

The Role of Zinc in the Development of Vascular Dementia and Parkinson’s Disease and the Potential of Carnosine as Their Therapeutic Agent
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Zn Neurotoxicity
2.1. Usefulness of GT1-7 Cells in the Study of Zn2+-Induced Neurotoxicity
2.2. Molecular Mechanism of Zn2+-Induced GT1-7 Cytotoxicity
2.2.1. Disruption of Calcium Homeostasis
2.2.2. Energy Deficiency and Mitochondrial Glycolysis Inhibition
2.2.3. ER Stress Pathway Involvement in Zn2+-Induced Cytotoxicity
2.2.4. Cu Enhances Zinc-Induced GT1-7 Cell Death
2.3. Role of Zn in the Development of VD and PD
2.3.1. Zn-Related Neurotoxicity
2.3.2. VD and Zn
2.3.3. PD and Zn
2.3.4. Carnosine Prevents Zn2+-Induced Neurotoxicity
3. Carnosine Can Be a Therapeutic Agent for Cerebrovascular Dementia
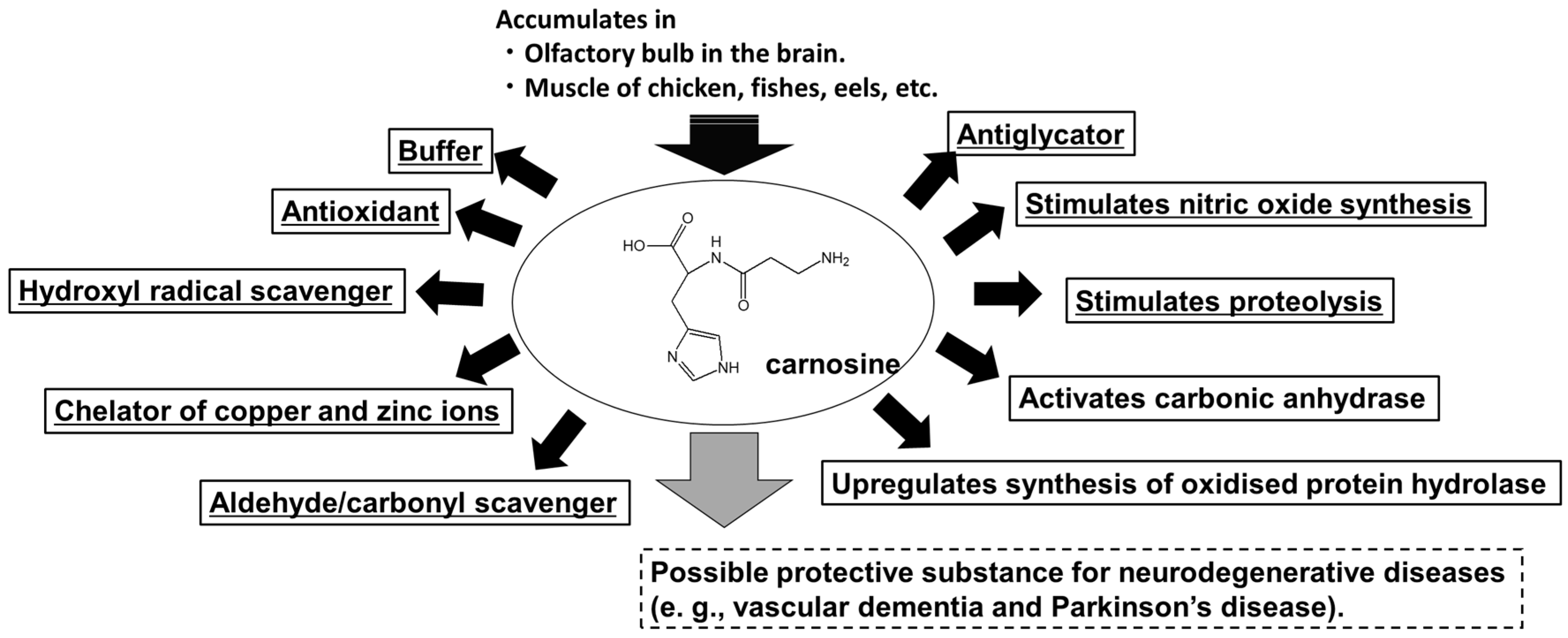
3.1. Carnosine
3.2. Carnosine in the Brain
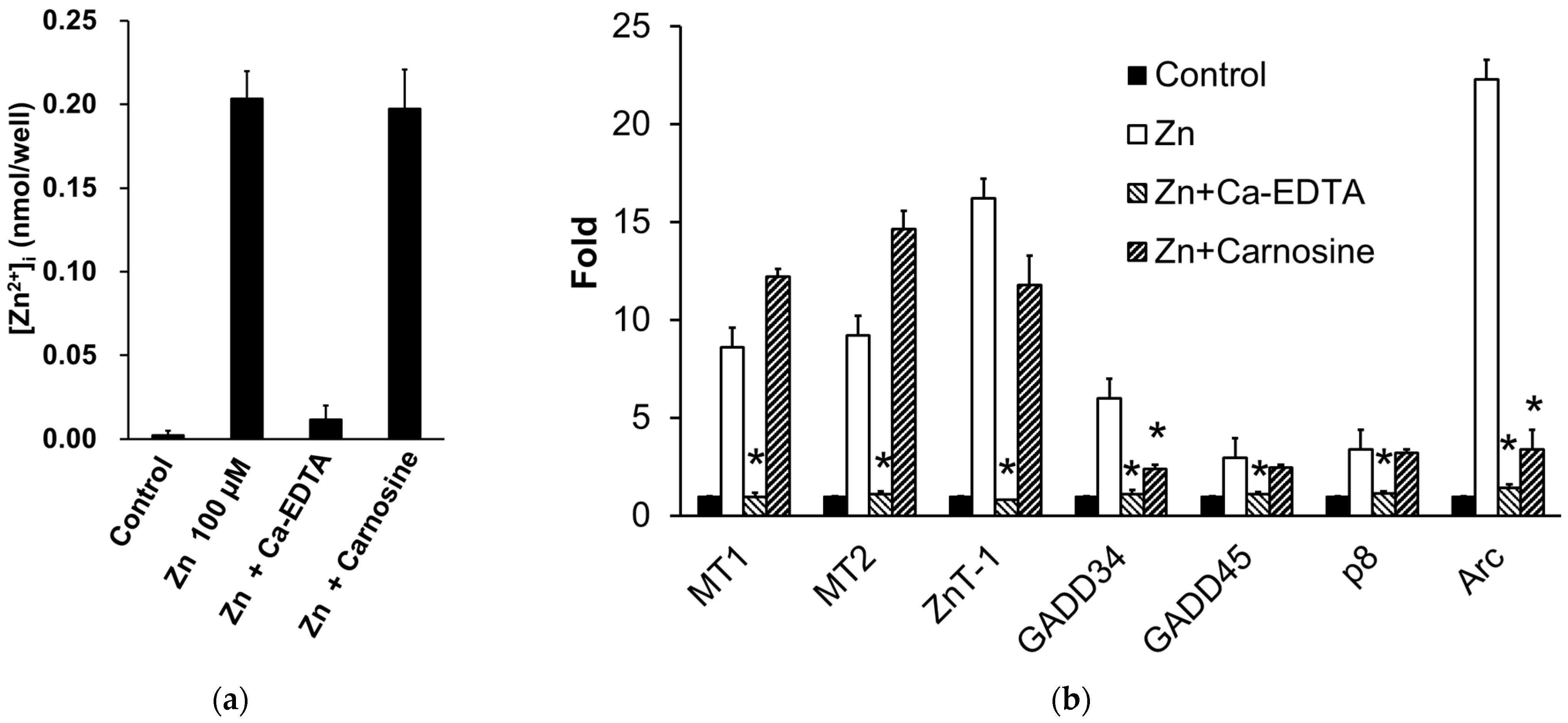
3.3. Carnosine Suppresses Zn-Induced Neuronal Death
3.4. Potential Uses of Carnosine and Its Derivative as Supplements
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hambidge, M. Human zinc deficiency. J. Nutr. 2000, 130, 1344S–1349S. [Google Scholar] [CrossRef]
- Weiss, J.H.; Sensi, S.L.; Koh, J.Y. Zn(2+): A novel ionic mediator of neural injury in brain disease. Trends Pharmacol. Sci. 2000, 21, 395–401. [Google Scholar] [CrossRef]
- Nishio, R.; Morioka, H.; Takeuchi, A.; Saeki, N.; Furuhata, R.; Katahira, M.; Chinenn, T.; Tamura, H.; Tamano, H.; Takeda, A. Intracellular hydrogen peroxide produced by 6-hydroxydopamine is a trigger for nigral dopaminergic degeneration of rats via rapid influx of extracellular Zn2+. Neurotoxicology 2022, 89, 1–8. [Google Scholar] [CrossRef]
- World Health Organization. Dementia. Fact Sheets of WHO. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 1 June 2023).
- Cao, Q.; Tan, C.-C.; Xu, W.; Hu, H.; Cao, X.-P.; Dong, Q.; Tan, L.; Yu, J.-T. The prevalence of dementia: A systematic review and meta-analysis. J. Alzheimer’s Dis. 2020, 73, 1157–1166. [Google Scholar] [CrossRef]
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Brás, I.C.; Dominguez-Meijide, A.; Gerhardt, E.; Koss, D.; Lázaro, D.F.; Santos, P.I.; Vasili, E.; Xylaki, M.; Outeiro, T.F. Synucleinopathies: Where we are and where we need to go. J. Neurochem. 2020, 153, 433–454. [Google Scholar] [CrossRef]
- Iadecola, C. The pathobiology of vascular dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef]
- Pagano, G.; Ferrara, N.; Brooks, D.J.; Pavese, N. Age at onset and Parkinson disease phenotype. Neurology 2016, 86, 1400–1407. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Erekat, N.S. Apoptosis and its Role in Parkinson’s Disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018; pp. 65–82. [Google Scholar]
- Macchi, B.; Di Paola, R.; Marino-Merlo, F.; Felice, M.R.; Cuzzocrea, S.; Mastino, A. Inflammatory and cell death pathways in brain and peripheral blood in Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2015, 14, 313–324. [Google Scholar] [CrossRef]
- Lees, A.J.; Hardy, J.; Revesz, T. Parkinson’s disease. Lancet 2009, 373, 2055–2066. [Google Scholar] [CrossRef]
- de Rijk, M.C.; Breteler, M.M.; Graveland, G.A.; Ott, A.; Grobbee, D.E.; van der Meché, F.G.; Hofman, A. Prevalence of Parkinson’s disease in the elderly: The Rotterdam Study. Neurology 1995, 45, 2143–2146. [Google Scholar] [CrossRef]
- Bower, J.H.; Maraganore, D.M.; McDonnell, S.K.; Rocca, W.A. Incidence and distribution of parkinsonism in Olmsted County, Minnesota, 1976–1990. Neurology 1999, 52, 1214–1220. [Google Scholar] [CrossRef]
- Kawahara, M.; Kato-Negishi, M.; Kuroda, Y. Pyruvate blocks zinc-induced neurotoxicity in immortalized hypothalamic neurons. Cell Mol. Neurobiol. 2002, 22, 87–93. [Google Scholar] [CrossRef]
- Koyama, H.; Konoha, K.; Sadakane, Y.; Ohkawara, S.; Kawahara, M. Zinc neurotoxicity and the pathogenesis of vascular-type dementia: Involvement of calcium dyshomeostasis and carnosine. J. Clin. Toxicol. 2012, S3, 002. [Google Scholar]
