

Glycosylation and Its Role in Immune Checkpoint Proteins: From Molecular Mechanisms to Clinical Implications
 ,
,
Abstract
:1. Introduction
2. The Glycosylation of PD-L1
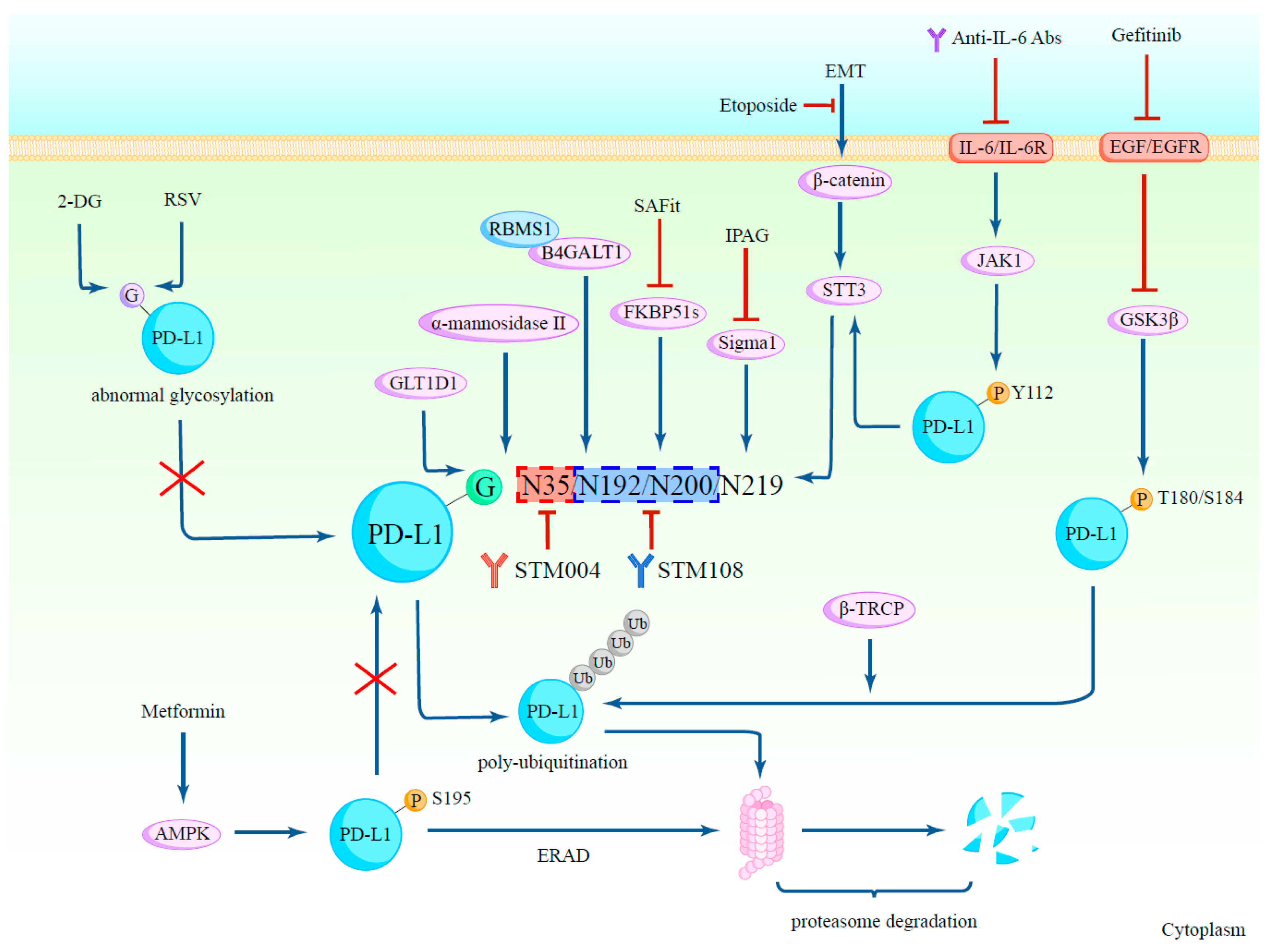
2.1. Glycosylation Regulates PD-L1 Protein Stability
2.2. The Glycosylation of PD-L1 Promotes Its Immunosuppressive Function
2.3. Relationships between PD-L1 Glycosylation and Other Post-Translational Modifications
2.4. Glycosylation in Clinical Diagnosis and PD-L1 Detection
2.5. Agents Targeting Glycosylated PD-L1
3. The Glycosylation of PD-1
3.1. Glycosylation Regulates PD-1 Protein Expression and Stability
3.2. Glycosylation-Specific Antibodies Targeting PD-1
4. Glycosylation Mediates PD-L1 and PD-1 Interaction
5. The Glycosylation of PD-L2
6. The Glycosylation of B7-H3
7. The Glycosylation of B7-H4
8. The Glycosylation of CTLA-4
9. Sialylation and Siglecs
9.1. Interaction between Siglecs and Its Sialoglycan Ligands
9.2. Agents Targeting Sialoglycan–Siglec Immune Checkpoint
10. Challenges and Future Perspectives
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, F.; Aebi, M. Mechanisms and principles of N-linked protein glycosylation. Curr. Opin. Struct. Biol. 2011, 21, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-H.; Wang, Y.-N.; Xia, W.; Chen, C.-H.; Rau, K.-M.; Ye, L.; Wei, Y.; Chou, C.-K.; Wang, S.-C.; Yan, M.; et al. Removal of N-Linked Glycosylation Enhances PD-L1 Detection and Predicts Anti-PD-1/PD-L1 Therapeutic Efficacy. Cancer Cell 2019, 36, 168–178.e4. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-W.; Lim, S.-O.; Xia, W.; Lee, H.-H.; Chan, L.-C.; Kuo, C.-W.; Khoo, K.-H.; Chang, S.-S.; Cha, J.-H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016, 7, 12632. [Google Scholar] [CrossRef] [PubMed]
- Breitling, J.; Aebi, M. N-Linked Protein Glycosylation in the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013359. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Canada, C.; Kelleher, D.J.; Gilmore, R. Cotranslational and Posttranslational N-Glycosylation of Polypeptides by Distinct Mammalian OST Isoforms. Cell 2009, 136, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.-M.; Xia, W.; Hsu, Y.-H.; Chan, L.-C.; Yu, W.-H.; Cha, J.-H.; Chen, C.-T.; Liao, H.-W.; Kuo, C.-W.; Khoo, K.-H.; et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 2018, 9, 1908. [Google Scholar] [CrossRef] [PubMed]
- D’Arrigo, P.; Russo, M.; Rea, A.; Tufano, M.; Guadagno, E.; Del Basso De Caro, M.L.; Pacelli, R.; Hausch, F.; Staibano, S.; Ilardi, G.; et al. A regulatory role for the co-chaperone FKBP51s in PD-L1 expression in glioma. Oncotarget 2017, 8, 68291–68304. [Google Scholar] [CrossRef]
- Maher, C.M.; Thomas, J.D.; Haas, D.A.; Longen, C.G.; Oyer, H.M.; Tong, J.Y.; Kim, F.J. Small-Molecule Sigma1 Modulator Induces Autophagic Degradation of PD-L1. Mol. Cancer Res. 2018, 16, 243–255. [Google Scholar] [CrossRef]
- Gaali, S.; Kirschner, A.; Cuboni, S.; Hartmann, J.; Kozany, C.; Balsevich, G.; Namendorf, C.; Fernandez-Vizarra, P.; Sippel, C.; Zannas, A.S.; et al. Selective inhibitors of the FK506-binding protein 51 by induced fit. Nat. Chem. Biol. 2015, 11, 33–37. [Google Scholar] [CrossRef]
