.; Prelle, A.; Sciacco, M.; Bordoni, A.; Fassone, E.; Fortunato, F.; et al. Mitochondrial Respiratory Chain Dysfunction in Muscle from Patients with Amyotrophic Lateral Sclerosis. Arch. Neurol. 2010, 67, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, F.R.; Winkler, K.; Kuznetsov, A.V.; Bartels, C.; Vielhaber, S.; Feistner, H.; Kunz, W.S. Impairment of Mitochondrial Function in Skeletal Muscle of Patients with Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 1998, 156, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.J.; Suh, Y.-L. Ultrastructural Changes of Mitochondria in the Skeletal Muscle of Patients with Amyotrophic Lateral Sclerosis. Ultrastruct. Pathol. 2002, 26, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Kubat, G.B.; Picone, P. Skeletal Muscle Dysfunction in Amyotrophic Lateral Sclerosis: A Mitochondrial Perspective and Therapeutic Approaches. Neurol. Sci. 2024; ahead of printing. [Google Scholar] [CrossRef]
- Curti, D.; Malaspina, A.; Facchetti, G.; Camana, C.; Mazzini, L.; Tosca, P.; Zerbi, F.; Ceroni, M. Amyotrophic Lateral Sclerosis: Oxidative Energy Metabolism and Calcium Homeostasis in Peripheral Blood Lymphocytes. Neurology 1996, 47, 1060–1064. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.P.; Duffy, L.M.; Shaw, P.J.; Grierson, A.J. Altered Age-Related Changes in Bioenergetic Properties and Mitochondrial Morphology in Fibroblasts from Sporadic Amyotrophic Lateral Sclerosis Patients. Neurobiol. Aging 2015, 36, 2893–2903. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Chen, P.; Li, M.; Zhu, Y.; He, Z.; Huang, X. Developing a Novel Immune Infiltration-Associated Mitophagy Prediction Model for Amyotrophic Lateral Sclerosis Using Bioinformatics Strategies. Front. Immunol. 2024, 15, 1360527. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.; Weinreich, M.; Lee, J.A.K.; Shaw, A.C.; Ferraiuolo, L.; Mortiboys, H.; Zhang, S.; Hop, P.J.; Zwamborn, R.A.J.; van Eijk, K.; et al. Rare and Common Genetic Determinants of Mitochondrial Function Determine Severity but Not Risk of Amyotrophic Lateral Sclerosis. Heliyon 2024, 10, e24975. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Adams, D.A.; Niedzwiecki, M.V.; Wong, M. Aberrant DNA and RNA Methylation Occur in Spinal Cord and Skeletal Muscle of Human SOD1 Mouse Models of ALS and in Human ALS: Targeting DNA Methylation Is Therapeutic. Cells 2022, 11, 3448. [Google Scholar] [CrossRef] [PubMed]
- Araujo, B.G.; Souza E Silva, L.F.; de Barros Torresi, J.L.; Siena, A.; Valerio, B.C.O.; Brito, M.D.; Rosenstock, T.R. Decreased Mitochondrial Function, Biogenesis, and Degradation in Peripheral Blood Mononuclear Cells from Amyotrophic Lateral Sclerosis Patients as a Potential Tool for Biomarker Research. Mol. Neurobiol. 2020, 57, 5084–5102. [Google Scholar] [CrossRef] [PubMed]
- Djafarzadeh, S.; Jakob, S.M. High-Resolution Respirometry to Assess Mitochondrial Function in Permeabilized and Intact Cells. J. Vis. Exp. 2017, 120, 54985. [Google Scholar] [CrossRef]
- Jang, D.H.; Greenwood, J.C.; Spyres, M.B.; Eckmann, D.M. Measurement of Mitochondrial Respiration and Motility in Acute Care: Sepsis, Trauma, and Poisoning. J. Intensive Care Med. 2017, 32, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.; Jiao, Y.; Ferrando, L.M.; Yablonska, S.; Li, F.; Horoszko, E.C.; Lacomis, D.; Friedlander, R.M.; Carlisle, D.L. Neuronal Mitochondrial Dysfunction in Sporadic Amyotrophic Lateral Sclerosis Is Developmentally Regulated. Sci. Rep. 2021, 11, 18916. [Google Scholar] [CrossRef] [PubMed]