- Konoha, K.; Sadakane, Y.; Kawahara, M. Effects of gadolinium and other metal on the neurotoxicity of immortalized hypothalamic neurons induced by zinc. Biomed. Res. Trace Elem. 2004, 15, 275–277. [Google Scholar]
- Tanaka, K.; Kawahara, M. Copper enhances zinc-induced neurotoxicity and the endoplasmic reticulum stress response in a neuronal model of vascular dementia. Front Neurosci. 2017, 11, 58. [Google Scholar] [CrossRef]
- Tanaka, K.; Shimoda, M.; Chuang, V.T.G.; Nishida, K.; Kawahara, M.; Ishida, T.; Otagiri, M.; Maruyama, T.; Ishima, Y. Thioredoxin-albumin fusion protein prevents copper enhanced zinc-induced neurotoxicity via its antioxidative activity. Int. J. Pharm. 2018, 535, 140–147. [Google Scholar] [CrossRef]
- Tanaka, K.; Shimoda, M.; Kawahara, M. Pyruvic acid prevents Cu2+/Zn2+-induced neurotoxicity by suppressing mitochondrial injury. Biochem. Biophys. Res. Commun. 2018, 495, 1335–1341. [Google Scholar] [CrossRef]
- Tanaka, K.; Shimoda, M.; Kasai, M.; Ikeda, M.; Ishima, Y.; Kawahara, M. Involvement of SAPK/JNK signaling pathway in copper enhanced zinc-induced neuronal cell death. Toxicol. Sci. 2019, 169, 293–302. [Google Scholar] [CrossRef]
- Kawahara, M.; Konoha, K.; Nagata, T.; Sadakane, Y. Protective substances against zinc-induced neuronal death after ischaemia: Carnosine as a target for drug of vascular type of dementia. Recent Pat. CNS Drug Discov. 2007, 2, 145–149. [Google Scholar] [CrossRef]
- Kubota, M.; Kobayashi, N.; Sugizaki, T.; Shimoda, M.; Kawahara, M.; Tanaka, K. Carnosine suppresses neuronal cell death and inflammation induced by 6-hydroxydopamine in an in vitro model of Parkinson’s disease. PLoS ONE 2020, 15, e0240448. [Google Scholar] [CrossRef] [PubMed]
- Sadakane, Y.; Konoha, K.; Nagata, T.; Kawahara, M. Protective activity of the extracts from Japanese eel (Anguilla japonica) against zinc-induced neuronal cell death: Carnosine and an unknown substance. Trace Nutr. Res. 2007, 24, 98–105. [Google Scholar]
- Hipkiss, A.R. On the relationship between energy metabolism, proteostasis, aging and Parkinson’s disease: Possible causative role of methylglyoxal and alleviative potential of carnosine. Aging Dis. 2017, 8, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Boldyrev, A.A.; Aldini, G.; Derave, W. Physiology and pathophysiology of carnosine. Physiol. Rev. 2013, 93, 1803–1845. [Google Scholar] [CrossRef] [PubMed]
- De Marchis, S.; Modena, C.; Peretto, P.; Migheli, A.; Margolis, F.L.; Fasolo, A. Carnosine-related dipeptides in neurons and glia. Biochemistry 2000, 65, 824–833. [Google Scholar] [PubMed]
- Mellon, P.L.; Windle, J.J.; Goldsmith, P.C.; Padula, C.A.; Roberts, J.L.; Weiner, R.I. Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron 1990, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Suh, S.W.; Silva, D.; Frederickson, C.J.; Thompson, R.B. Importance of zinc in the central nervous system: The zinc-containing neuron. J. Nutr. 2000, 130, 1471S–1483S. [Google Scholar] [CrossRef] [PubMed]
- Mahesh, V.B.; Zamorano, P.; De Sevilla, L.; Lewis, D.; Brann, D.W. Characterization of ionotropic glutamate receptors in rat hypothalamus, pituitary and immortalized gonadotropin-releasing hormone (GnRH) neurons (GT1-7 cells). Neuroendocrinology 1999, 69, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Sadakane, Y.; Koyama, H.; Konoha, K.; Ohkawara, S. D-histidine and L-histidine attenuate zinc-induced neuronal death in GT1-7 cells. Metallomics 2013, 5, 453–460. [Google Scholar] [CrossRef]
- Mizuno, D.; Konoha-Mizuno, D.; Mori, M.; Sadakane, Y.; Koyama, H.; Ohkawara, S.; Kawahara, M. Protective activity of carnosine and anserine against zinc-induced neurotoxicity: A possible treatment for vascular dementia. Metallomics 2015, 7, 1233–1239. [Google Scholar] [CrossRef]
- Kawahara, M.; Sadakane, Y.; Mizuno, K.; Kato-Negishi, M.; Tanaka, K.I. Carnosine as a possible drug for zinc-induced neurotoxicity and vascular dementia. Int. J. Mol. Sci. 2020, 21, 2570. [Google Scholar] [CrossRef]