- Hanßke, B.; Thiel, C.; Lübke, T.; Hasilik, M.; Höning, S.; Peters, V.; Heidemann, P.H.; Hoffmann, G.F.; Berger, E.G.; Von Figura, K.; et al. Deficiency of UDP-galactose:N-acetylglucosamine β-1,4-galactosyltransferase I causes the congenital disorder of glycosylation type IId. J. Clin. Investig. 2002, 109, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, G.; Zhang, W.; Bai, L.; Wang, L.; Li, T.; Yan, L.; Xu, Y.; Chen, D.; Gao, W.; et al. Loss of RBMS1 promotes anti-tumor immunity through enabling PD-L1 checkpoint blockade in triple-negative breast cancer. Cell Death Differ. 2022, 29, 2247–2261. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Li, J.; Zhang, P.; Yin, D.; Wang, Z.; Dai, J.; Wang, W.; Zhang, E.; Guo, R. B4GALT1 promotes immune escape by regulating the expression of PD-L1 at multiple levels in lung adenocarcinoma. J. Exp. Clin. Cancer Res. 2023, 42, 146. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-W.; Lim, S.-O.; Chung, E.M.; Kim, Y.-S.; Park, A.H.; Yao, J.; Cha, J.-H.; Xia, W.; Chan, L.-C.; Kim, T.; et al. Eradication of Triple-Negative Breast Cancer Cells by Targeting Glycosylated PD-L1. Cancer Cell 2018, 33, 187–201.e10. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Y.; Han, Y.; Lu, W.; Yang, J.; Tian, J.; Sun, P.; Yu, T.; Hu, Y.; Zhang, H.; et al. Overexpression of GLT1D1 induces immunosuppression through glycosylation of PD-L1 and predicts poor prognosis in B-cell lymphoma. Mol. Oncol. 2020, 14, 1028–1044. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Deng, J.; Xia, W.; Xu, J.; Li, Y.M.; Gunduz, M.; Hung, M.-C. Dual regulation of Snail by GSK-3β-mediated phosphorylation in control of epithelial–mesenchymal transition. Nat. Cell Biol. 2004, 6, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.-H.; Yang, W.-H.; Xia, W.; Wei, Y.; Chan, L.-C.; Lim, S.-O.; Li, C.-W.; Kim, T.; Chang, S.-S.; Lee, H.-H.; et al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol. Cell 2018, 71, 606–620.e7. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.-C.; Li, C.-W.; Xia, W.; Hsu, J.-M.; Lee, H.-H.; Cha, J.-H.; Wang, H.-L.; Yang, W.-H.; Yen, E.-Y.; Chang, W.-C.; et al. IL-6/JAK1 pathway drives PD-L1 Y112 phosphorylation to promote cancer immune evasion. J. Clin. Investig. 2019, 129, 3324–3338. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Marianna Zahurak, M.S.; Stephen, C.; Yang, M.D.; David, R.; et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N. Engl. J. Med. 2018, 379, e14. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Lichinitser, M.; Khattak, A.; Carlino, M.S.; et al. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N. Engl. J. Med. 2018, 378, 1789–1801. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef] [PubMed]
- Nishino, M.; Ramaiya, N.H.; Hatabu, H.; Hodi, F.S. Monitoring immune-checkpoint blockade: Response evaluation and biomarker development. Nat. Rev. Clin. Oncol. 2017, 14, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Yang, X.; Liu, D.; Ye, S.; Yang, W.; Xie, Z.; Lei, X. Research progress of PD-L1 non-glycosylation in cancer immunotherapy. Scand. J. Immunol. 2022, 96, e13205. [Google Scholar] [CrossRef]
- Morales-Betanzos, C.A.; Lee, H.; Gonzalez Ericsson, P.I.; Balko, J.M.; Johnson, D.B.; Zimmerman, L.J.; Liebler, D.C. Quantitative Mass Spectrometry Analysis of PD-L1 Protein Expression, N-glycosylation and Expression Stoichiometry with PD-1 and PD-L2 in Human Melanoma. Mol. Cell. Proteom. 2017, 16, 1705–1717. [Google Scholar] [CrossRef]
