
Exploring the Therapeutic Potential of Gene Therapy in Arrhythmogenic Right Ventricular Cardiomyopathy


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
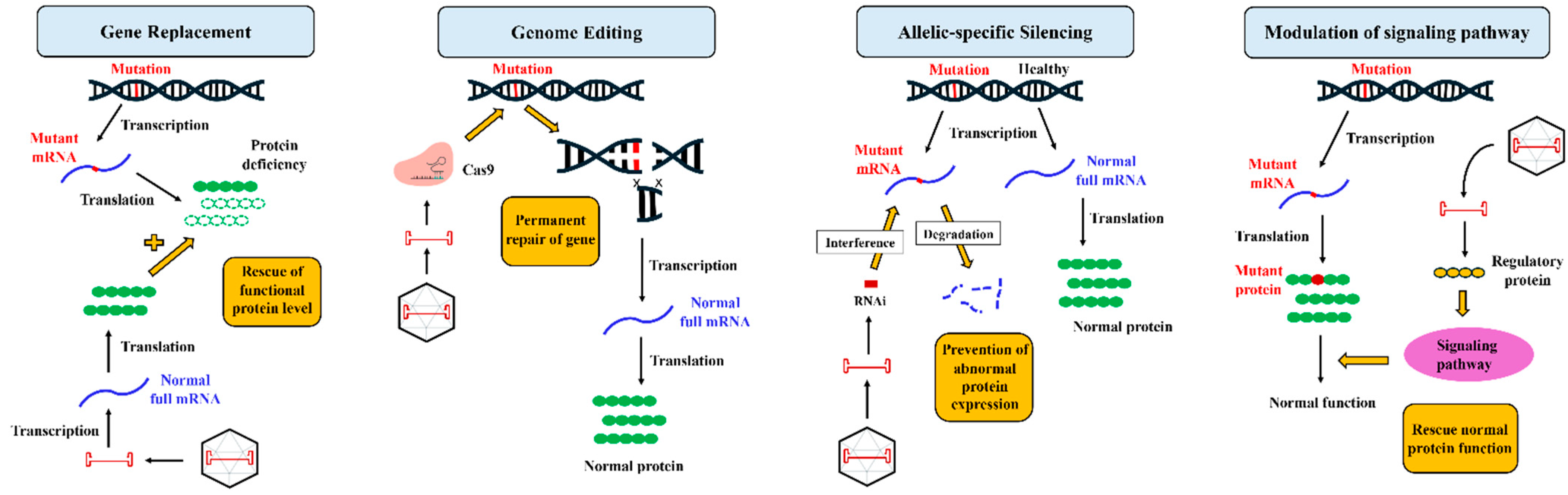
2. Gene Therapy: Understanding the Fundamentals
3. Gene Therapy Breakthroughs in ARVC Treatment
3.1. Plakophilin-2 (PKP2) Pathogenic Variant
3.2. Phospholamban (PLN) Pathogenic Variant
3.3. Desmoglein-2 (DSG2) Pathogenic Variant
4. Challenges in Translation from Bench to Bedside
4.1. Recombinant AAV Vector Design
4.2. Mode of AAV Delivery
4.3. Host Immune System
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm 2019, 16, e301–e372. [Google Scholar] [CrossRef] [PubMed]
- Helms, A.S.; Thompson, A.D.; Day, S.M. Translation of New and Emerging Therapies for Genetic Cardiomyopathies. JACC Basic. Transl. Sci. 2022, 7, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Te Riele, A.S.; James, C.A.; Philips, B.I.N.U.; Rastegar, N.E.D.A.; Bhonsale, A.; Groeneweg, J.A.; Murray, B.; Tichnell, C.; Judge, D.P.; Van Der Heijden, J.F.; et al. Mutation-positive arrhythmogenic right ventricular dysplasia/cardiomyopathy: The triangle of dysplasia displaced. J. Cardiovasc. Electrophysiol. 2013, 24, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- James, C.A.; Jongbloed, J.D.; Hershberger, R.E.; Morales, A.; Judge, D.P.; Syrris, P.; Pilichou, K.; Domingo, A.M.; Murray, B.; Cadrin-Tourigny, J.; et al. International Evidence Based Reappraisal of Genes Associated With Arrhythmogenic Right Ventricular Cardiomyopathy Using the Clinical Genome Resource Framework. Circ. Genom. Precis Med. 2021, 14, e003273. [Google Scholar] [CrossRef] [PubMed]
- McNally, E.; MacLeod, H.; Dellefave-Castillo, L. Arrhythmogenic Right Ventricular Cardiomyopathy Overview. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2023. [Google Scholar]
- Krahn Andrew, D.; Wilde Arthur, A.M.; Calkins, H.; La Gerche, A.; Cadrin-Tourigny, J.; Roberts Jason, D.; Han, H.-C. Arrhythmogenic Right Ventricular Cardiomyopathy. JACC Clin. Electrophysiol. 2022, 8, 533–553. [Google Scholar] [CrossRef] [PubMed]