- Büsselberg, D.; Platt, B.; Haas, H.L.; Carpenter, D.O. Voltage gated calcium channel currents of rat dorsal root ganglion (DRG) cells are blocked by Al3+. Brain Res. 1993, 622, 163–168. [Google Scholar] [CrossRef]
- Kawahara, M.; Kato-Negishi, M.; Hosoda, R.; Imamura, L.; Tsuda, M.; Kuroda, Y. Brain-derived neurotrophic factor protects cultured rat hippocampal neurons from aluminum maltolate neurotoxicity. J. Inorg. Biochem. 2003, 97, 124–131. [Google Scholar] [CrossRef]
- Sheline, C.T.; Behrens, M.M.; Choi, D.W. Zinc-induced cortical neuronal death: Contribution of energy failure attributable to loss of NAD(+) and inhibition of glycolysis. J. Neurosci. 2000, 20, 3139–3154. [Google Scholar] [CrossRef]
- Cai, A.L.; Zipfel, G.J.; Sheline, C.T. Zinc neurotoxicity is dependent on intracellular NAD+ levels and the sirtuin pathway. Eur. J. Neurosci. 2006, 24, 2169–2176. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, Y.H.; Koh, J.Y. Protection by pyruvate against transient forebrain ischaemia in rats. J. Neurosci. 2001, 21, RC171. [Google Scholar] [CrossRef]
- Sensi, S.L.; Ton-That, D.; Sullivan, P.G.; Jonas, E.A.; Gee, K.R.; Kaczmarek, L.K.; Weiss, J.H. Modulation of mitochondrial function by endogenous Zn2+ pools. Proc. Natl. Acad. Sci. USA 2003, 100, 6157–6162. [Google Scholar] [CrossRef]
- Malaiyandi, L.M.; Honick, A.S.; Rintoul, G.L.; Wang, Q.J.; Reynolds, I.J. Zn2+ inhibits mitochondrial movement in neurons by phosphatidylinositol 3-kinase activation. J. Neurosci. 2005, 25, 9507–9514. [Google Scholar] [CrossRef]
- Higo, T.; Kozo, H.; Hisatsune, C.; Nukina, N.; Hashikawa, T.; Hattori, M.; Nakamura, T.; Mikoshiba, K. Mechanism of ER stress-induced brain damage by IP(3) receptor. Neuron 2010, 68, 865–878. [Google Scholar] [CrossRef]
- Kaneko, M.; Imaizumi, K.; Saito, A.; Kanemoto, S.; Asada, R.; Matsuhisa, K.; Ohtake, Y. ER stress and disease: Toward prevention and treatment. Biol. Pharm. Bull. 2017, 40, 1337–1343. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef]
- Becker, J.S.; Matusch, A.; Palm, C.; Salber, D.; Morton, K.A.; Becker, J.S. Bioimaging of metals in brain tissue by laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) and metallomics. Metallomics 2010, 2, 104–111. [Google Scholar] [CrossRef]
- Kawahara, M.; Kato-Negishi, M.; Tanaka, K. Cross talk between neurometals and amyloidogenic proteins at the synapse and the pathogenesis of neurodegenerative diseases. Metallomics 2017, 9, 619–633. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, L.; Naik, I.; Braunstein, Z.; Zhong, J.; Ren, B. Transcription factor C/EBP homologous protein in health and diseases. Front. Immunol. 2017, 8, 1612. [Google Scholar] [CrossRef]
- Lucke-Wold, B.P.; Turner, R.C.; Logsdon, A.F.; Nguyen, L.; Bailes, J.E.; Lee, J.M.; Robson, M.J.; Omalu, B.I.; Huber, J.D.; Rosen, C.L. Endoplasmic reticulum stress implicated in chronic traumatic encephalopathy. J. Neurosurg. 2016, 124, 687–702. [Google Scholar] [CrossRef]
- Dent, P.; Yacoub, A.; Contessa, J.; Caron, R.; Amorino, G.; Valerie, K.; Hagan, M.P.; Grant, S.; Schmidt-Ullrich, R. Stress and radiation-induced activation of multiple intracellular signaling pathways. Radiat. Res. 2003, 159, 283–300. [Google Scholar] [CrossRef]
- Dandekar, A.; Mendez, R.; Zhang, K. Cross talk between ER stress, oxidative stress, and inflammation in health and disease. Methods Mol. Biol. 2015, 1292, 205–214. [Google Scholar]
- Papaconstantinou, J. The role of signaling pathways of inflammation and oxidative stress in development of senescence and aging phenotypes in cardiovascular disease. Cells 2019, 8, 1383. [Google Scholar] [CrossRef]
- Konno, T.; Melo, E.P.; Chambers, J.E.; Avezov, E. Intracellular sources of ROS/H2O2 in health and neurodegeneration: Spotlight on endoplasmic reticulum. Cells 2021, 10, 233. [Google Scholar] [CrossRef]
- Nakano, Y.; Shimoda, M.; Okudomi, S.; Kawaraya, S.; Kawahara, M.; Tanaka, K. Seleno-L-methionine suppresses copper-enhanced zinc-induced neuronal cell death via induction of glutathione peroxidase. Metallomics 2020, 12, 1693–1701. [Google Scholar] [CrossRef] [PubMed]