- Boussiotis, V.A. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N. Engl. J. Med. 2016, 375, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Hsu, J.-M.; Yang, W.-H.; Hung, M.-C. Mechanisms regulating PD-L1 expression in cancers and associated opportunities for novel small-molecule therapeutics. Nat. Rev. Clin. Oncol. 2022, 19, 287–305. [Google Scholar] [CrossRef] [PubMed]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef]
- Chen, D.S.; Irving, B.A.; Hodi, F.S. Molecular Pathways: Next-Generation Immunotherapy—Inhibiting Programmed Death-Ligand 1 and Programmed Death-1. Clin. Cancer Res. 2012, 18, 6580–6587. [Google Scholar] [CrossRef]
- Dahan, R.; Sega, E.; Engelhardt, J.; Selby, M.; Korman, A.J.; Ravetch, J.V. FcγRs Modulate the Anti-tumor Activity of Antibodies Targeting the PD-1/PD-L1 Axis. Cancer Cell 2015, 28, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Jefferis, R.; Lund, J.; Pound, J.D. IgG-Fc-mediated effector functions: Molecular definition of interaction sites for effector ligands and the role of glycosylation. Immunol. Rev. 1998, 163, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Masuda, K.; Kubota, T.; Kaneko, E.; Iida, S.; Wakitani, M.; Kobayashi-Natsume, Y.; Kubota, A.; Shitara, K.; Nakamura, K. Enhanced binding affinity for FcγRIIIa of fucose-negative antibody is sufficient to induce maximal antibody-dependent cellular cytotoxicity. Mol. Immunol. 2007, 44, 3122–3131. [Google Scholar] [CrossRef] [PubMed]
- Goletz, C.; Lischke, T.; Harnack, U.; Schiele, P.; Danielczyk, A.; Rühmann, J.; Goletz, S. Glyco-Engineered Anti-Human Programmed Death-Ligand 1 Antibody Mediates Stronger CD8 T Cell Activation Than Its Normal Glycosylated and Non-Glycosylated Counterparts. Front. Immunol. 2018, 9, 1614. [Google Scholar] [CrossRef]
- Shao, B.; Li, C.-W.; Lim, S.-O.; Sun, L.; Lai, Y.-J.; Hou, J.; Liu, C.; Qiu, Y.; Hsu, J.-M.; Chan, L.-C.; et al. Deglycosylation of PD-L1 by 2-deoxyglucose reverses PARP inhibitor-induced immunosuppression in triple-negative breast cancer. Am. J. Cancer Res. 2018, 8, 1837. [Google Scholar] [PubMed]
- Kim, B.; Sun, R.; Oh, W.; Kim, A.M.J.; Schwarz, J.R.; Lim, S. Saccharide analog, 2-deoxy- D -glucose enhances 4-1BB-mediated antitumor immunity via PD-L1 deglycosylation. Mol. Carcinog. 2020, 59, 691–700. [Google Scholar] [CrossRef]
- Verdura, S.; Cuyàs, E.; Cortada, E.; Brunet, J.; Lopez-Bonet, E.; Martin-Castillo, B.; Bosch-Barrera, J.; Encinar, J.A.; Menendez, J.A. Resveratrol targets PD-L1 glycosylation and dimerization to enhance antitumor T-cell immunity. Aging 2020, 12, 8–34. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Zhang, H.; Chai, Y.; Song, H.; Tong, Z.; Wang, Q.; Qi, J.; Wong, G.; Zhu, X.; Liu, W.J.; et al. An unexpected N-terminal loop in PD-1 dominates binding by nivolumab. Nat. Commun. 2017, 8, 14369. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Zhang, C.W.-H.; Gao, G.F. Seeing is believing: Anti-PD-1/PD-L1 monoclonal antibodies in action for checkpoint blockade tumor immunotherapy. Sig Transduct. Target. Ther. 2016, 1, 16029. [Google Scholar] [CrossRef]
- Xi, X.; Zhao, W. Anti-Tumor Potential of Post-Translational Modifications of PD-1. Curr. Issues Mol. Biol. 2024, 46, 2119–2132. [Google Scholar] [CrossRef]
