
The Role of Gut Microbiota in Thromboangiitis Obliterans: Cohort and Mendelian Randomization Study

Abstract
:1. Introduction
2. Materials and Methods
2.1. Assumptions of MR
2.2. Selection of Instrumental Variables
2.3. Data Sources of Outcomes
2.4. Case–Control Study
2.5. MR Analysis and Statistical Analysis
3. Results
3.1. Genetic Associations between Gut Microbiota and TAO
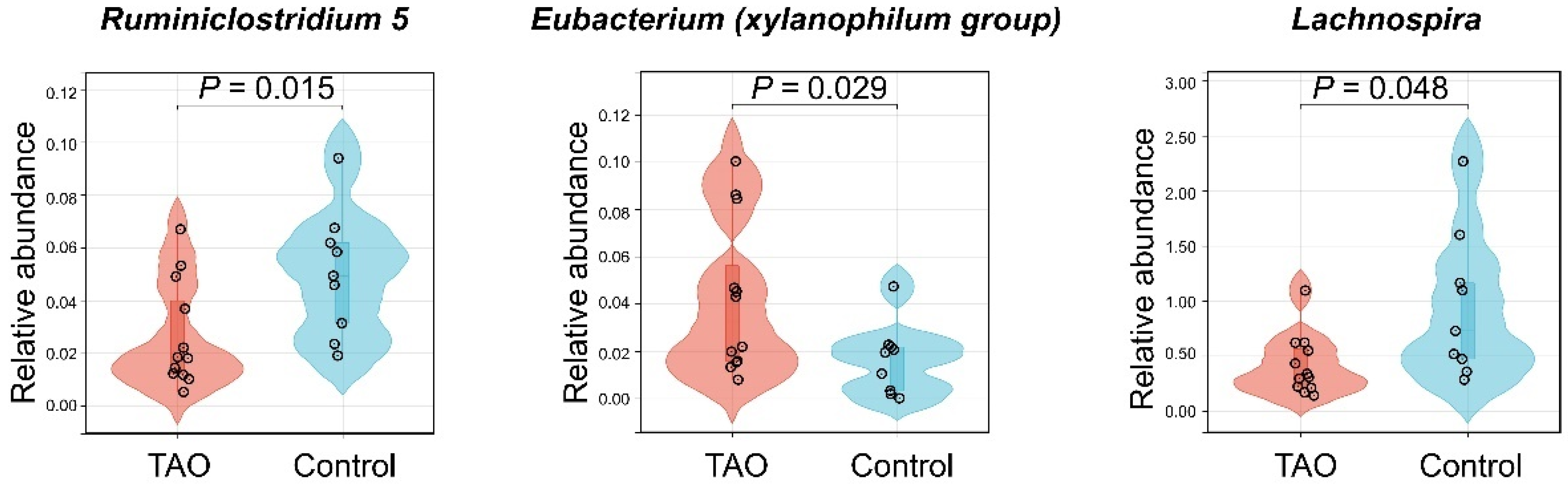
3.2. Case–Control Study
4. Discussion
4.1. Principal Findings
4.2. Comparison with Other Studies and Possible Mechanisms
4.3. Strengths and Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TAO | Thromboangiitis obliterans |
| MR | Mendelian randomization |
| GWAS | Genome-wide association study |
| SNPs | Single nucleotide polymorphisms |
| IVs | Instrumental variables |
| IVW | Inverse-variance weighted |
| ML | Maximum likelihood |
| LPS | Lipopolysaccharide |
| mbQTL | Microbiota quantitative trait loci |
| LD | Linkage disequilibrium |
| ICD-10 | International Classification of Diseases 10th version |
| InSIDE | Instrument strength independent of direct effect |
| OR | Odds ratio |
| CI | Confidence interval |
| PCoA | Principal coordinates analysis |
| MAMPs | Microbiota-associated molecular patterns |
| TLR4 | Toll-like receptor 4 |
| EA | Metabolite ethanolamine |
| SCFAs | Short-chain-fatty acids |
References
- Piazza, G.; Creager, M.A. Thromboangiitis obliterans. Circulation 2010, 121, 1858–1861. [Google Scholar] [CrossRef] [PubMed]
- Li, M.D.; Wang, Y.F.; Yang, M.W.; Hong, F.F.; Yang, S.L. Risk Factors, Mechanisms and Treatments of Thromboangiitis Obliterans: An Overview of Recent Research. Curr. Med. Chem. 2020, 27, 6057–6072. [Google Scholar] [CrossRef] [PubMed]
- Shepard, Z.; Skorupa, T.; Espinoza, L.; Erlandson, K.; Damioli, L. Coxiella burnetii Infection Associated With Thromboangiitis Obliterans-like Phenomena With Digital Autoamputation: A Case Report and Review of Q Fever-Associated Autoimmunity. Open Forum Infect. Dis. 2022, 9, ofab637. [Google Scholar] [CrossRef] [PubMed]
- Galyfos, G.; Liakopoulos, D.; Chamzin, A.; Sigala, F.; Filis, K. A systematic review and meta-analysis of early and late outcomes after endovascular angioplasty among patients with thromboangiitis obliterans and chronic limb ischemia. J. Vasc. Surg. 2022, 77, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Cacione, D.G.; Macedo, C.R.; do Carmo Novaes, F.; Baptista-Silva, J.C. Pharmacological treatment for Buerger’s disease. Cochrane Database Syst. Rev. 2020, 5, CD011033. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J. Gut microbial metabolites in health and disease. Gut Microbes 2016, 7, 187–188. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Kitai, T.; Hazen, S.L. Gut Microbiota in Cardiovascular Health and Disease. Circ. Res. 2017, 120, 1183–1196. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Xu, J.; Deng, X.; Zhou, S. A Mendelian Randomization Study: Roles of Gut Microbiota in Sepsis—Who is the Angle? Pol. J. Microbiol. 