- Ehinger, J.K.; Morota, S.; Hansson, M.J.; Paul, G.; Elmér, E. Mitochondrial Dysfunction in Blood Cells from Amyotrophic Lateral Sclerosis Patients. J. Neurol. 2015, 262, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Ghiasi, P.; Hosseinkhani, S.; Noori, A.; Nafissi, S.; Khajeh, K. Mitochondrial Complex I Deficiency and ATP/ADP Ratio in Lymphocytes of Amyotrophic Lateral Sclerosis Patients. Neurol. Res. 2012, 34, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Sakae, N.; Bieniek, K.F.; Zhang, Y.-J.; Ross, K.; Gendron, T.F.; Murray, M.E.; Rademakers, R.; Petrucelli, L.; Dickson, D.W. Poly-GR Dipeptide Repeat Polymers Correlate with Neurodegeneration and Clinicopathological Subtypes in C9ORF72-Related Brain Disease. Acta Neuropathol. Commun. 2018, 6, 63. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liu, H.; Itoh, K.; Oh, S.; Zhao, L.; Murata, D.; Sesaki, H.; Hartung, T.; Na, C.H.; Wang, J. C9orf72 Regulates Energy Homeostasis by Stabilizing Mitochondrial Complex I Assembly. Cell Metab. 2021, 33, 531–546.e9. [Google Scholar] [CrossRef] [PubMed]
- Sverdlov, A.L.; Elezaby, A.; Qin, F.; Behring, J.B.; Luptak, I.; Calamaras, T.D.; Siwik, D.A.; Miller, E.J.; Liesa, M.; Shirihai, O.S.; et al. Mitochondrial Reactive Oxygen Species Mediate Cardiac Structural, Functional, and Mitochondrial Consequences of Diet-Induced Metabolic Heart Disease. J. Am. Heart Assoc. 2016, 5, e002555. [Google Scholar] [CrossRef] [PubMed]
- Mourier, A.; Larsson, N.-G. Tracing the Trail of Protons through Complex I of the Mitochondrial Respiratory Chain. PLoS Biol. 2011, 9, e1001129. [Google Scholar] [CrossRef] [PubMed]
- Browne, S.E.; Bowling, A.C.; Baik, M.J.; Gurney, M.; Brown, R.H.; Beal, M.F. Metabolic Dysfunction in Familial, but Not Sporadic, Amyotrophic Lateral Sclerosis. J. Neurochem. 1998, 71, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Duranti, E.; Villa, C. Muscle Involvement in Amyotrophic Lateral Sclerosis: Understanding the Pathogenesis and Advancing Therapeutics. Biomolecules 2023, 13, 1582. [Google Scholar] [CrossRef] [PubMed]
- Hor, J.-H.; Santosa, M.M.; Lim, V.J.W.; Ho, B.X.; Taylor, A.; Khong, Z.J.; Ravits, J.; Fan, Y.; Liou, Y.-C.; Soh, B.-S.; et al. ALS Motor Neurons Exhibit Hallmark Metabolic Defects That Are Rescued by SIRT3 Activation. Cell Death Differ. 2021, 28, 1379–1397. [Google Scholar] [CrossRef] [PubMed]
- Connolly, N.M.C.; Theurey, P.; Adam-Vizi, V.; Bazan, N.G.; Bernardi, P.; Bolaños, J.P.; Culmsee, C.; Dawson, V.L.; Deshmukh, M.; Duchen, M.R.; et al. Guidelines on Experimental Methods to Assess Mitochondrial Dysfunction in Cellular Models of Neurodegenerative Diseases. Cell Death Differ. 2018, 25, 542–572. [Google Scholar] [CrossRef] [PubMed]
- Desler, C.; Hansen, T.L.; Frederiksen, J.B.; Marcker, M.L.; Singh, K.K.; Juel Rasmussen, L. Is There a Link between Mitochondrial Reserve Respiratory Capacity and Aging? J. Aging Res. 2012, 2012, 192503. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Porras, G.; Arribas-Blázquez, M.; Maestro, I.; Borrego-Hernández, D.; Boya, P.; Cerdán, S.; García-Redondo, A.; Martínez, A.; Martin-Requero, Á. Molecular Alterations in Sporadic and SOD1-ALS Immortalized Lymphocytes: Towards a Personalized Therapy. Int. J. Mol. Sci. 2021, 22, 3007. [Google Scholar] [CrossRef] [PubMed]
- Goetzman, E.; Gong, Z.; Zhang, B.; Muzumdar, R. Complex II Biology in Aging, Health, and Disease. Antioxidants 2023, 12, 1477. [Google Scholar] [CrossRef] [PubMed]