- Sensi, S.L.; Paoletti, P.; Bush, A.I.; Sekler, I. Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 2009, 10, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Song, N.; Jiao, Q.; Shi, L.; Du, X. Iron pathophysiology in parkinson diseases. Adv. Exp. Med. Biol. 2019, 1173, 45–66. [Google Scholar] [PubMed]
- Kawahara, M.; Mizuno, D.; Koyama, H.; Konoha, K.; Ohkawara, S.; Sadakane, Y. Disruption of zinc homeostasis and the pathogenesis of senile dementia. Metallomics 2014, 6, 209–219. [Google Scholar] [CrossRef]
- Stork, C.J.; Li, Y.V. Elevated cytoplasmic free zinc and increased reactive oxygen species generation in the context of brain injury. Acta Neurochir. Suppl. 2016, 121, 347–353. [Google Scholar]
- Kalaria, R.N. The pathology and pathophysiology of vascular dementia. Neuropharmacology 2018, 134, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Pendlebury, S.T.; Rothwell, P.M. Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: A systematic review and meta-analysis. Lancet Neurol. 2009, 8, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Klitenick, M.A.; Manton, W.I.; Kirkpatrick, J.B. Cytoarchitectonic distribution of zinc in the hippocampus of man and the rat. Brain Res. 1983, 27, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.Y.; Suh, S.W.; Gwag, B.J.; He, Y.Y.; Hsu, C.Y.; Choi, D.W. The role of zinc in selective neuronal death after transient global cerebral ischaemia. Science 1996, 272, 1013–1016. [Google Scholar] [CrossRef]
- Kitamura, Y.; Iida, Y.; Abe, J.; Mifune, M.; Kasuya, F.; Ohta, M.; Igarashi, K.; Saito, Y.; Saji, H. Release of vesicular Zn2+ in a rat transient middle cerebral artery occlusion model. Brain Res. Bull. 2006, 69, 622–625. [Google Scholar] [CrossRef]
- Qi, Z.; Shi, W.; Zhao, Y.; Ji, X.; Liu, K.J. Zinc accumulation in mitochondria promotes ischaemia-induced BBB disruption through Drp1-dependent mitochondria fission. Toxicol. Appl. Pharmacol. 2019, 377, 114601. [Google Scholar] [CrossRef]
- Kawahara, M.; Tanaka, K.; Kato-Negishi, M. Copper as a collaborative partner of zinc-induced neurotoxicity in the pathogenesis of vascular dementia. Int. J. Mol. Sci. 2021, 22, 7242. [Google Scholar] [CrossRef]
- Konnova, E.A.; Swanberg, M. Animal Models of Parkinson’s Disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018; pp. 83–106. [Google Scholar]
- Rodriguez-Pallares, J.; Parga, J.A.; Joglar, B.; Guerra, M.J.; Labandeira-Garcia, J.L. The mitochondrial ATP-sensitive potassium channel blocker 5-hydroxydecanoate inhibits toxicity of 6-hydroxydopamine on dopaminergic neurons. Neurotox. Res. 2009, 15, 82–95. [Google Scholar] [CrossRef]
- Jonsson, G. Chemical neurotoxins as denervation tools in neurobiology. Annu. Rev. Neurosci. 1980, 3, 169–187. [Google Scholar] [CrossRef]
- Tamano, H.; Morioka, H.; Nishio, R.; Takeuchi, A.; Takeda, A. Blockade of rapid influx of extracellular Zn2+ into nigral dopaminergic neurons overcomes Paraquat-induced Parkinson’s disease in rats. Mol. Neurobiol. 2019, 56, 4539–4548. [Google Scholar] [CrossRef]
- Tamano, H.; Nishio, R.; Morioka, H.; Furuhata, R.; Komata, Y.; Takeda, A. Paraquat as an environmental risk factor in Parkinson’s disease accelerates age-related degeneration via rapid influx of extracellular Zn2+ into nigral dopaminergic neurons. Mol. Neurobiol. 2019, 56, 7789–7799. [Google Scholar] [CrossRef]
- Fisher, A.B. Redox signaling across cell membranes. Antioxid. Redox Signal. 2009, 11, 1349–1356. [Google Scholar] [CrossRef]
- Hara, Y.; Wakamori, M.; Ishii, M.; Maeno, E.; Nishida, M.; Yoshida, T.; Yamada, H.; Shimizu, S.; Mori, E.; Kudoh, J.; et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol. Cell 2002, 9, 163–173. [Google Scholar] [CrossRef]
- Calderone, A.; Jover, T.; Mashiko, T.; Noh, K.M.; Tanaka, H.; Bennett, M.V.; Zukin, R.S. Late calcium EDTA rescues hippocampal CA1 neurons from global ischaemia-induced death. J. Neurosci. 2004, 24, 9903–9913. [Google Scholar] [CrossRef]