2024, 73, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Verhaar, B.J.H.; Prodan, A.; Nieuwdorp, M.; Muller, M. Gut Microbiota in Hypertension and Atherosclerosis: A Review. Nutrients 2020, 12, 2982. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.L.; Shen, B.; Yuan, Y.; Liu, C.; Xie, Q.W.; Hu, T.T.; Yao, Q.; Wu, Q.Q.; Tang, Q.Z. The effect of HMGA1 in LPS-induced Myocardial Inflammation. Int. J. Biol. Sci. 2020, 16, 1798–1810. [Google Scholar] [CrossRef] [PubMed]
- Cook, M.D.; Hogue, T. Exercise and the Microbiome: Mechanistic Perspectives of the Impact of Exercise on the Gut-Vascular Axis. mSystems 2021, 6, e0065021. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Li, H.; Zhou, H.; Zhang, X.; Zhang, A.; Xie, Y.; Li, Y.; Lv, S.; Zhang, J. Role and Effective Therapeutic Target of Gut Microbiota in Heart Failure. Cardiovasc. Ther. 2019, 2019, 5164298. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Zhai, W.; Yang, C.; Li, Z.; Mao, L.; Zhao, M.; Wu, X. The Relationship among Physical Activity, Intestinal Flora, and Cardiovascular Disease. Cardiovasc. Ther. 2021, 2021, 3364418. [Google Scholar] [CrossRef] [PubMed]
- Iwai, T.; Inoue, Y.; Umeda, M.; Huang, Y.; Kurihara, N.; Koike, M.; Ishikawa, I. Oral bacteria in the occluded arteries of patients with Buerger disease. J. Vasc. Surg. 2005, 42, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Ling, Z.; Liu, X.; Cheng, Y.; Yan, X.; Wu, S. Gut microbiota and aging. Crit. Rev. Food Sci. Nutr. 2022, 62, 3509–3534. [Google Scholar] [CrossRef] [PubMed]
- Schoeler, M.; Caesar, R. Dietary lipids, gut microbiota and lipid metabolism. Rev. Endocr. Metab. Disord. 2019, 20, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Zmora, N.; Suez, J.; Elinav, E. You are what you eat: Diet, health and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 35–56. [Google Scholar] [CrossRef] [PubMed]
- Kurilshikov, A.; Medina-Gomez, C.; Bacigalupe, R.; Radjabzadeh, D.; Wang, J.; Demirkan, A.; Le Roy, C.I.; Raygoza Garay, J.A.; Finnicum, C.T.; Liu, X.; et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat. Genet. 2021, 53, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Davenport, E.R.; Beaumont, M.; Jackson, M.A.; Knight, R.; Ober, C.; Spector, T.D.; Bell, J.T.; Clark, A.G.; Ley, R.E. Genetic Determinants of the Gut Microbiome in UK Twins. Cell Host Microbe 2016, 19, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human genetics shape the gut microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Greenland, S. An introduction to instrumental variables for epidemiologists. Int. J. Epidemiol. 2000, 29, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Sanna, S.; van Zuydam, N.R.; Mahajan, A.; Kurilshikov, A.; Vich Vila, A.; Võsa, U.; Mujagic, Z.; Masclee, A.A.M.; Jonkers, D.; Oosting, M.; et al. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat. Genet. 2019, 51, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Ni, J.J.; Han, B.X.; Yan, S.S.; Wei, X.T.; Feng, G.J.; Zhang, H.; Zhang, L.; Li, B.; Pei, Y.F. Causal Relationship Between Gut Microbiota and Autoimmune Diseases: A Two-Sample Mendelian Randomization Study. Front. Immunol. 2021, 12, 746998. [Google Scholar] [CrossRef] [PubMed]
