
Exploring the Role of Neutrophil-Related Genes in Osteosarcoma via an Integrative Analysis of Single-Cell and Bulk Transcriptome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Database Selection
2.2. Single-Cell Sequencing Analysis
2.3. Screening of Neutrophil-Associated DEmRNAs
2.4. Functional Enrichment Analysis for scRNA seq
2.5. Construction and Evaluation of a Neutrophil-Related Prognostic Signature
2.6. Establishment and Validation of a Predictive Nomogram
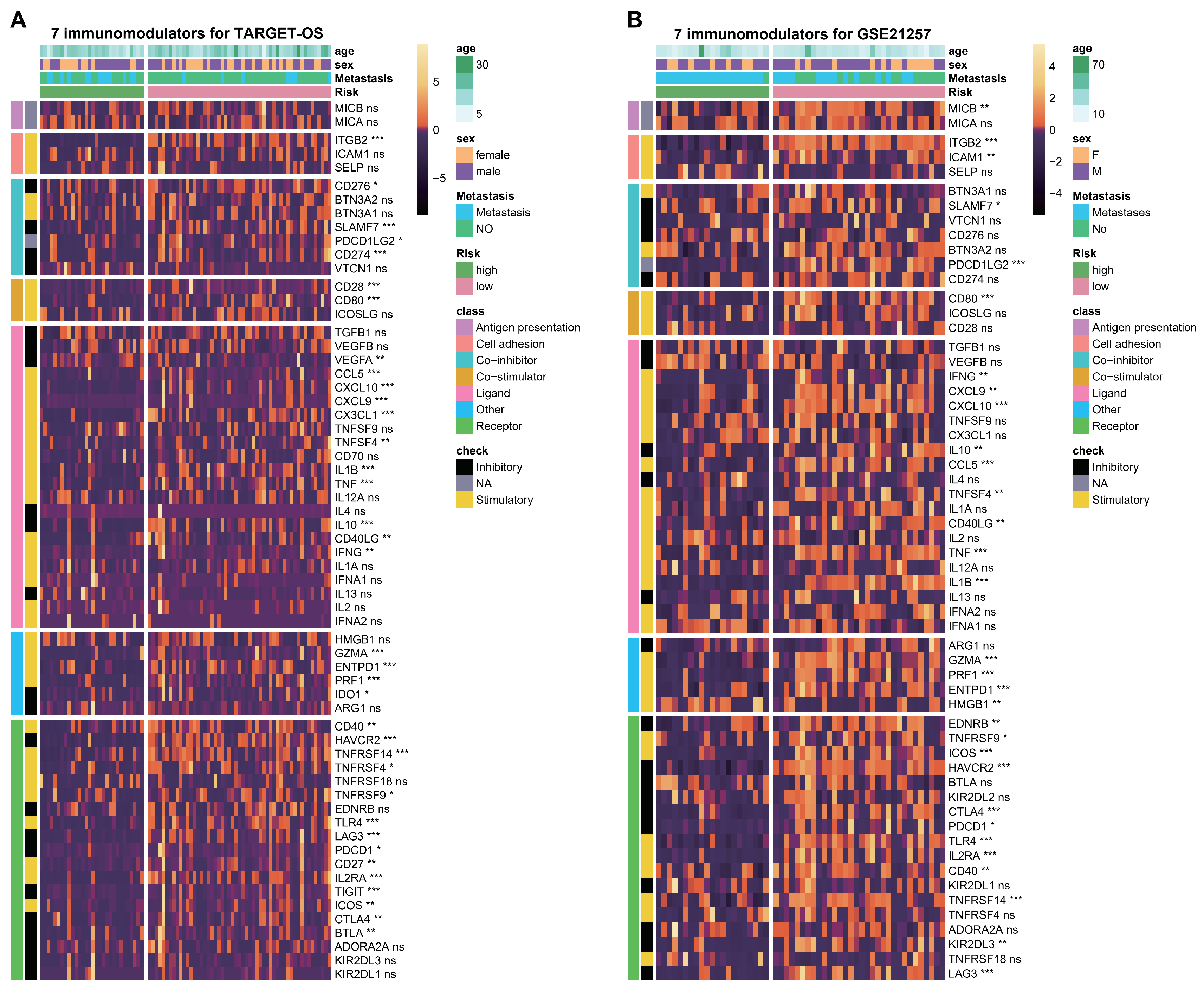
2.7. Analysis of TME, Immunomodulators and Signal Pathways
2.8. Cell Culture
2.9. Osteosarcoma Mouse Model Construction
2.10. Quantitative Real-Time PCR (RT-qPCR)
2.11. Immunohistochemistry (IHC)
2.12. Statistical Analysis