- Ranganayaki, S.; Jamshidi, N.; Aiyaz, M.; Rashmi, S.-K.; Gayathri, N.; Harsha, P.K.; Padmanabhan, B.; Srinivas Bharath, M.M. Inhibition of Mitochondrial Complex II in Neuronal Cells Triggers Unique Pathways Culminating in Autophagy with Implications for Neurodegeneration. Sci. Rep. 2021, 11, 1483. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Sharma, B.; Bell, E.; Chandel, N.S.; Schumacker, P.T. Loss of the SdhB, but Not the SdhA, Subunit of Complex II Triggers Reactive Oxygen Species-Dependent Hypoxia-Inducible Factor Activation and Tumorigenesis. Mol. Cell. Biol. 2008, 28, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Hajek, P.; Lin, C.; Shin, S.K.; Attardi, G.; Chomyn, A. Mitochondrial Outer Membrane Permeability Change and Hypersensitivity to Digitonin Early in Staurosporine-Induced Apoptosis. J. Biol. Chem. 2003, 278, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Schirinzi, T.; Salvatori, I.; Zenuni, H.; Grillo, P.; Valle, C.; Martella, G.; Mercuri, N.B.; Ferri, A. Pattern of Mitochondrial Respiration in Peripheral Blood Cells of Patients with Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 10863. [Google Scholar] [CrossRef] [PubMed]
- Annesley, S.J.; Lay, S.T.; De Piazza, S.W.; Sanislav, O.; Hammersley, E.; Allan, C.Y.; Francione, L.M.; Bui, M.Q.; Chen, Z.-P.; Ngoei, K.R.W.; et al. Immortalized Parkinson’s Disease Lymphocytes Have Enhanced Mitochondrial Respiratory Activity. Dis. Model. Mech. 2016, 9, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Arduíno, D.M.; Esteves, A.R.; Swerdlow, R.H.; Cardoso, S.M. A Cybrid Cell Model for the Assessment of the Link Between Mitochondrial Deficits and Sporadic Parkinson’s Disease. Methods Mol. Biol. 2015, 1265, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Bindoff, L.A.; Birch-Machin, M.A.; Cartlidge, N.E.F.; Parker, W.D.; Turnbull, D.M. Respiratory Chain Abnormalities in Skeletal Muscle from Patients with Parkinson’s Disease. J. Neurol. Sci. 1991, 104, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Fišar, Z.; Jirák, R.; Zvěřová, M.; Setnička, V.; Habartová, L.; Hroudová, J.; Vaníčková, Z.; Raboch, J. Plasma Amyloid Beta Levels and Platelet Mitochondrial Respiration in Patients with Alzheimer’s Disease. Clin. Biochem. 2019, 72, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Feldhaus, P.; Fraga, D.B.; Ghedim, F.V.; De Luca, R.D.; Bruna, T.D.; Heluany, M.; Matos, M.P.; Ferreira, G.K.; Jeremias, I.C.; Heluany, C.; et al. Evaluation of Respiratory Chain Activity in Lymphocytes of Patients with Alzheimer Disease. Metab. Brain Dis. 2011, 26, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Rabinovici, G.D.; Miller, B.L. Frontotemporal Lobar Degeneration. CNS Drugs 2010, 24, 375–398. [Google Scholar] [CrossRef] [PubMed]
- Perier, C.; Vila, M. Mitochondrial Biology and Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009332. [Google Scholar] [CrossRef]
- Bayliak, M.M.; Gospodaryov, D.V.; Lushchak, V.I. Homeostasis of Carbohydrates and Reactive Oxygen Species Is Critically Changed in the Brain of Middle-Aged Mice: Molecular Mechanisms and Functional Reasons. BBA Adv. 2023, 3, 100077. [Google Scholar] [CrossRef] [PubMed]
- Kuzma-Kozakiewicz, M.; Kwiecinski, H. New Therapeutic Targets for Amyotrophic Lateral Sclerosis. Expert Opin. Ther. Targets 2011, 15, 127–143. [Google Scholar] [CrossRef] [PubMed]
- Matsuhashi, T.; Hishiki, T.; Zhou, H.; Ono, T.; Kaneda, R.; Iso, T.; Yamaguchi, A.; Endo, J.; Katsumata, Y.; Atsushi, A.; et al. Activation of Pyruvate Dehydrogenase by Dichloroacetate Has the Potential to Induce Epigenetic Remodeling in the Heart. J. Mol. Cell. Cardiol. 2015, 82, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine Transport and Fatty Acid Oxidation. Biochim. Biophys. Acta 2016, 1863, 2422–2435. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Oliveira, T.; Montezinho, L.; Simões, R.F.; Carvalho, M.; Ferreiro, E.; Silva, F.S.G. Mitochondria: A Promising Convergent Target for the Treatment of Amyotrophic Lateral Sclerosis. Cells 2024, 13, 248. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).