- Bhonsale, A.; te Riele, A.S.J.M.; Sawant, A.C.; Groeneweg, J.A.; James, C.A.; Murray, B.; Tichnell, C.; Mast, T.P.; van der Pols, M.J.; Cramer, M.J.M.; et al. Cardiac phenotype and long-term prognosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia patients with late presentation. Heart Rhythm 2017, 14, 883–891. [Google Scholar] [CrossRef]
- Gilotra, N.A.; Bhonsale, A.; James, C.A.; te Riele, A.S.J.; Murray, B.; Tichnell, C.; Sawant, A.; Ong, C.S.; Judge, D.P.; Russell, S.D.; et al. Heart Failure Is Common and Under-Recognized in Patients with Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia. Circ. Heart Fail. 2017, 10, e003819. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, D.; Kok, C.; Farraha, M.; Selvakumar, D.; Clayton, Z.E.; Kumar, S.; Chong, J.; Kizana, E. Gene and Cell Therapy for Cardiac Arrhythmias. Clin. Ther. 2020, 42, 1911–1922. [Google Scholar] [CrossRef] [PubMed]
- Gushchina, L.V.; Frair, E.C.; Rohan, N.; Bradley, A.J.; Simmons, T.R.; Chavan, H.D.; Chou, H.J.; Eggers, M.; Waldrop, M.A.; Wein, N.; et al. Lack of Toxicity in Nonhuman Primates Receiving Clinically Relevant Doses of an AAV9.U7snRNA Vector Designed to Induce DMD Exon 2 Skipping. Hum. Gene Ther. 2021, 32, 882–894. [Google Scholar] [CrossRef]
- Greenberg, B.; Butler, J.; Felker, G.M.; Ponikowski, P.; Voors, A.A.; Desai, A.S.; Barnard, D.; Bouchard, A.; Jaski, B.; Lyon, A.R.; et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): A randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet 2016, 387, 1178–1186. [Google Scholar] [CrossRef]
- Rossano, J.; Taylor, M.; Lin, K.; Epstein, S.; Battiprolu, P.; Ricks, D.; Waldron, A.; Schwartz, J.; Greenberg, B. Abstract 11117: Phase 1 Danon Disease Results: The First Single Dose Intravenous (IV) Gene Therapy (RP-A501) With Recombinant Adeno-Associated Virus (AAV9:LAMP2B) for a Monogenic Cardiomyopathy. Circulation 2022, 146, A11117-A. [Google Scholar] [CrossRef]
- AskBio. AskBio Presents Preliminary Data from Phase 1 Trial of Gene Therapy for Congestive Heart Failure (CHF) at the 2023 American Heart Association Scientific Sessions 2023. Available online: https://www.bayer.com/media/en-us/askbio-presents-preliminary-data-from-phase-1-trial-of-gene-therapy-for-congestive-heart-failure-chf-at-the-2023-american-heart-association-scientific-sessions/ (accessed on 7 June 2024).