3. Results
3.1. ScRNA Sequencing Data Analysis
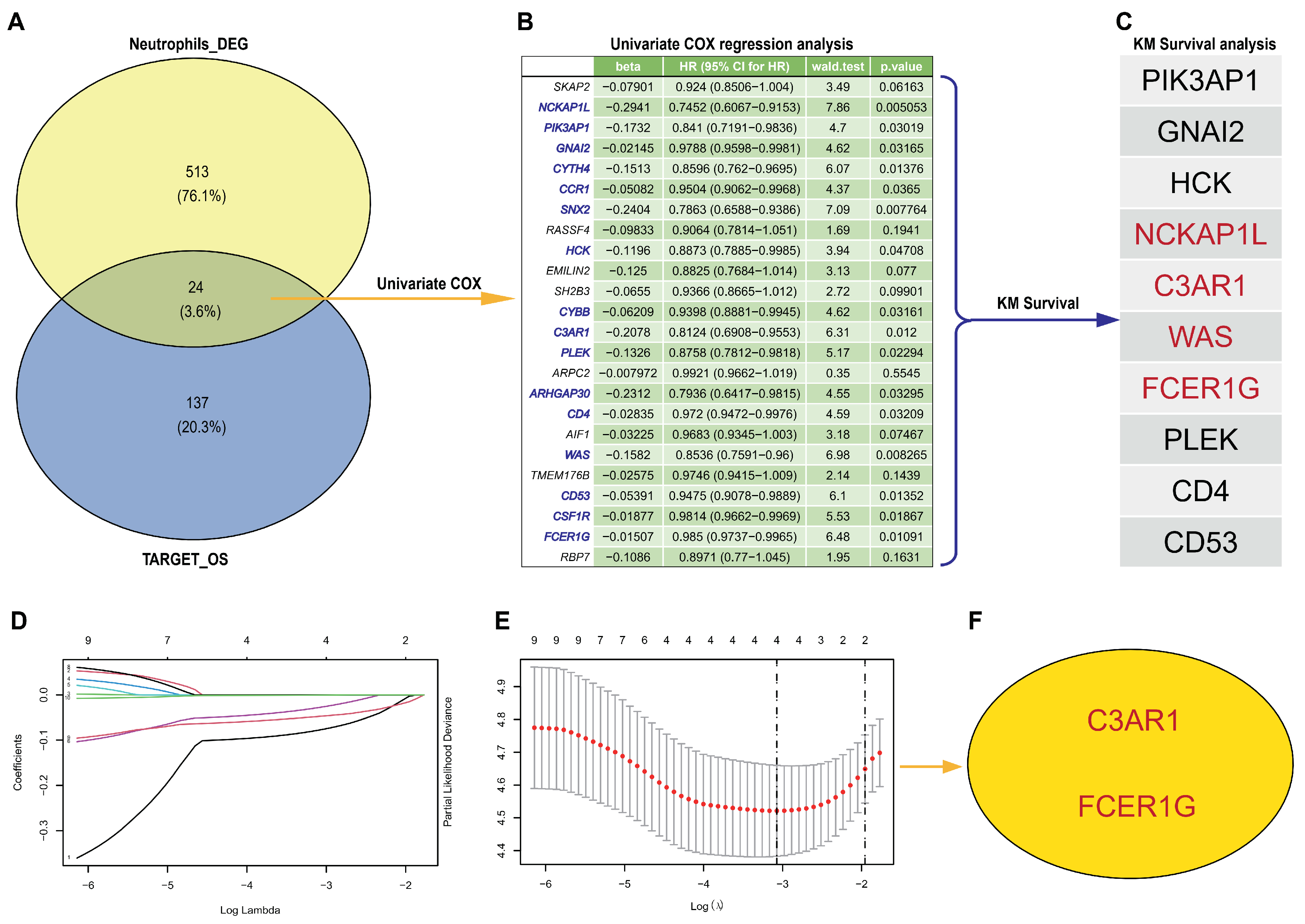
3.2. Hub Genes Detection from scRNA-Seq and TARGET-OS
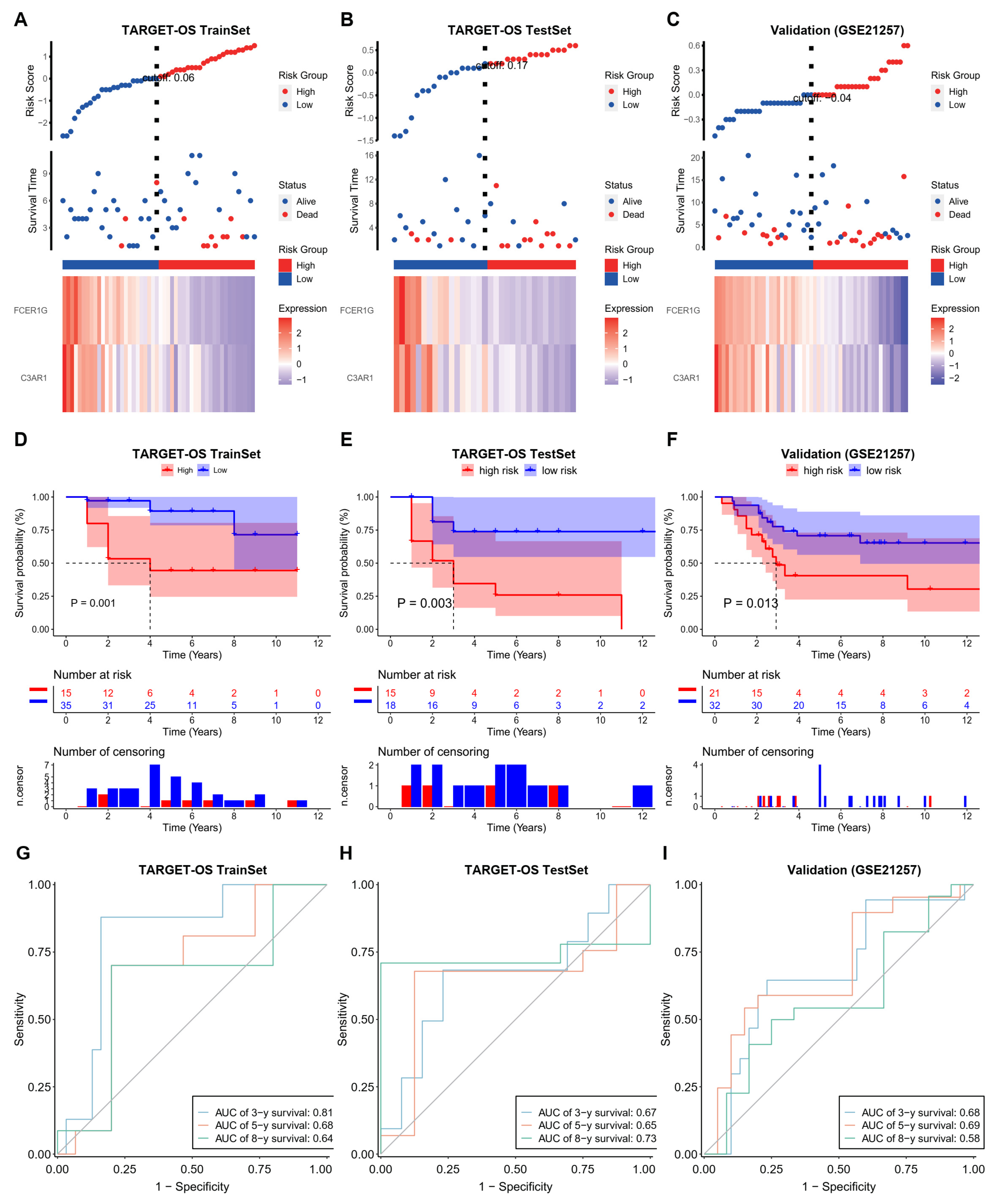
3.3. Establish an Effective Prognostic Risk Model
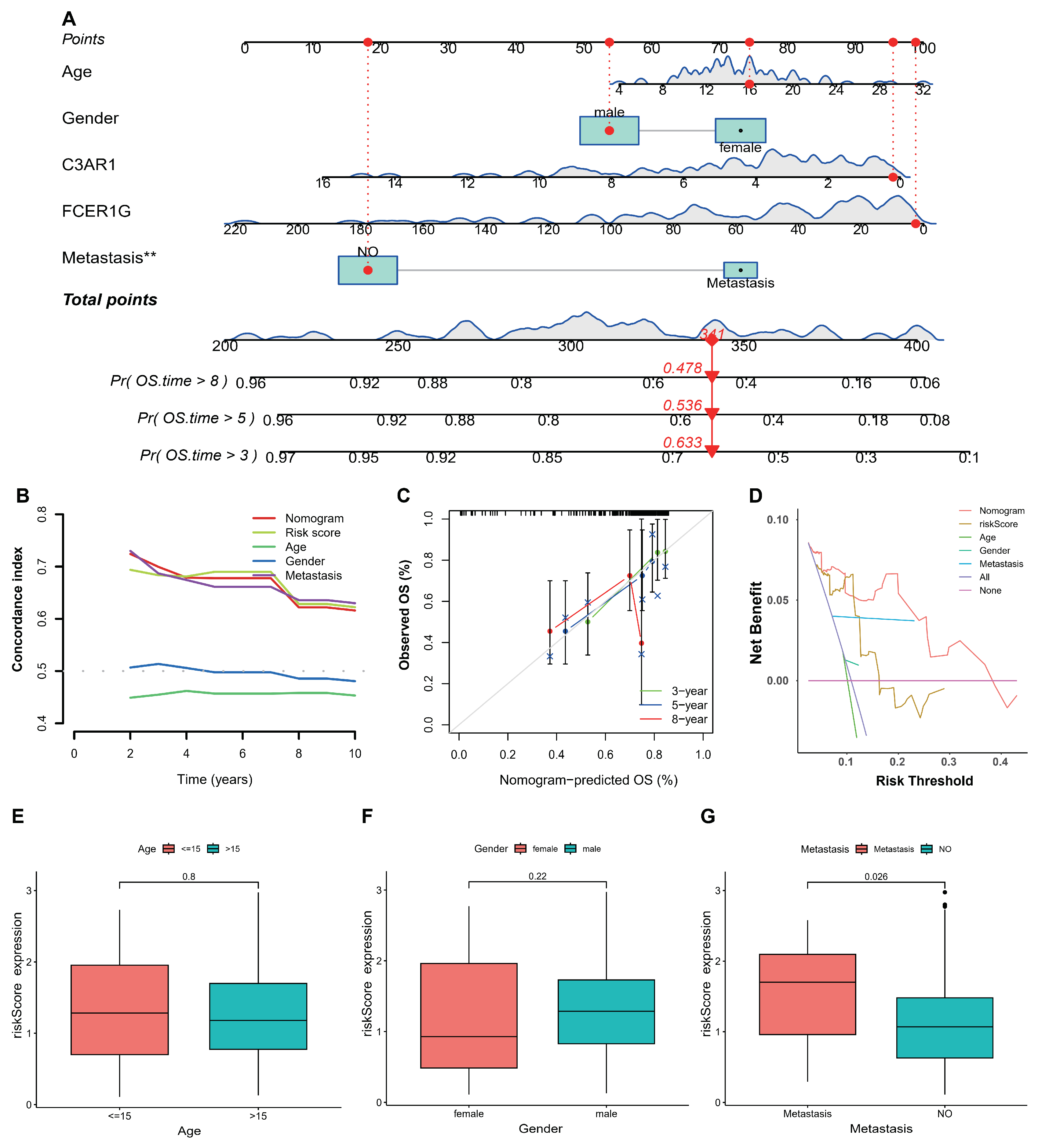
3.4. Establishment and Validation of Nomogram
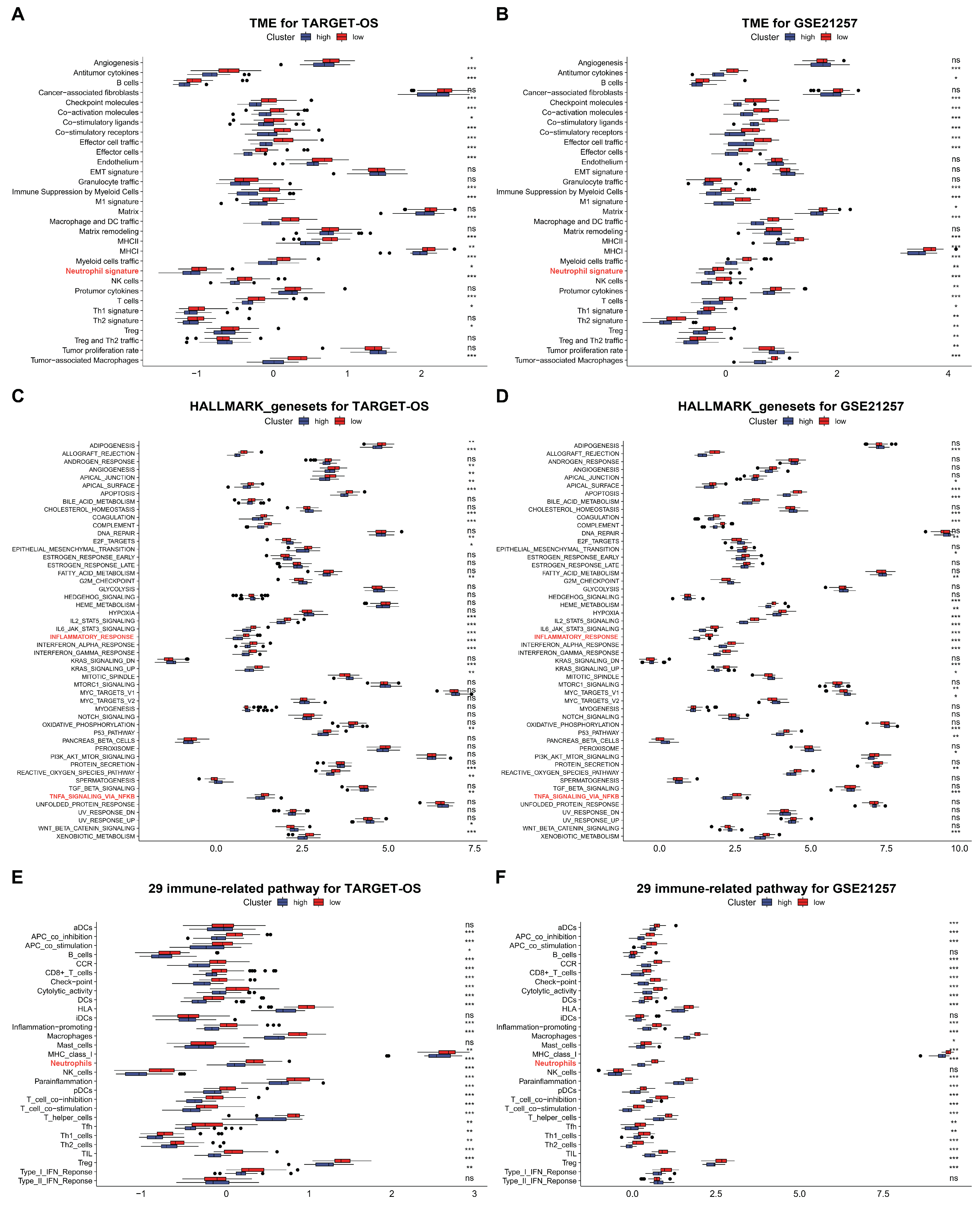
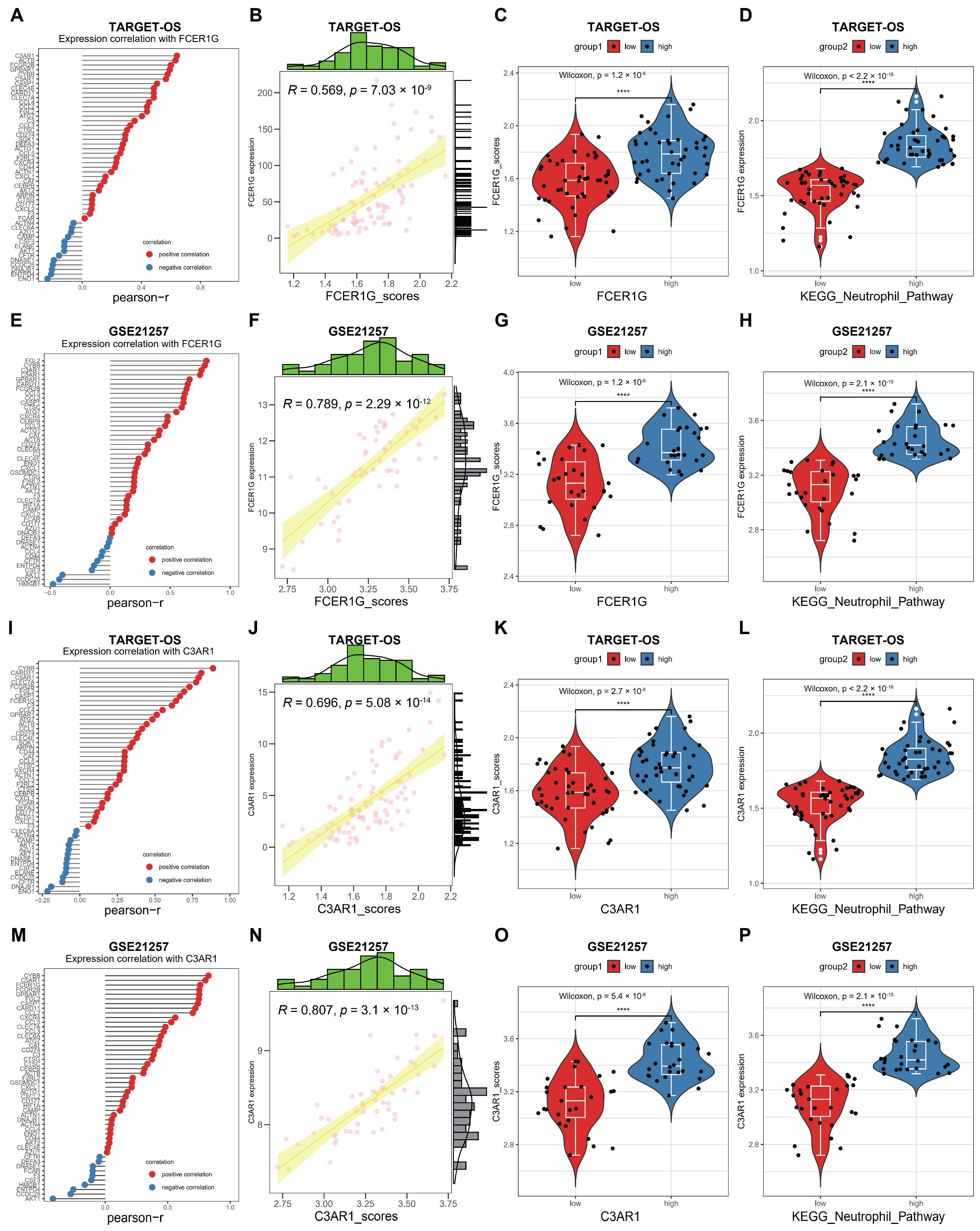
3.5. C3AR1 and FCER1G Reshapes the TME and Immune Signal Pathways of OS
3.6. ESTIMATE Evaluation and Immunomodulators Analysis Based on the Two Hub Genes
3.7. Correlation between FCER1G and C3AR1 and KEGG Neutrophil Signaling Pathway in OS
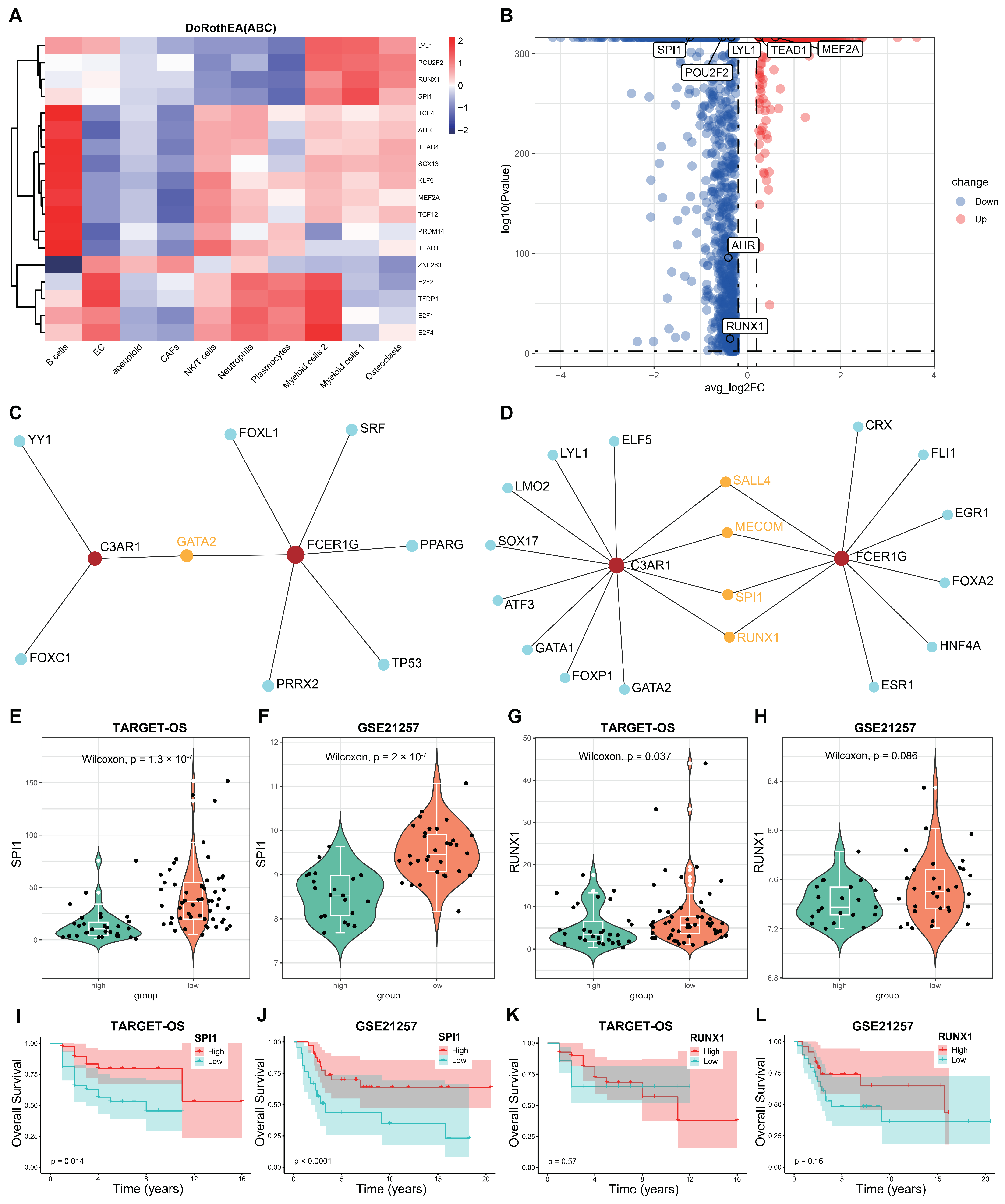
3.8. Screening the Key Transcription Factors (TFs) Regulating the Two Hub Genes