- Sadakane, Y.; Konoha, K.; Kawahara, M. Protective activity of mango (Mangifera indica L.) fruit against a zinc-induced neuronal cell death is independent of its antioxidant activity. Trace Nutr. Res. 2005, 22, 73–79. [Google Scholar]
- Everaert, I.; Mooyaart, A.; Baguet, A.; Zutinic, A.; Baelde, H.; Achten, E.; Taes, Y.; De Heer, E.; Derav, W. Vegetarianism, female gender and increasing age, but not CNDP1 genotype, are associated with reduced muscle carnosine levels in humans. Amino Acids 2011, 40, 1221–1229. [Google Scholar] [CrossRef]
- Derave, W.; Everaert, I.; Beeckman, S.; Baguet, A. Muscle carnosine metabolism and beta-alanine supplementation in relation to exercise and training. Sports Med. 2010, 40, 247–263. [Google Scholar] [CrossRef]
- Bakardjiev, A.; Bauer, K. Biosynthesis, release, and uptake of carnosine in primary cultures. Biochemistry 2000, 65, 779–782. [Google Scholar]
- Harris, R.C.; Wise, J.A.; Price, K.A.; Kim, H.J.; Kim, C.K.; Sale, C. Determinants of muscle carnosine content. Amino Acids 2012, 43, 5–12. [Google Scholar] [CrossRef]
- Gariballa, A.E.; Sinclair, A.J. Carnosine: Physiological properties and therapeutic potential. Age Ageing 2000, 29, 207–210. [Google Scholar] [CrossRef]
- Mori, M.; Mizuno, D.; Konoha-Mizuno, K.; Sadakane, Y.; Kawahara, M. Carnosine concentration in the muscle of thoroughbred horses and its implications in exercise performance. Trace Nutr. Res. 2015, 32, 49–53. [Google Scholar]
- Kawai, M.; Minami, Y.; Sayama, Y.; Kuwano, A.; Hiraga, A.; Miyata, H. Muscle Fiber Population and Biochemical Properties of Whole Body Muscles in Thoroughbred Horses. Anat. Rec. 2009, 292, 1663–1669. [Google Scholar] [CrossRef]
- Abe, H. Role of histidine-related compounds as intracellular proton buffering constituents in vertebrate muscle. Biochemistry 2000, 65, 757–765. [Google Scholar]
- Sale, C.; Artioli, G.G.; Gualano, B.; Saunders, B.; Hobson, R.M.; Harris, R.C. Carnosine: From exercise performance to health. Amino Acids 2013, 44, 1477–1491. [Google Scholar] [CrossRef]
- Quesnele, J.J.; Laframboise, M.A.; Wong, J.J.; Kim, P.; Wells, G.D. The effects of beta-alanine supplementation on performance: A systematic review of the literature. Int. J. Sport. Nutr. Exerc. Metab. 2014, 2014. 24, 14–27. [Google Scholar] [CrossRef]
- Guney, Y.; Turkcu, U.O.; Hicsonmez, A.; Andrieu, M.N.; Guney, H.Z.; Bilgihan, A.; Kurtman, C. Carnosine may reduce lung injury caused by radiation therapy. Med. Hypotheses 2006, 66, 957–959. [Google Scholar] [CrossRef]
- Hipkiss, A.R. Carnosine and its possible roles in nutrition and health. Adv. Food. Nutr. Res. 2009, 57, 87–154. [Google Scholar]
- Dubois, V.D.; Bastawrous, A. N-acetylcarnosine (NAC) drops for age-related cataract. Cochrane Database Syst. Rev. 2017, 2, CD009493. [Google Scholar] [CrossRef]
- Matsukura, T.; Tanaka, H. Applicability of zinc complex of L-carnosine for medical use. Biochemistry 2000, 65, 817–823. [Google Scholar]
- Kimura, K.; Nakano, Y.; Sugizaki, T.; Shimoda, M.; Kobayashi, N.; Kawahara, M.; Tanaka, K. Protective effect of polaprezinc on cadmium-induced injury of lung epithelium. Metallomics 2019, 11, 1310–1320. [Google Scholar] [CrossRef]
- Baslow, M.H.; Suckow, R.F.; Berg, M.J.; Marks, N.; Saito, M.; Bhakoo, K.K. Differential expression of carnosine, homocarnosine and N-acetyl-L-histidine hydrolytic activities in cultured rat macroglial cells. J. Mol. Neurosci. 2001, 17, 351–359. [Google Scholar] [CrossRef]
- Tiedje, K.E.; Stevens, K.; Barnes, S.; Weaver, D.F. β-Alanine as a small molecule neurotransmitter. Neurochem. Int. 2010, 57, 177–188. [Google Scholar] [CrossRef]
- Wang, H.; Fei, Y.J.; Ganapathy, V.; Leibach, F.H. Electrophysiological characteristics of the proton-coupled peptide transporter PEPT2 cloned from rat brain. Am. J. Physiol. 1998, 275, C967–C975. [Google Scholar] [CrossRef] [PubMed]
- Lopachev, A.V.; Abaimov, D.A.; Filimonov, I.S.; Kulichenkova, K.N.; Fedorova, T.N. An assessment of the transport mechanism and intraneuronal stability of L-carnosine. Amino Acids 2022, 54, 1115–1122. [Google Scholar] [CrossRef]