- Papanikolaou, E.; Bosio, A. The Promise and the Hope of Gene Therapy. Front. Genome Ed. 2021, 3, 618346. [Google Scholar] [CrossRef]
- Hajjar, R.J. Potential of gene therapy as a treatment for heart failure. J. Clin. Investig. 2013, 123, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. Viral Vectors in Gene Therapy. Diseases 2018, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Kim, H. Therapeutic application of the CRISPR system: Current issues and new prospects. Human Genet. 2019, 138, 563–590. [Google Scholar] [CrossRef] [PubMed]
- Castanotto, D.; Rossi, J.J. The promises and pitfalls of RNA-interference-based therapeutics. Nature 2009, 457, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Bezzerides, V.J.; Prondzynski, M.; Carrier, L.; Pu, W.T. Gene therapy for inherited arrhythmias. Cardiovasc. Res. 2020, 116, 1635–1650. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Nakamura, S.; Higo, S.; Shiba, M.; Kohama, Y.; Kondo, T.; Kameda, S.; Tabata, T.; Okuno, S.; Ikeda, Y.; et al. Modeling reduced contractility and impaired desmosome assembly due to plakophilin-2 deficiency using isogenic iPS cell-derived cardiomyocytes. Stem Cell Rep. 2022, 17, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Wu, I.; Zeng, A.; Greer-Short, A.; Aycinena, J.A.; Tefera, A.E.; Shenwai, R.; Farshidfar, F.; Van Pell, M.; Xu, E.; Reid, C.; et al. AAV9:PKP2 improves heart function and survival in a Pkp2-deficient mouse model of arrhythmogenic right ventricular cardiomyopathy. Commun. Med. 2024, 4, 38. [Google Scholar] [CrossRef]
- Kyriakopoulou, E.; Versteeg, D.; de Ruiter, H.; Perini, I.; Seibertz, F.; Döring, Y.; Zentilin, L.; Tsui, H.; van Kampen, S.J.; Tiburcy, M.; et al. Therapeutic efficacy of AAV-mediated restoration of PKP2 in arrhythmogenic cardiomyopathy. Nat. Cardiovasc. Res. 2023, 2, 1262–1276. [Google Scholar] [CrossRef]
- Tiburcy, M.; Meyer, T.; Satin, P.L.; Zimmermann, W.H. Defined Engineered Human Myocardium for Disease Modeling, Drug Screening, and Heart Repair. Methods Mol. Biol. 2022, 2485, 213–225. [Google Scholar] [PubMed]
- Tsui, H.; van Kampen, S.J.; Han, S.J.; Meraviglia, V.; van Ham, W.B.; Casini, S.; van der Kraak, P.; Vink, A.; Yin, X.; Mayr, M.; et al. Desmosomal protein degradation as an underlying cause of arrhythmogenic cardiomyopathy. Sci. Transl. Med. 2023, 15, eadd4248. [Google Scholar] [CrossRef] [PubMed]
- Bradford, W.H.; Zhang, J.; Gutierrez-Lara, E.J.; Liang, Y.; Do, A.; Wang, T.-M.; Nguyen, L.; Mataraarachchi, N.; Wang, J.; Gu, Y.; et al. Plakophilin 2 gene therapy prevents and rescues arrhythmogenic right ventricular cardiomyopathy in a mouse model harboring patient genetics. Nat. Cardiovasc. Res. 2023, 2, 1246–1261. [Google Scholar] [CrossRef]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; Taylor, J.C.; et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 2017, 19, 192–203. [Google Scholar] [CrossRef] [PubMed]