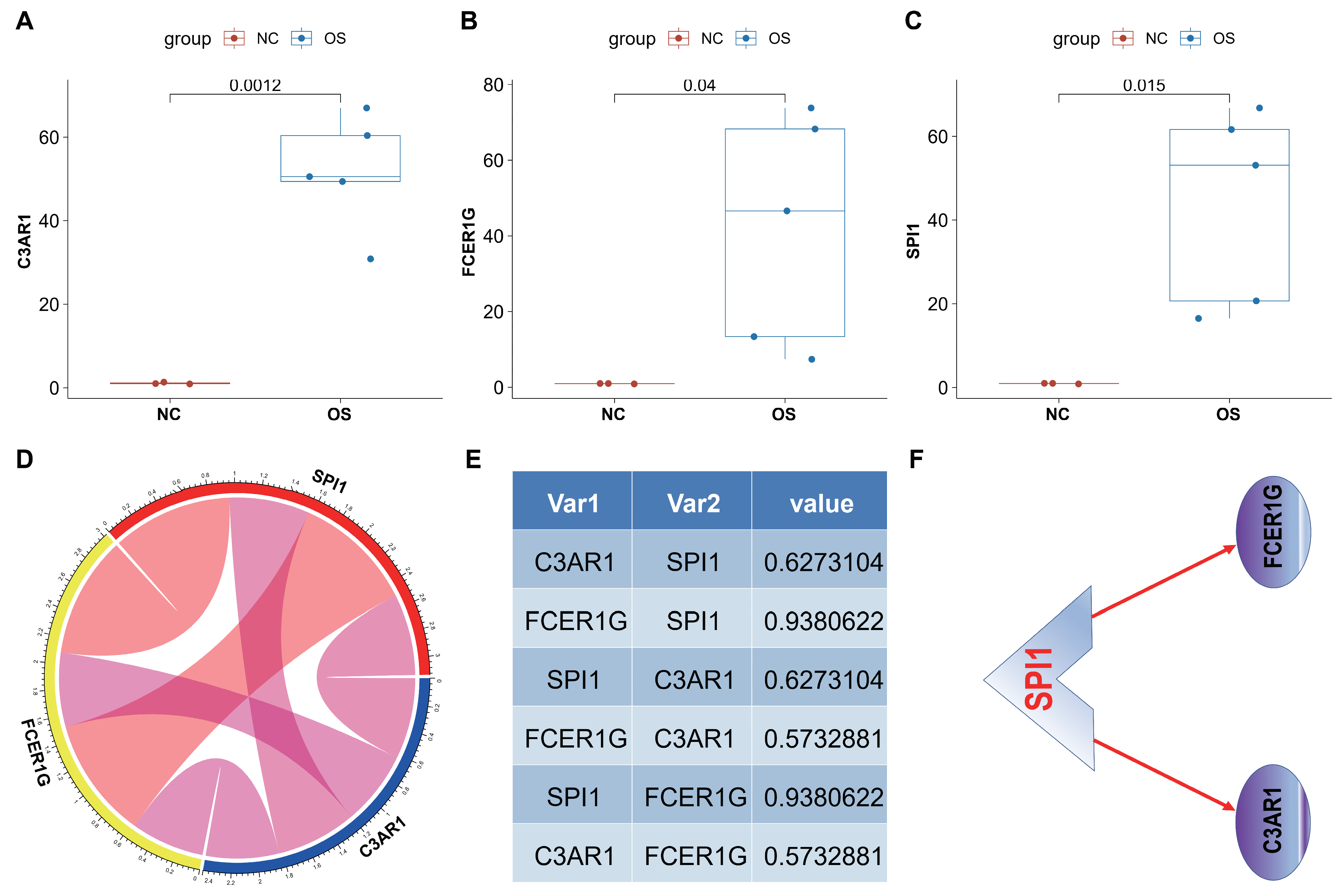
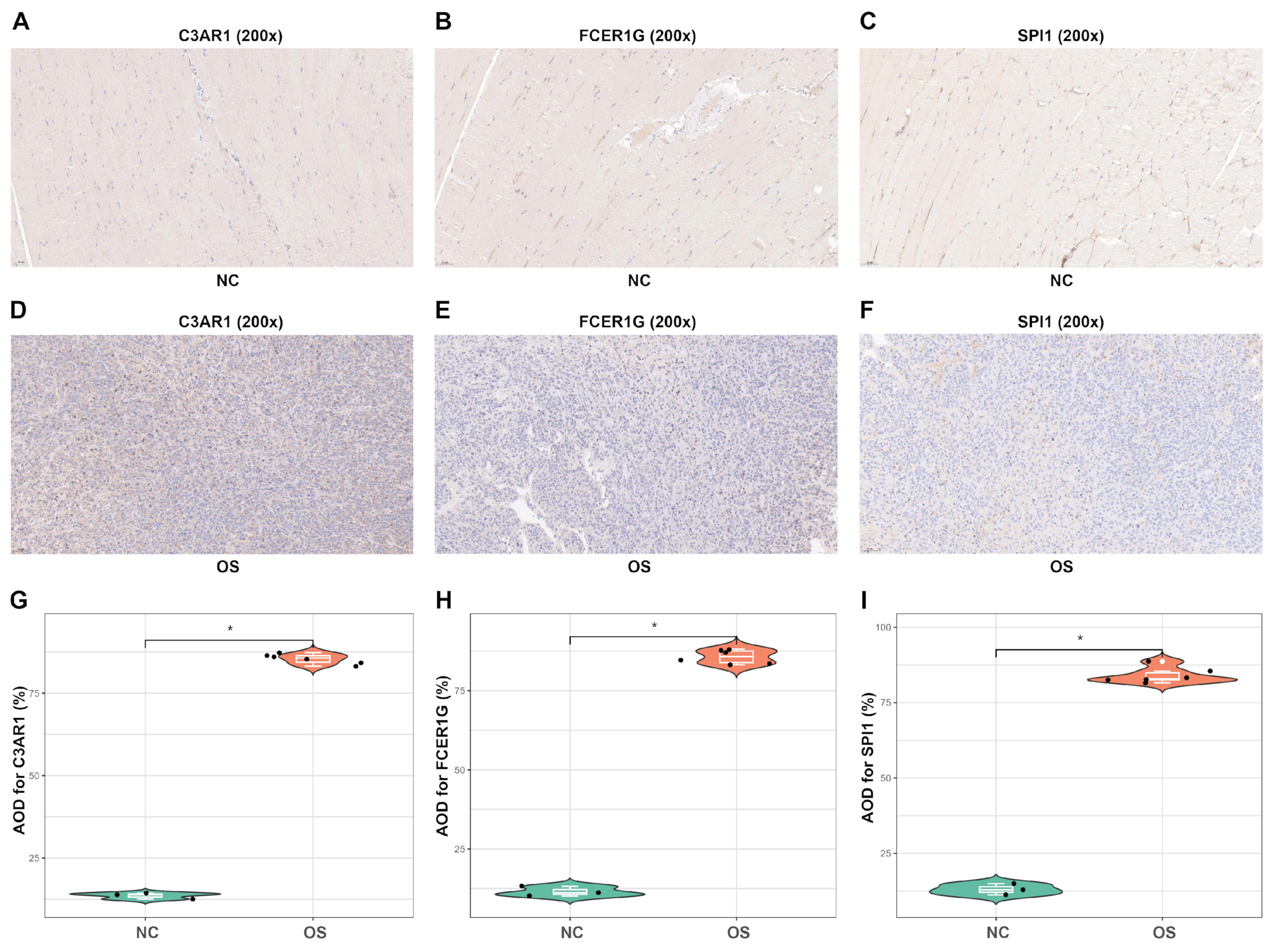
3.9. In Vivo Analyses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, F.; Sun, G.; Peng, Z.; Liu, J.; Li, Z.; Yan, J. Cannabidiol promotes apoptosis of osteosarcoma cells in vitro and in vivo by activating the SP1-CBX2 axis. Am. J. Transl. Res. 2022, 14, 1188–1203. [Google Scholar]
- Rojas, G.A.; Hubbard, A.K.; Diessner, B.J.; Ribeiro, K.B.; Spector, L.G. International trends in incidence of osteosarcoma (1988–2012). Int. J. Cancer 2021, 149, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Lei, T.; Qian, H.; Lei, P.; Hu, Y. Ferroptosis-related gene signature associates with immunity and predicts prognosis