- Bonfanti, L.; Peretto, P.; De Marchis, S.; Fasolo, A. Carnosine-related dipeptides in the mammalian brain. Prog. Neurobiol. 1999, 59, 333–353. [Google Scholar] [CrossRef] [PubMed]
- Bakardjiev, A. Carnosine and beta-alanine release is stimulated by glutamatergic receptors in cultured rat oligodendrocytes. Glia 1998, 24, 346–351. [Google Scholar] [CrossRef]
- Mori, M.; Mizuno, D.; Konoha-Mizuno, K.; Sadakane, Y.; Kawahara, M. Quantitative analysis of carnosine and anserine in foods by performing high performance liquid chromatography. Biomed. Res. Trace Elem. 2015, 26, 147–152. [Google Scholar]
- Margolis, F.L.; Grillo, M.; Kawano, T.; Farbman, A.I. Carnosine synthesis in olfactory tissue during ontogeny: Effect of exogenous beta-alanine. J. Neurochem. 1985, 44, 1459–1464. [Google Scholar] [CrossRef] [PubMed]
- Biffo, S.; Grillo, M.; Margolis, F.L. Cellular localization of carnosine-like and anserine-like immunoreactivities in rodent and avian central nervous system. Neuroscience 1990, 35, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Margolis, F.L.; Grillo, M. Axoplasmic transport of carnosine (β-alanyl-L-histidine) in the mouse olfactory pathway. Neurochem. Res. 1977, 2, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.; Graziadei, P.P.; Monti Graziadei, G.A.; Margolis, F.L. Denervation in the primary olfactory pathway of mice. IV. Biochemical and morphological evidence for neuronal replacement following nerve section. Brain Res. 1977, 132, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.; Margolis, F.L. Denervation in the primary olfactory pathway of mice. III. Effect on enzymes of carnosine metabolism. Brain Res. 1976, 110, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Baran, E.J. Metal complexes of carnosine. Biochemistry 2000, 65, 789–797. [Google Scholar] [PubMed]
- Trombley, P.Q.; Horning, M.S.; Blakemore, L.J. Interactions between carnosine and zinc and copper: Implications for neuromodulation and neuroprotection. Biochemistry 2000, 65, 807–816. [Google Scholar]
- Hobart, L.J.; Seibel, I.; Yeargans, G.S.; Seidler, M.W. Anti-crosslinking properties of carnosine: Significance of histidine. Life Sci. 2004, 75, 1379–1389. [Google Scholar] [CrossRef]
- Kang, J.H.; Kim, K.S. Enhanced oligomerization of the alpha-synuclein mutant by the Cu, Zn-superoxide dismutase and hydrogen peroxide system. Mol. Cells 2003, 15, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Aloisi, A.; Barca, A.; Romano, A.; Guerrieri, S.; Storelli, C.; Rinaldi, R.; Verri, T. Anti-aggregating effect of the naturally occurring dipeptide carnosine on Aβ1-42 fibril formation. PLoS ONE 2013, 8, e68159. [Google Scholar] [CrossRef] [PubMed]
- Attanasio, F.; Convertino, M.; Magno, A.; Caflisch, A.; Corazza, A.; Haridas, H.; Esposito, G.; Cataldo, S.; Pignataro, B.; Milardi, D.; et al. Carnosine inhibits Aβ42 aggregation by perturbing the H-bond network in and around the central hydrophobic cluster. ChemBioChem 2013, 14, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Corona, C.; Frazzini, V.; Silvestri, E.; Lattanzio, R.; La Sorda, R.; Piantelli, M.; Canzoniero, L.M.; Ciavardelli, D.; Rizzarelli, E.; Sensi, S.L. Effects of dietary supplementation of carnosine on mitochondrial dysfunction, amyloid pathology, and cognitive deficits in 3xTg-AD mice. PLoS ONE 2011, 6, e17971. [Google Scholar] [CrossRef] [PubMed]
- Caruso, G.; Fresta, C.G.; Musso, N.; Giambirtone, M.; Grasso, M.; Spampinato, S.F.; Merlo, S.; Drago, F.; Lazzarino, G.; Sortinom, M.A.; et al. Carnosine prevents Aβ-induced oxidative stress and inflammation in microglial cells: A key role of TGF-β. Cells 2019, 8, 64. [Google Scholar] [CrossRef]
- Kawahara, M.; Koyama, H.; Nagata, T.; Sadakane, Y. Zinc, copper, and carnosine attenuate neurotoxicity of prion fragment PrP106-126. Metallomics 2011, 3, 726–734. [Google Scholar] [CrossRef]