- van Opbergen, C.J.M.; Narayanan, B.; Sacramento, C.B.; Stiles, K.M.; Mishra, V.; Frenk, E.; Ricks, D.; Chen, G.; Zhang, M.; Yarabe, P.; et al. AAV-Mediated Delivery of Plakophilin-2a Arrests Progression of Arrhythmogenic Right Ventricular Cardiomyopathy in Murine Hearts: Preclinical Evidence Supporting Gene Therapy in Humans. Circ. Genom. Precis Med. 2024, 17, e004305. [Google Scholar] [CrossRef] [PubMed]
- van der Zwaag, P.A.; van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Karakikes, I.; Stillitano, F.; Nonnenmacher, M.; Tzimas, C.; Sanoudou, D.; Termglinchan, V.; Kong, C.W.; Rushing, S.; Hansen, J.; Ceholski, D.; et al. Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat. Commun. 2015, 6, 6955. [Google Scholar] [CrossRef] [PubMed]
- Dave, J.; Raad, N.; Mittal, N.; Zhang, L.; Fargnoli, A.; Oh, J.G.; Savoia, M.E.; Hansen, J.; Fava, M.; Yin, X.; et al. Gene editing reverses arrhythmia susceptibility in humanized PLN-R14del mice: Modelling a European cardiomyopathy with global impact. Cardiovasc. Res. 2022, 118, 3140–3150. [Google Scholar] [CrossRef] [PubMed]
- Shiba, M.; Higo, S.; Kondo, T.; Li, J.; Liu, L.; Ikeda, Y.; Kohama, Y.; Kameda, S.; Tabata, T.; Inoue, H.; et al. Phenotypic recapitulation and correction of desmoglein-2-deficient cardiomyopathy using human-induced pluripotent stem cell-derived cardiomyocytes. Human Mol. Genet. 2021, 30, 1384–1397. [Google Scholar] [CrossRef]
- Renovacor. Renovacor Announces Pipeline Expansion with New Research Program for Multiple Genetic Segments of Arrhythmogenic Cardiomyopathy; Renovacor, Inc.: Greenwich, CT, USA, 2022; Available online: https://www.biospace.com/article/releases/renovacor-announces-pipeline-expansion-with-new-research-program-for-multiple-genetic-segments-of-arrhythmogenic-cardiomyopathy/ (accessed on 7 June 2024).
- Büning, H.; Srivastava, A. Capsid Modifications for Targeting and Improving the Efficacy of AAV Vectors. Mol. Ther. Methods Clin. Dev. 2019, 12, 248–265. [Google Scholar] [CrossRef]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L., 3rd; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef] [PubMed]
- Kok, C.Y.; Tsurusaki, S.; Cabanes-Creus, M.; Igoor, S.; Rao, R.; Skelton, R.; Liao, S.H.Y.; Ginn, S.L.; Knight, M.; Scott, S.; et al. Development of new adeno-associated virus capsid variants for targeted gene delivery to human cardiomyocytes. Mol. Ther. Methods Clin. Dev. 2023, 30, 459–473. [Google Scholar] [CrossRef]
- Jaski, B.E.; Jessup, M.L.; Mancini, D.M.; Cappola, T.P.; Pauly, D.F.; Greenberg, B.; Borow, K.; Dittrich, H.; Zsebo, K.M.; Hajjar, R.J. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J. Card Fail. 2009, 15, 171–181. [Google Scholar] [CrossRef]
- Pupo, A.; Fernández, A.; Low, S.H.; François, A.; Suárez-Amarán, L.; Samulski, R.J. AAV vectors: The Rubik’s cube of human gene therapy. Mol. Ther. 2022, 30, 3515–3541. [Google Scholar] [CrossRef] [PubMed]
- Asokan, A.; Conway, J.C.; Phillips, J.L.; Li, C.; Hegge, J.; Sinnott, R.; Yadav, S.; DiPrimio, N.; Nam, H.J.; Agbandje-McKenna, M.; et al. Reengineering a receptor footprint of adeno-associated virus enables selective and systemic gene transfer to muscle. Nat. Biotechnol. 2010, 28, 79–82. [Google Scholar] [CrossRef]