- Inamo, J. Non-causal association of gut microbiome on the risk of rheumatoid arthritis: A Mendelian randomisation study. Ann. Rheum. Dis. 2021, 80, e103. [Google Scholar] [CrossRef] [PubMed]
- Skrivankova, V.W.; Richmond, R.C.; Woolf, B.A.R.; Yarmolinsky, J.; Davies, N.M.; Swanson, S.A.; VanderWeele, T.J.; Higgins, J.P.T.; Timpson, N.J.; Dimou, N.; et al. Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization: The STROBE-MR Statement. JAMA 2021, 326, 1614–1621. [Google Scholar] [CrossRef] [PubMed]
- Sekula, P.; Del Greco, M.F.; Pattaro, C.; Köttgen, A. Mendelian Randomization as an Approach to Assess Causality Using Observational Data. J. Am. Soc. Nephrol. 2016, 27, 3253–3265. [Google Scholar] [CrossRef] [PubMed]
- Walker, V.M.; Davies, N.M.; Hemani, G.; Zheng, J.; Haycock, P.C.; Gaunt, T.R.; Davey Smith, G.; Martin, R.M. Using the MR-Base platform to investigate risk factors and drug targets for thousands of phenotypes. Wellcome Open Res. 2019, 4, 113. [Google Scholar] [CrossRef] [PubMed]
- Stock, J.; Staiger, D. Instrumental Variables Regression with Weak Instruments. Econometrica 1997, 65, 557–586. [Google Scholar]
- Staley, J.R.; Blackshaw, J.; Kamat, M.A.; Ellis, S.; Surendran, P.; Sun, B.B.; Paul, D.S.; Freitag, D.; Burgess, S.; Danesh, J.; et al. PhenoScanner: A database of human genotype-phenotype associations. Bioinformatics 2016, 32, 3207–3209. [Google Scholar] [CrossRef] [PubMed]
- Kurki, M.I.; Karjalainen, J.; Palta, P.; Sipilä, T.P.; Kristiansson, K.; Donner, K.; Reeve, M.P.; Laivuori, H.; Aavikko, M.; Kaunisto, M.A.; et al. FinnGen: Unique genetic insights from combining isolated population and national health register data. medRxiv 2022. [Google Scholar] [CrossRef]
- Fairley, S.; Lowy-Gallego, E.; Perry, E.; Flicek, P. The International Genome Sample Resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Res. 2020, 48, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Qiu, P.; Ishimoto, T.; Fu, L.; Zhang, J.; Zhang, Z.; Liu, Y. The Gut Microbiota in Inflammatory Bowel Disease. Front. Cell Infect. Microbiol. 2022, 12, 733992. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.; Davey Smith, G.; Haycock, P.C.; Burgess, S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet. Epidemiol. 2016, 40, 304–314. [Google Scholar] [CrossRef]
- Pierce, B.L.; Burgess, S. Efficient design for Mendelian randomization studies: Subsample and 2-sample instrumental variable estimators. Am. J. Epidemiol. 2013, 178, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, F.P.; Davey Smith, G.; Bowden, J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int. J. Epidemiol. 2017, 46, 1985–1998. [Google Scholar] [CrossRef] [PubMed]