- Ommati, M.M.; Heidari, R.; Ghanbarinejad, V.; Aminian, A.; Abdoli, N.; Niknahad, H. The neuroprotective properties of carnosine in a mouse model of manganism is mediated via mitochondria regulating and antioxidative mechanisms. Nutr. Neurosci. 2020, 23, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Berezhnoy, D.S.; Stvolinsky, S.L.; Lopachev, A.V.; Devyatov, A.A.; Lopacheva, O.M.; Kulikova, O.I.; Abaimov, D.A.; Fedorova, T.N. Carnosine as an effective neuroprotector in brain pathology and potential neuromodulator in normal conditions. Amino Acids 2019, 51, 139–150. [Google Scholar] [CrossRef]
- Konoha, K.; Sadakane, Y.; Kawahara, M. Carnosine protects GT1–7 cells against zinc-induced neurotoxicity: A possible candidate for treatment for vascular type of dementia. Trace Nutr. Res. 2006, 23, 56–62. [Google Scholar]
- Ferreiro, E.; Baldeiras, I.; Ferreira, I.L.; Costa, R.O.; Rego, A.C.; Pereira, C.F.; Oliveira, C.R. Mitochondrial- and endoplasmic reticulum-associated oxidative stress in Alzheimer’s disease: From pathogenesis to biomarkers. Int. J. Cell Biol. 2012, 2012, 735206. [Google Scholar] [CrossRef]
- Moskalev, A.A.; Smit-McBride, Z.; Shaposhnikov, M.V.; Plyusnina, E.N.; Zhavoronkov, A.; Budovsky, A.; Tacutu, R.; Fraifeld, V.E. Gadd45 proteins: Relevance to aging, longevity and age-related pathologies. Ageing Res. Rev. 2012, 11, 51–66. [Google Scholar] [CrossRef]
- Korb, E.; Finkbeiner, S. Arc in synaptic plasticity: From gene to behavior. Trends Neurosci. 2011, 34, 591–598. [Google Scholar] [CrossRef]
- Zhang, X.; Song, L.; Cheng, X.; Yang, Y.; Luan, B.; Jia, L.; Xu, F.; Zhang, Z. Carnosine pretreatment protects against hypoxia-ischaemia brain damage in the neonatal rat model. Eur. J. Pharmacol. 2011, 667, 202–207. [Google Scholar] [CrossRef]
- Park, H.S.; Han, K.H.; Shin, J.A.; Park, J.H.; Song, K.Y.; Kim, D.H. The neuroprotective effects of carnosine in early stage of focal ischaemia rodent model. J. Korean Neurosurg. Soc. 2014, 55, 125–130. [Google Scholar] [CrossRef]
- Noguchi, K.; Ali, T.F.S.; Miyoshi, J.; Orito, K.; Negoto, T.; Biswas, T.; Taira, N.; Koga, R.; Okamoto, Y.; Fujita, M.; et al. Neuroprotective effects of a novel carnosine-hydrazide derivative on hippocampal CA1 damage after transient cerebral ischaemia. Eur. J. Med. Chem. 2019, 163, 207–214. [Google Scholar] [CrossRef]
- Davis, C.K.; Laud, P.J.; Bahor, Z.; Rajanikant, G.K.; Majid, A. Systematic review and stratified meta-analysis of the efficacy of carnosine in animal models of ischemic stroke. J. Cereb. Blood Flow Metab. 2016, 36, 1686–1694. [Google Scholar] [CrossRef] [PubMed]
- Afshin-Majd, S.; Khalili, M.; Roghani, M.; Mehranmehr, N.; Baluchnejadmojarad, T. Carnosine exerts neuroprotective effect against 6-hydroxydopamine toxicity in hemiparkinsonian rat. Mol. Neurobiol. 2015, 51, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Sadikali, F.; Darwish, R.; Watson, W.C. Carnosinase activity of human gastrointestinal mucosa. Gut 1975, 16, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Asatoor, A.M.; Bandoh, J.K.; Lant, A.F.; Milne, M.D.; Navab, F. Intestinal absorption of carnosine and its constituent amino acids in man. Gut 1970, 11, 250–254. [Google Scholar] [CrossRef]
- Harris, R.C.; Tallon, M.J.; Dunnett, M.; Boobis, L.; Coakley, J.; Kim, H.J.; Fallowfield, J.L.; Hill, C.A.; Sale, C.; Wise, J.A. The absorption of orally supplied beta-alanine and its effect on muscle carnosine synthesis in human vastus lateralis. Amino Acids 2006, 30, 279–289. [Google Scholar] [CrossRef]
- Drozak, J.; Veiga-da-Cunha, M.; Vertommen, D.; Stroobant, V.; Van Schaftingen, E. Molecular identification of carnosine synthase as ATP-grasp domain-containing protein 1 (ATPGD1). J. Biol. Chem. 2010, 285, 9346–9356. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.W.; O’Fallon, J.V. The subcellular distribution of carnosine, carnosine synthetase, and carnosinase in mouse olfactory tissues. Brain Res. 1979, 173, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Tomonaga, S.; Hayakawa, T.; Yamane, H.; Maemura, H.; Sato, M.; Takahata, Y.; Morimatsu, F.; Furuse, M. Oral administration of chicken breast extract increases brain carnosine and anserine concentrations in rats. Nutr. Neurosci. 