- Liu, K.; Tan, S.; Jin, W.; Guan, J.; Wang, Q.; Sun, H.; Qi, J.; Yan, J.; Chai, Y.; Wang, Z.; et al. N-glycosylation of PD-1 promotes binding of camrelizumab. EMBO Rep. 2020, 21, e51444. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, E.; Moriwaki, K.; Nakagawa, T. Biological Function of Fucosylation in Cancer Biology. J. Biochem. 2007, 143, 725–729. [Google Scholar] [CrossRef]
- Fujita, K.; Hatano, K.; Hashimoto, M.; Tomiyama, E.; Miyoshi, E.; Nonomura, N.; Uemura, H. Fucosylation in Urological Cancers. Int. J. Mol. Sci. 2021, 22, 13333. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Chikuma, S.; Kondo, T.; Hibino, S.; Machiyama, H.; Yokosuka, T.; Nakano, M.; Yoshimura, A. Blockage of Core Fucosylation Reduces Cell-Surface Expression of PD-1 and Promotes Anti-tumor Immune Responses of T Cells. Cell Rep. 2017, 20, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Bastian, K.; Scott, E.; Elliott, D.J.; Munkley, J. FUT8 Alpha-(1,6)-Fucosyltransferase in Cancer. Int. J. Mol. Sci. 2021, 22, 455. [Google Scholar] [CrossRef]
- Zhang, N.; Li, M.; Xu, X.; Zhang, Y.; Liu, Y.; Zhao, M.; Li, P.; Chen, J.; Fukuda, T.; Gu, J.; et al. Loss of core fucosylation enhances the anticancer activity of cytotoxic T lymphocytes by increasing PD-1 degradation. Eur. J. Immunol. 2020, 50, 1820–1833. [Google Scholar] [CrossRef]
- Meng, X.; Liu, X.; Guo, X.; Jiang, S.; Chen, T.; Hu, Z.; Liu, H.; Bai, Y.; Xue, M.; Hu, R.; et al. FBXO38 mediates PD-1 ubiquitination and regulates anti-tumour immunity of T cells. Nature 2018, 564, 130–135. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Sun, L.; Li, C.-W.; Chung, E.M.; Yang, R.; Kim, Y.-S.; Park, A.H.; Lai, Y.-J.; Yang, Y.; Wang, Y.-H.; Liu, J.; et al. Targeting Glycosylated PD-1 Induces Potent Antitumor Immunity. Cancer Res. 2020, 80, 2298–2310. [Google Scholar] [CrossRef]
- Na, Z.; Yeo, S.P.; Bharath, S.R.; Bowler, M.W.; Balıkçı, E.; Wang, C.-I.; Song, H. Structural basis for blocking PD-1-mediated immune suppression by therapeutic antibody pembrolizumab. Cell Res. 2017, 27, 147–150. [Google Scholar] [CrossRef]
- Markham, A.; Keam, S.J. Camrelizumab: First Global Approval. Drugs 2019, 79, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Gruber, D.R.; Zeng, W.; Thurman, B.; O’Connor, B.P.; Gardai, S.J.; Smith, A.J. Abstract 6361: A preclinical model of acquired anti-PD-1 resistance is responsive to SEA-TGT, an effector-function enhanced anti-TIGIT monoclonal antibody. Cancer Res. 2023, 83, 6361. [Google Scholar] [CrossRef]
- Yeh, J.-C.; Hiraoka, N.; Petryniak, B.; Nakayama, J.; Ellies, L.G.; Rabuka, D.; Hindsgaul, O.; Marth, J.D.; Lowe, J.B.; Fukuda, M. Novel Sulfated Lymphocyte Homing Receptors and Their Control by a Core1 Extension. Cell 2001, 105, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Chui, D.; Oh-Eda, M.; Liao, Y.-F.; Panneerselvam, K.; Lal, A.; Marek, K.W.; Freeze, H.H.; Moremen, K.W.; Fukuda, M.N.; Marth, J.D. Alpha-Mannosidase-II Deficiency Results in Dyserythropoiesis and Unveils an Alternate Pathway in Oligosaccharide Biosynthesis. Cell 1997, 90, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Gu, S.; Han, T.; Zhang, W.; Huang, L.; Li, Z.; Pan, D.; Fu, J.; Ge, J.; Brown, M.; et al. Inhibition of MAN2A1 Enhances the Immune Response to Anti–PD-L1 in Human Tumors. Clin. Cancer Res. 2020, 26, 5990–6002. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Gao, Z.; Hu, R.; Wang, Y.; Wang, Y.; Su, Z.; Zhang, X.; Yang, J.; Mei, M.; Ren, Y.; et al. PD-L2 glycosylation promotes immune evasion and predicts anti-EGFR efficacy. J. Immunother. Cancer 2021, 9, e002699. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yao, H.; Li, C.; Liang, L.; Zhang, Y.; Shi, H.; Zhou, C.; Chen, Y.; Fang, J.-Y.; Xu, J. PD-L2 expression in colorectal cancer: Independent prognostic effect and targetability by deglycosylation. OncoImmunology 2017, 6, e1327494. [Google Scholar] [CrossRef] [PubMed]
- Kontos, F.; Michelakos, T.; Kurokawa, T.; Sadagopan, A.; Schwab, J.H.; Ferrone, C.R.; Ferrone, S. B7-H3: An Attractive Target for Antibody-based Immunotherapy. Clin. Cancer Res. 2021, 27, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Picarda, E.; Ohaegbulam, K.C.; Zang, X. Molecular Pathways: Targeting B7-H3 (CD276) for Human Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 3425–3431. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, H.-L.; Li, Z.-L.; Du, T.; Chen, Y.-H.; Wang, Y.; Ni, H.-H.; Zhang, K.-M.; Mai, J.; Hu, B.-X.; et al. FUT8-mediated aberrant N-glycosylation of B7H3 suppresses the immune response in triple-negative breast cancer. Nat. Commun. 2021, 12, 2672. [Google Scholar] [CrossRef]
- Chen, J.-T.; Chen, C.-H.; Ku, K.-L.; Hsiao, M.; Chiang, C.-P.; Hsu, T.-L.; Chen, M.-H.; Wong, C.-H. Glycoprotein B7-H3 overexpression and aberrant glycosylation in oral cancer and immune response. Proc. Natl. Acad. Sci. USA 2015, 112, 13057–13062. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, M.; Kaur, J.; Gunjan Kathpalia, M.; Kaur, N. Clinical relevance of glycosylation in triple negative breast cancer: A review. Glycoconj. J. 2024, 41, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Prasad DV, R.; Richards, S.; Mai, X.M.; Dong, C. B7S1, a Novel B7 Family Member that Negatively Regulates T Cell Activation. Immunity 2003, 18, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Zang, X.; Loke, P.; Kim, J.; Murphy, K.; Waitz, R.; Allison, J.P. B7x: A widely expressed B7 family member that inhibits T cell activation. Proc. Natl. Acad. Sci. USA 2003, 100, 10388–10392. [Google Scholar] [CrossRef] [PubMed]
- Sica, G.L.; Choi, I.-H.; Zhu, G.; Tamada, K.; Wang, S.-D.; Tamura, H.; Chapoval, A.I.; Flies, D.B.; Bajorath, J.; Chen, L. B7-H4, a Molecule of the B7 Family, Negatively Regulates T Cell Immunity. Immunity 2003, 18, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Podojil, J.R.; Miller, S.D. Potential targeting of B7-H4 for the treatment of cancer. Immunol. Rev. 2017, 276, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zhou, Z.; Li, H.; Xue, Y.; Lu, X.; Bahar, I.; Kepp, O.; Hung, M.-C.; Kroemer, G.; Wan, Y. Pharmacologic Suppression of B7-H4 Glycosylation Restores Antitumor Immunity in Immune-Cold Breast Cancers. Cancer Discov. 2020, 10, 1872–1893. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Y.; Jiang, X.; Xu, M.; Wang, M.; Wang, R.; Zheng, B.; Chen, M.; Ke, Q.; Long, J. Unleashing the power of immune checkpoints: Post-translational modification of novel molecules and clinical applications. Cancer Lett. 2024, 588, 216758. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.-C.D.; Zhang, X.; Fedorov, A.A.; Nathenson, S.G.; Almo, S.C. Structural basis for co-stimulation by the human CTLA-4/B7-2 complex. Nature 2001, 410, 604–608. [Google Scholar] [CrossRef]