- Verbanck, M.; Chen, C.Y.; Neale, B.; Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 2018, 50, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S. Sample size and power calculations in Mendelian randomization with a single instrumental variable and a binary outcome. Int. J. Epidemiol. 2014, 43, 922–929. [Google Scholar] [CrossRef] [PubMed]
- Hemani, G.; Tilling, K.; Davey Smith, G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 2017, 13, e1007081. [Google Scholar] [CrossRef]
- Liu, B.; Zhou, Z.; Jin, Y.; Lu, J.; Feng, D.; Peng, R.; Sun, H.; Mu, X.; Li, C.; Chen, Y. Hepatic stellate cell activation and senescence induced by intrahepatic microbiota disturbances drive progression of liver cirrhosis toward hepatocellular carcinoma. J. Immunother. Cancer 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Galperin, M.Y.; Kristensen, D.M.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Microbial genome analysis: The COG approach. Brief. Bioinform. 2019, 20, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Hong, Z.; Huang, L.S.; Tsukasaki, Y.; Nepal, S.; Di, A.; Zhong, M.; Wu, W.; Ye, Z.; Gao, X.; et al. IL-1β suppression of VE-cadherin transcription underlies sepsis-induced inflammatory lung injury. J. Clin. Investig. 2020, 130, 3684–3698. [Google Scholar] [CrossRef] [PubMed]
- Oettgen, P. Regulation of vascular inflammation and remodeling by ETS factors. Circ. Res. 2006, 99, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Heintz-Buschart, A.; Wilmes, P. Human Gut Microbiome: Function Matters. Trends Microbiol. 2018, 26, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D. Human gut microbiome: Hopes, threats and promises. Gut 2018, 67, 1716–1725. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Neish, A.S. Gut Microbiota in Intestinal and Liver Disease. Annu. Rev. Pathol. 2021, 16, 251–275. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Helmink, B.A.; Spencer, C.N.; Reuben, A.; Wargo, J.A. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell 2018, 33, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Berding, K.; Long-Smith, C.M.; Carbia, C.; Bastiaanssen, T.F.S.; van de Wouw, M.; Wiley, N.; Strain, C.R.; Fouhy, F.; Stanton, C.; Cryan, J.F.; et al. A specific dietary fibre supplementation improves cognitive performance-an exploratory randomised, placebo-controlled, crossover study. Psychopharmacology 2021, 238, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.S.; Gaffney, M.; Hopkins, S.; Kelley, T.; Gonzalez, A.; Bowers, S.J.; Vitaterna, M.H.; Turek, F.W.; Foxx, C.L.; Lowry, C.A.; et al. Ruminiclostridium 5, Parabacteroides distasonis, and bile acid profile are modulated by prebiotic diet and associate with facilitated sleep/clock realignment after chronic disruption of rhythms. Brain Behav. Immun. 2021, 97, 150–166. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, X.; Du, Y.; Liu, X.; Chen, G.; Xiang, P.; Wu, H.; Liu, C.; Wang, D. Brussels Chicory Stabilizes Unstable Atherosclerotic Plaques and Reshapes the Gut Microbiota in Apoe-/- Mice. J. Nutr. 2022, 152, 2209–2217. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Greten, T.F. Gut microbiome in HCC—Mechanisms, diagnosis and therapy. J. Hepatol. 2020, 72, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Sharebiani, H.; Fazeli, B.; Maniscalco, R.; Ligi, D.; Mannello, F. The Imbalance among Oxidative Biomarkers and Antioxidant Defense Systems in Thromboangiitis Obliterans (Winiwarter-Buerger Disease). J. Clin. Med. 2020, 9, 1036. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Sun, L.; Ling, S.; Xu, J.W. Levistilide A Ameliorates NLRP3 Expression Involving the Syk-p38/JNK Pathway and Peripheral Obliterans in Rats. Mediators Inflamm. 2018, 2018, 7304096. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Takahashi, M.; Naruse, T.; Nakajima, T.; Chen, Y.W.; Inoue, Y.; Ishikawa, I.; Iwai, T.; Kimura, A. Synergistic contribution of CD14 and HLA loci in the susceptibility to Buerger disease. Hum. Genet. 2007, 122, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Mohareri, M.; Mirhosseini, A.; Mehraban, S.; Fazeli, B. Thromboangiitis obliterans episode: Autoimmune flare-up or reinfection? Vasc. Health Risk Manag. 2018, 14, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Naderpoor, N.; Mousa, A.; Fernanda Gomez Arango, L.; Barrett, H.L.; Dekker Nitert, M.; de Courten, B. Effect of Vitamin D Supplementation on Faecal Microbiota: A Randomised Clinical Trial. Nutrients 2019, 11, 2888. [Google Scholar] [CrossRef] [PubMed]
- Fazeli, B.; Ligi, D.; Keramat, S.; Maniscalco, R.; Sharebiani, H.; Mannello, F. Recent Updates and Advances in Winiwarter-Buerger Disease (Thromboangiitis Obliterans): Biomolecular Mechanisms, Diagnostics and Clinical Consequences. Diagnostics 2021, 11, 1736. [Google Scholar] [CrossRef]
- Forbes, J.D.; Chen, C.Y.; Knox, N.C.; Marrie, R.A.; El-Gabalawy, H.; de Kievit, T.; Alfa, M.; Bernstein, C.N.; Van Domselaar, G. A comparative study of the gut microbiota in immune-mediated inflammatory diseases-does a common dysbiosis exist? Microbiome 2018, 6, 221. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Ma, Y.; Liang, X.; Zhang, Y.; Hong, D.; Wang, Y.; Bai, D. Efficacy and Mechanism of Qianshan Huoxue Gao in Acute Coronary Syndrome via Regulation of Intestinal Flora and Metabolites. Drug Des. Devel Ther. 2023, 17, 579–595. [Google Scholar] [CrossRef] [PubMed]
- Hamer, H.M.; Jonkers, D.M.; Bast, A.; Vanhoutvin, S.A.; Fischer, M.A.; Kodde, A.; Troost, F.J.; Venema, K.; Brummer, R.J. Butyrate modulates oxidative stress in the colonic mucosa of healthy humans. Clin. Nutr. 2009, 28, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Guilloteau, P.; Martin, L.; Eeckhaut, V.; Ducatelle, R.; Zabielski, R.; Van Immerseel, F. From the gut to the peripheral tissues: The multiple effects of butyrate. Nutr. Res. Rev. 2010, 23, 366–384. [Google Scholar] [CrossRef] [PubMed]