- Gacita, A.M.; Fullenkamp, D.E.; Ohiri, J.; Pottinger, T.; Puckelwartz, M.J.; Nobrega, M.A.; McNally, E.M. Genetic Variation in Enhancers Modifies Cardiomyopathy Gene Expression and Progression. Circulation 2021, 143, 1302–1316. [Google Scholar] [CrossRef]
- Qiao, C.; Yuan, Z.; Li, J.; He, B.; Zheng, H.; Mayer, C.; Li, J.; Xiao, X. Liver-specific microRNA-122 target sequences incorporated in AAV vectors efficiently inhibits transgene expression in the liver. Gene Ther. 2011, 18, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Duan, D. Systemic delivery of adeno-associated viral vectors. Curr. Opin Virol. 2016, 21, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Byrne, B.J.; Corti, M.; Muntoni, F. Considerations for Systemic Use of Gene Therapy. Mol. Ther. 2021, 29, 422–423. [Google Scholar] [CrossRef]
- A Phase 1/2 Trial of the Safety and Efficacy of SRD-001 (AAV1/SERCA2a) in Subjects with Heart Failure with Reduced Ejection Fraction. ClinicalTrials.gov Identifier: NCT04703842. Available online: https://www.clinicaltrials.gov/study/NCT04703842?intr=A%20phase%201%2F2%20trial%20of%20the%20safety%20and%20efficacy%20of%20SRD-001%20(AAV1%2FSERCA2a)%20in%20subjects%20with%20heart%20failure%20with%20reduced%20ejection%20fraction&rank=1 (accessed on 7 June 2024).
- ASGCT 27th Annual Meeting Late-Breaking Abstracts. Mol. Ther. 2024, 32 (Suppl. S1), 1–17. [CrossRef]
- Mingozzi, F.; High, K.A. Overcoming the Host Immune Response to Adeno-Associated Virus Gene Delivery Vectors: The Race Between Clearance, Tolerance, Neutralization, and Escape. Annu. Rev. Virol. 2017, 4, 511–534. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Wu, D.M.; Xue, Y.; Wang, S.K.; Chung, M.J.; Ji, X.; Rana, P.; Zhao, S.R.; Mai, S.; Cepko, C.L. AAV cis-regulatory sequences are correlated with ocular toxicity. Proc. Natl. Acad. Sci. USA 2019, 116, 5785–5794. [Google Scholar] [CrossRef] [PubMed]
- Lek, A.; Atas, E.; Hesterlee, S.E.; Byrne, B.J.; Bönnemann, C.G. Meeting Report: 2022 Muscular Dystrophy Association Summit on ‘Safety and Challenges in Gene Transfer Therapy’. J. Neuromuscul. Dis. 2023, 10, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Lek, A.; Wong, B.; Keeler, A.; Blackwood, M.; Ma, K.; Huang, S.; Sylvia, K.; Batista, A.R.; Artinian, R.; Kokoski, D.; et al. Death after High-Dose rAAV9 Gene Therapy in a Patient with Duchenne’s Muscular Dystrophy. N. Engl. J. Med. 2023, 389, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Louis Jeune, V.; Joergensen, J.A.; Hajjar, R.J.; Weber, T. Pre-existing anti-adeno-associated virus antibodies as a challenge in AAV gene therapy. Human Gene Ther. Methods 2013, 24, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Philippidis, A. Novartis Confirms Deaths of Two Patients Treated with Gene Therapy Zolgensma. Human Gene Ther. 2022, 33, 842–844. [Google Scholar] [CrossRef]
- Mundisugih, J.; Kizana, E. Crossing the Threshold of Therapeutic Hope for Patients with PKP2 Arrhythmogenic Cardiomyopathy. Circ. Genom. Precis. Med. 2024, 17, e004572. [Google Scholar] [CrossRef]