accurately in patients with osteosarcoma. Cancer Sci. 2021, 112, 4785–4798. [Google Scholar] [CrossRef] [PubMed]
- Mirabello, L.; Troisi, R.J.; Savage, S.A. Osteosarcoma incidence and survival rates from 1973 to 2004: Data from the Surveillance, Epidemiology, and End Results Program. Cancer 2009, 115, 1531–1543. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.E. Update on Survival in Osteosarcoma. Orthop. Clin. N. Am. 2016, 47, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Kager, L.; Zoubek, A.; Pötschger, U.; Kastner, U.; Flege, S.; Kempf-Bielack, B.; Branscheid, D.; Kotz, R.; Salzer-Kuntschik, M.; Winkelmann, W.; et al. Primary metastatic osteosarcoma: Presentation and outcome of patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. J. Clin. Oncol. 2003, 21, 2011–2018. [Google Scholar] [CrossRef]
- Gorlick, R.; Janeway, K.; Lessnick, S.; Randall, R.L.; Marina, N.; COG Bone Tumor Committee. Children’s Oncology Group’s 2013 blueprint for research: Bone tumors. Pediatr. Blood Cancer 2013, 60, 1009–1015. [Google Scholar] [CrossRef]
- Liu, J.; Wu, S.; Xie, X.; Wang, Z.; Lei, Q. Identification of potential crucial genes and key pathways in osteosarcoma. Hereditas 2020, 157, 29. [Google Scholar] [CrossRef] [PubMed]
- Maurizi, G.; Verma, N.; Gadi, A.; Mansukhani, A.; Basilico, C. Sox2 is required for tumor development and cancer cell proliferation in osteosarcoma. Oncogene 2018, 37, 4626–4632. [Google Scholar] [CrossRef]