- Linsley, P.S.; Bradshaw, J.; Greene, J.; Peach, R.; Bennett, K.L.; Mittler, R.S. Intracellular Trafficking of CTLA-4 and Focal Localization Towards Sites of TCR Engagement. Immunity 1996, 4, 535–543. [Google Scholar] [CrossRef]
- Grigorian, A.; Mkhikian, H.; Demetriou, M. Interleukin-2, Interleukin-7, T cell-mediated autoimmunity, and N-glycosylation. Ann. N. Y. Acad. Sci. 2012, 1253, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Anjos, S.; Nguyen, A.; Ounissi-Benkalha, H.; Tessier, M.-C.; Polychronakos, C. A Common Autoimmunity Predisposing Signal Peptide Variant of the Cytotoxic T-lymphocyte Antigen 4 Results in Inefficient Glycosylation of the Susceptibility Allele. J. Biol. Chem. 2002, 277, 46478–46486. [Google Scholar] [CrossRef] [PubMed]
- Mkhikian, H.; Grigorian, A.; Li, C.F.; Chen, H.-L.; Newton, B.; Zhou, R.W.; Beeton, C.; Torossian, S.; Tatarian, G.G.; Lee, S.-U.; et al. Genetics and the environment converge to dysregulate N-glycosylation in multiple sclerosis. Nat. Commun. 2011, 2, 334. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.S.; Partridge, E.A.; Grigorian, A.; Silvescu, C.I.; Reinhold, V.N.; Demetriou, M.; Dennis, J.W. Complex N-Glycan Number and Degree of Branching Cooperate to Regulate Cell Proliferation and Differentiation. Cell 2007, 129, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Bongers, J.; Devincentis, J.; Fu, J.; Huang, P.; Kirkley, D.H.; Leister, K.; Liu, P.; Ludwig, R.; Rumney, K.; Tao, L.; et al. Characterization of glycosylation sites for a recombinant IgG1 monoclonal antibody and a CTLA4-Ig fusion protein by liquid chromatography–mass spectrometry peptide mapping. J. Chromatogr. A 2011, 1218, 8140–8149. [Google Scholar] [CrossRef] [PubMed]
- Crocker, P.R.; Paulson, J.C.; Varki, A. Siglecs and their roles in the immune system. Nat. Rev. Immunol. 2007, 7, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Macauley, M.S.; Crocker, P.R.; Paulson, J.C. Siglec-mediated regulation of immune cell function in disease. Nat. Rev. Immunol. 2014, 14, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Angata, T. Possible Influences of Endogenous and Exogenous Ligands on the Evolution of Human Siglecs. Front. Immunol. 2018, 9, 2885. [Google Scholar] [CrossRef] [PubMed]
- Bochner, B.S.; Zimmermann, N. Role of siglecs and related glycan-binding proteins in immune responses and immunoregulation. J. Allergy Clin. Immunol. 2015, 135, 598–608. [Google Scholar] [CrossRef]
- Pearce, O.M.T.; Läubli, H. Sialic acids in cancer biology and immunity. Glycobiology 2016, 26, 111–128. [Google Scholar] [CrossRef]
- Rodriguez, E.; Boelaars, K.; Brown, K.; Eveline Li, R.J.; Kruijssen, L.; Bruijns, S.C.M.; Van Ee, T.; Schetters, S.T.T.; Crommentuijn, M.H.W.; Van Der Horst, J.C.; et al. Sialic acids in pancreatic cancer cells drive tumour-associated macrophage differentiation via the Siglec receptors Siglec-7 and Siglec-9. Nat. Commun. 2021, 12, 1270. [Google Scholar] [CrossRef] [PubMed]