- Coutinho, A.E.; Chapman, K.E. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol. Cell Endocrinol. 2011, 335, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, B.A.; Wright, J.F. Challenges Posed by Immune Responses to AAV Vectors: Addressing Root Causes. Front. Immunol. 2021, 12, 675897. [Google Scholar] [CrossRef]
- Konkle, B.A.; Walsh, C.E.; Escobar, M.A.; Josephson, N.C.; Young, G.; von Drygalski, A.; McPhee, S.W.J.; Samulski, R.J.; Bilic, I.; de la Rosa, M.; et al. BAX 335 hemophilia B gene therapy clinical trial results: Potential impact of CpG sequences on gene expression. Blood 2021, 137, 763–774. [Google Scholar] [CrossRef]
- Chu, W.S.; Ng, J. Immunomodulation in Administration of rAAV: Preclinical and Clinical Adjuvant Pharmacotherapies. Front. Immunol. 2021, 12, 658038. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Kole, T.P.; Zheng, Y.; Zarek, P.E.; Matthews, K.L.; Xiao, B.; Worley, P.F.; Kozma, S.C.; Powell, J.D. The mTOR Kinase Differentially Regulates Effector and Regulatory T Cell Lineage Commitment. Immunity 2009, 30, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R. Rituximab (monoclonal anti-CD20 antibody): Mechanisms of action and resistance. Oncogene 2003, 22, 7359–7368. [Google Scholar] [CrossRef] [PubMed]
- Corti, M.; Cleaver, B.; Clément, N.; Conlon, T.J.; Faris, K.J.; Wang, G.; Benson, J.; Tarantal, A.F.; Fuller, D.; Herzog, R.W.; et al. Evaluation of Readministration of a Recombinant Adeno-Associated Virus Vector Expressing Acid Alpha-Glucosidase in Pompe Disease: Preclinical to Clinical Planning. Human Gene Ther. Clin. Dev. 2015, 26, 185–193. [Google Scholar] [CrossRef]
- Byrne, B.J.; Fuller, D.D.; Smith, B.K.; Clement, N.; Coleman, K.; Cleaver, B.; Vaught, L.; Falk, D.J.; McCall, A.; Corti, M. Pompe disease gene therapy: Neural manifestations require consideration of CNS directed therapy. Ann. Transl. Med. 2019, 7, 290. [Google Scholar] [CrossRef]
- SADS Foundation. Gene Therapy in PKP2 ACM Clinical Trials. 2024. Available online: https://www.youtube.com/watch?v=bBPbzalXLRc (accessed on 7 June 2024).


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mundisugih, J.; Ravindran, D.; Kizana, E. Exploring the Therapeutic Potential of Gene Therapy in Arrhythmogenic Right Ventricular Cardiomyopathy. Biomedicines 2024, 12, 1351. https://doi.org/10.3390/biomedicines12061351
Mundisugih J, Ravindran D, Kizana E. Exploring the Therapeutic Potential of Gene Therapy in Arrhythmogenic Right Ventricular Cardiomyopathy. Biomedicines. 2024; 12(6):1351. https://doi.org/10.3390/biomedicines12061351
Chicago/Turabian StyleMundisugih, Juan, Dhanya Ravindran, and Eddy Kizana. 2024. "Exploring the Therapeutic Potential of Gene Therapy in Arrhythmogenic Right Ventricular Cardiomyopathy" Biomedicines 12, no. 6: 1351. https://doi.org/10.3390/biomedicines12061351
APA StyleMundisugih, J., Ravindran, D., & Kizana, E. (2024). Exploring the Therapeutic Potential of Gene Therapy in Arrhythmogenic Right Ventricular Cardiomyopathy. Biomedicines, 12(6), 1351. https://doi.org/10.3390/biomedicines12061351