2007, 10, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.R.; Rathmacher, J.A.; Robinson, J.; Gepner, Y.; Cohen, H. Effect of β-alanine supplementation on carnosine and histidine content in the hippocampus of 14-month-old rats. Appl. Physiol. Nutr. Metab. 2019, 44, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Araminia, B.; Shalbafan, M.; Mortezaei, A.; Shirazi, E.; Ghaffari, S.; Sahebolzamani, E.; Mortazavi, S.H.; Shariati, B.; Ardebili, M.E.; Aqamolaei, A.; et al. L-Carnosine combination therapy for major depressive disorder: A randomized, double-blind, placebo-controlled trial. J. Affect. Disord. 2020, 267, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Hisatsune, T.; Kaneko, J.; Kurashige, H.; Cao, Y.; Satsu, H.; Totsuka, M.; Katakura, Y.; Imabayashi, E.; Matsuda, H. Effect of anserine/carnosine supplementation on verbal episodic memory in elderly people. J. Alzheimer’s Dis. 2016, 50, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Masuoka, N.; Yoshimine, C.; Hori, M.; Tanaka, M.; Asada, T.; Abe, K.; Hisatsune, T. Effects of anserine/carnosine supplementation on mild cognitive impairment with APOE4. Nutrients 2019, 11, 1626. [Google Scholar] [CrossRef] [PubMed]
- Oppermann, H.; Heinrich, M.; Birkemeyer, C.; Meixensberger, J.; Gaunitz, F. The proton-coupled oligopeptide transporters PEPT2, PHT1 and PHT2 mediate the uptake of carnosine in glioblastoma cells. Amino Acids 2019, 51, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Hata, J.; Ohara, T.; Katakura, Y.; Shimizu, K.; Yamashita, S.; Yoshida, D.; Honda, T.; Hirakawa, Y.; Shibata, M.; Sakata, S.; et al. Association between serum β-alanine and risk of dementia. Am. J. Epidemiol. 2019, 188, 1637–1645. [Google Scholar] [CrossRef]
- Stuerenburg, H.J. The roles of carnosine in aging of skeletal muscle and in neuromuscular diseases. Biochemistry 2000, 65, 862–865. [Google Scholar]
- Anderson, E.J.; Vistoli, G.; Katunga, L.A.; Funai, K.; Regazzoni, L.; Monroe, T.B.; Gilardoni, E.; Cannizzaro, L.; Colzani, M.; De Maddis, D.; et al. A carnosine analog mitigates metabolic disorders of obesity by reducing carbonyl stress. J. Clin. Investig. 2018, 128, 5280–5293. [Google Scholar] [CrossRef]
- de Jager, S.; Vermeulen, A.; De Baere, S.; Van der Stede, T.; Lievens, E.; Croubels, S.; Jäger, R.; Purpura, M.; Bourgois, J.G.; Derave, W. Acute balenine supplementation in humans as a natural carnosinase-resistant alternative to carnosine. Sci. Rep. 2023, 13, 6484. [Google Scholar] [CrossRef]
- Kawahara, M.; Sadakane, Y.; Konoha, K. Preventive or Therapeutic Agent for Cerebrovascular Dementia [Translated from Japanese]. JP5382633, 11 October 2013. [Google Scholar]
- Kawahara, M.; Sadakane, Y.; Konoha, K. Drugs for Prevention or Treatment of Vascular Dementia [Translated from Japanese]. JP5294194, 21 June 2013. [Google Scholar]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mizuno, D.; Kawahara, M.; Konoha-Mizuno, K.; Hama, R.; Ogawara, T. The Role of Zinc in the Development of Vascular Dementia and Parkinson’s Disease and the Potential of Carnosine as Their Therapeutic Agent. Biomedicines 2024, 12, 1296. https://doi.org/10.3390/biomedicines12061296
Mizuno D, Kawahara M, Konoha-Mizuno K, Hama R, Ogawara T. The Role of Zinc in the Development of Vascular Dementia and Parkinson’s Disease and the Potential of Carnosine as Their Therapeutic Agent. Biomedicines. 2024; 12(6):1296. https://doi.org/10.3390/biomedicines12061296
Chicago/Turabian StyleMizuno, Dai, Masahiro Kawahara, Keiko Konoha-Mizuno, Ryoji Hama, and Terumasa Ogawara. 2024. "The Role of Zinc in the Development of Vascular Dementia and Parkinson’s Disease and the Potential of Carnosine as Their Therapeutic Agent" Biomedicines 12, no. 6: 1296. https://doi.org/10.3390/biomedicines12061296
APA StyleMizuno, D., Kawahara, M., Konoha-Mizuno, K., Hama, R., & Ogawara, T. (2024). The Role of Zinc in the Development of Vascular Dementia and Parkinson’s Disease and the Potential of Carnosine as Their Therapeutic Agent. Biomedicines, 12(6), 1296. https://doi.org/10.3390/biomedicines12061296