- Jandus, C.; Boligan, K.F.; Chijioke, O.; Liu, H.; Dahlhaus, M.; Démoulins, T.; Schneider, C.; Wehrli, M.; Hunger, R.E.; Baerlocher, G.M.; et al. Interactions between Siglec-7/9 receptors and ligands influence NK cell–dependent tumor immunosurveillance. J. Clin. Investig. 2014, 124, 1810–1820. [Google Scholar] [CrossRef] [PubMed]
- Stanczak, M.A.; Siddiqui, S.S.; Trefny, M.P.; Thommen, D.S.; Boligan, K.F.; Von Gunten, S.; Tzankov, A.; Tietze, L.; Lardinois, D.; Heinzelmann-Schwarz, V.; et al. Self-associated molecular patterns mediate cancer immune evasion by engaging Siglecs on T cells. J. Clin. Investig. 2018, 128, 4912–4923. [Google Scholar] [CrossRef] [PubMed]
- Haas, Q.; Boligan, K.F.; Jandus, C.; Schneider, C.; Simillion, C.; Stanczak, M.A.; Haubitz, M.; Seyed Jafari, S.M.; Zippelius, A.; Baerlocher, G.M.; et al. Siglec-9 Regulates an Effector Memory CD8+ T-cell Subset That Congregates in the Melanoma Tumor Microenvironment. Cancer Immunol. Res. 2019, 7, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Beatson, R.; Tajadura-Ortega, V.; Achkova, D.; Picco, G.; Tsourouktsoglou, T.-D.; Klausing, S.; Hillier, M.; Maher, J.; Noll, T.; Crocker, P.R.; et al. The mucin MUC1 modulates the tumor immunological microenvironment through engagement of the lectin Siglec-9. Nat. Immunol. 2016, 17, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Barkal, A.A.; Brewer, R.E.; Markovic, M.; Kowarsky, M.; Barkal, S.A.; Zaro, B.W.; Krishnan, V.; Hatakeyama, J.; Dorigo, O.; Barkal, L.J.; et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019, 572, 392–396. [Google Scholar] [CrossRef]
- O’Reilly, M.K.; Paulson, J.C. Siglecs as targets for therapy in immune-cell-mediated disease. Trends Pharmacol. Sci. 2009, 30, 240–248. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef]
- Spiegel, J.Y.; Patel, S.; Muffly, L.; Hossain, N.M.; Oak, J.; Baird, J.H.; Frank, M.J.; Shiraz, P.; Sahaf, B.; Craig, J.; et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: A phase 1 trial. Nat. Med. 2021, 27, 1419–1431. [Google Scholar] [CrossRef]
- Castaigne, S.; Pautas, C.; Terré, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.-N.; Legrand, O.; Thomas, X.; Turlure, P.; Reman, O.; et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet 2012, 379, 1508–1516. [Google Scholar] [CrossRef]
- Büll, C.; Boltje, T.J.; Balneger, N.; Weischer, S.M.; Wassink, M.; Van Gemst, J.J.; Bloemendal, V.R.; Boon, L.; Van Der Vlag, J.; Heise, T.; et al. Sialic Acid Blockade Suppresses Tumor Growth by Enhancing T-cell–Mediated Tumor Immunity. Cancer Res. 2018, 78, 3574–3588. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Woods, E.C.; Vukojicic, P.; Bertozzi, C.R. Precision glycocalyx editing as a strategy for cancer immunotherapy. Proc. Natl. Acad. Sci. USA 2016, 113, 10304–10309. [Google Scholar] [CrossRef] [PubMed]
- Ibarlucea-Benitez, I.; Weitzenfeld, P.; Smith, P.; Ravetch, J.V. Siglecs-7/9 function as inhibitory immune checkpoints in vivo and can be targeted to enhance therapeutic antitumor immunity. Proc. Natl. Acad. Sci. USA 2021, 118, e2107424118. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, J.; Liu, L.N.; Flies, D.B.; Nie, X.; Toki, M.; Zhang, J.; Song, C.; Zarr, M.; Zhou, X.; et al. Siglec-15 as an immune suppressor and potential target for normalization cancer immunotherapy. Nat. Med. 2019, 25, 656–666. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Xu, Z.; Zhang, D.; Jiang, M.; Liu, K.; He, J.; Ma, D.; Ma, X.; Tan, S.; Gao, G.F.; et al. PD-1 N58-Glycosylation-Dependent Binding of Monoclonal Antibody Cemiplimab for Immune Checkpoint Therapy. Front. Immunol. 2022, 13, 826045. [Google Scholar] [CrossRef]