- Haycock, P.C.; Burgess, S.; Nounu, A.; Zheng, J.; Okoli, G.N.; Bowden, J.; Wade, K.H.; Timpson, N.J.; Evans, D.M.; Willeit, P.; et al. Association Between Telomere Length and Risk of Cancer and Non-Neoplastic Diseases: A Mendelian Randomization Study. JAMA Oncol. 2017, 3, 636–651. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Bäck, M.; Rees, J.M.B.; Mason, A.M.; Burgess, S. Body mass index and body composition in relation to 14 cardiovascular conditions in UK Biobank: A Mendelian randomization study. Eur. Heart J. 2020, 41, 221–226. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Taxa (Exposure) | MR Method | No. of SNP | F-Statistic | Power | OR | 95% CI | p-Value |
|---|---|---|---|---|---|---|---|
| Ruminiclostridium 5 | IVW | 11 | 12.03 | 0.13 | 0.119 | 0.021–0.688 | 0.017 |
| ML | 11 | 0.121 | 0.020–0.742 | 0.022 | |||
| MR-Egger | 11 | 0.385 | 0.000–572.786 | 0.804 | |||
| Weighted median | 11 | 0.074 | 0.007–0.752 | 0.028 | |||
| Weighted mode | 11 | 0.020 | 0.000–1.549 | 0.108 | |||
| Eubacterium (xylanophilum group) | IVW | 9 | 24.67 | 1.00 | 5.383 | 1.128–25.693 | 0.035 |
| ML | 9 | 5.658 | 1.142–28.021 | 0.034 | |||
| MR-Egger | 9 | 6.043 | 0.055–660.248 | 0.477 | |||
| Weighted median | 9 | 5.545 | 0.718–42.852 | 0.101 | |||
| Weighted mode | 9 | 6.942 | 0.364–132.271 | 0.234 | |||
| Lachnospira | IVW | 6 | 26.97 | 0.12 | 0.064 | 0.003–1.176 | 0.064 |
| ML | 6 | 0.055 | 0.004–0.755 | 0.030 | |||
| MR-Egger | 6 | 0.000 | 0.000–15,532.199 | 0.410 | |||
| Weighted median | 6 | 0.189 | 0.006–5.775 | 0.339 | |||
| Weighted mode | 6 | 0.309 | 0.004–25.026 | 0.623 |
| TAO (n = 12) | Controls (n = 9) | |
|---|---|---|
| Age, median (IQR), y | 39 (32.25, 44.25) | 42 (32.00, 45.50) |
| Gender, No. (%) | ||
| Male | 12 (100) | 9 (100) |
| Female | 0 (0) | 0 (0) |
| Smoking history, No. (%) | ||
| Yes | 12 (100) | 7 (78) |
| No | 0 (0) | 2 (22) |
| Intermittent claudication, No. (%) | ||
| Yes | 12 (100) | 0 (0) |
| No | 0 (0) | 0 (0) |
| Resting pain, No. (%) | ||
| Yes | 9 (75) | 0 (0) |
| No | 3 (25) | 0 (0) |
| Ulcer or gangrene, No. (%) | ||
| Yes | 10 (83) | 0 (0) |
| No | 2 (17) | 0 (0) |
| Migratory superficial phlebitis, No. (%) | ||
| Yes | 6 (50) | 0 (0) |
| No | 6 (50) | 0 (0) |
| Reynolds phenomenon, No. (%) | ||
| Yes | 4 (33) | 0 (0) |
| No | 8 (67) | 0 (0) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheng, C.; Huang, W.; Liao, M.; Yang, P. The Role of Gut Microbiota in Thromboangiitis Obliterans: Cohort and Mendelian Randomization Study. Biomedicines 2024, 12, 1459. https://doi.org/10.3390/biomedicines12071459
Sheng C, Huang W, Liao M, Yang P. The Role of Gut Microbiota in Thromboangiitis Obliterans: Cohort and Mendelian Randomization Study. Biomedicines. 2024; 12(7):1459. https://doi.org/10.3390/biomedicines12071459
Chicago/Turabian StyleSheng, Chang, Weihua Huang, Mingmei Liao, and Pu Yang. 2024. "The Role of Gut Microbiota in Thromboangiitis Obliterans: Cohort and Mendelian Randomization Study" Biomedicines 12, no. 7: 1459. https://doi.org/10.3390/biomedicines12071459