- Zheng, L.; Yang, Q.; Li, F.; Zhu, M.; Yang, H.; Tan, T.; Wu, B.; Liu, M.; Xu, C.; Yin, J.; et al. The Glycosylation of Immune Checkpoints and Their Applications in Oncology. Pharmaceuticals 2022, 15, 1451. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Immune Checkpoints | Glycosylation Sites | Related Enzymes | Biological Effects | Potential Clinical Applications |
|---|---|---|---|---|
| PD-L1 | N35, N192, N200, N219 | STT3 Sigma1 FKBP51s B4GALT1 GLT1D1 B3GNT3 GSK3β | protect PD-L1 from degradation and enhance its protein stability facilitate binding with PD-1 and mediate immunosuppressive function | biomarker for clinical diagnosis and PD-L1 detection small molecular agents targeting PD-L1 glycosylation can improve antitumor immunity and can be used as combination therapy |
| PD-1 | N49, N58, N74, N116 | B3GNT2 FUT8 | upregulate PD-1 expression and protein stability | the fucosylation inhibitor can increase T-cell activation and inhibit PD-1 expression glycosylation-specific antibodies can inhibit PD-1/PD-L1 interaction and increase treatment efficacy |
| PD-L2 | N64, N157, N163, N189 | FUT8 | stabilize PD-L2 and mediate PD-1/PD-L2 interaction | have the potential to become a novel therapeutic target |
| B7-H3 | N91, N309, N104, N322, N189, N407, N215, N433 | FUT8 | stabilize B7-H3 and mediate immunosuppressive function | use fucosylation inhibitors to restore cancer cells’ sensitivity to immune response and functions as a combination agent with immunotherapy |
| B7-H4 | N112, N140, N156, N160, N255 | UGGG1 RPN1/2 STT3A | stabilize the structure of the B7-H4 protein inhibit the immunogenicity of cancer cells | an OST inhibitor can be used as a triple combination with camsirubicin and the PD-L1 blockade to suppress tumor growth in TNBC |
| CTLA-4 | N113, N145 | Mgat1 | inhibit CTLA-4 internalization and surface expression | is related to autoimmune diseases and its treatment |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Xu, X.; Zhong, H.; Yu, M.; Abuduaini, N.; Zhang, S.; Yang, X.; Feng, B. Glycosylation and Its Role in Immune Checkpoint Proteins: From Molecular Mechanisms to Clinical Implications. Biomedicines 2024, 12, 1446. https://doi.org/10.3390/biomedicines12071446
Liu J, Xu X, Zhong H, Yu M, Abuduaini N, Zhang S, Yang X, Feng B. Glycosylation and Its Role in Immune Checkpoint Proteins: From Molecular Mechanisms to Clinical Implications. Biomedicines. 2024; 12(7):1446. https://doi.org/10.3390/biomedicines12071446
Chicago/Turabian StyleLiu, Jingyi, Ximo Xu, Hao Zhong, Mengqin Yu, Naijipu Abuduaini, Sen Zhang, Xiao Yang, and Bo Feng. 2024. "Glycosylation and Its Role in Immune Checkpoint Proteins: From Molecular Mechanisms to Clinical Implications" Biomedicines 12, no. 7: 1446. https://doi.org/10.3390/biomedicines12071446
APA StyleLiu, J., Xu, X., Zhong, H., Yu, M., Abuduaini, N., Zhang, S., Yang, X., & Feng, B. (2024). Glycosylation and Its Role in Immune Checkpoint Proteins: From Molecular Mechanisms to Clinical Implications. Biomedicines, 12(7), 1446. https://doi.org/10.3390/biomedicines12071446