- Duan, G.; Zhang, C.; Xu, C.; Xu, C.; Zhang, L.; Zhang, Y. Knockdown of MALAT1 inhibits osteosarcoma progression via regulating the miR-34a/cyclin D1 axis. Int. J. Oncol. 2019, 54, 17–28. [Google Scholar] [CrossRef]
- Shimizu, T.; Sugihara, E.; Yamaguchi-Iwai, S.; Tamaki, S.; Koyama, Y.; Kamel, W.; Ueki, A.; Ishikawa, T.; Chiyoda, T.; Osuka, S.; et al. IGF2 preserves osteosarcoma cell survival by creating an autophagic state of dormancy that protects cells against chemotherapeutic stress. Cancer Res. 2014, 74, 6531–6541. [Google Scholar] [CrossRef]
- Wei, R.; Thanindratarn, P.; Dean, D.C.; Hornicek, F.J.; Guo, W.; Duan, Z. Cyclin E1 is a prognostic biomarker and potential therapeutic target in osteosarcoma. J. Orthop. Res. 2020, 38, 1952–1964. [Google Scholar] [CrossRef]
- Zou, T.; Liu, W.; Wang, Z.; Chen, J.; Lu, S.; Huang, K.; Li, W. C3AR1 mRNA as a Potential Therapeutic Target Associates With Clinical Outcomes and Tumor Microenvironment in Osteosarcoma. Front. Med. 2021, 8, 642615. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Luo, K.; Liu, Y.; Liao, S.; He, J.; He, M.; Xie, T.; Jiang, X.; Li, B.; Liu, H.; et al. ATG16L1 is a Potential Prognostic Biomarker and Immune Signature for Osteosarcoma: A Study Based on Bulk RNA and Single-Cell RNA-Sequencing. Int. J. Gen. Med. 2022, 15, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Liu, S.; Zhu, P.; Tang, M.; Wang, Y.; Tian, Y.; Li, D.; Zhu, X.; Yan, D.; Zhu, Z. Cross-Species Gene Expression Analysis Reveals Gene Modules Implicated in Human Osteosarcoma. Front. Genet. 2019, 10, 697. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Yan, T.; Guo, W.; Wang, W.; Zhao, Z.; Ren, T.; Huang, Y.; Zhang, H.; Yu, Y.; Liang, X. Identification of Potential Therapeutic Targets and Immune Cell Infiltration Characteristics in Osteosarcoma Using Bioinformatics Strategy. Front. Oncol. 2020, 10, 1628. [Google Scholar] [CrossRef]
- Jia, Y.; Liu, Y.; Han, Z.; Tian, R. Identification of potential gene signatures associated with osteosarcoma by integrated bioinformatics analysis. PeerJ 2021, 9, e11496. [Google Scholar] [CrossRef] [PubMed]
- Yao, N.; Ren, K.; Gu, X.J.; Wu, S.J.; Shi, X.; Chang, Q.; Li, Y.G.; Gao, Z.X.; Jin, Q.M.; Zhang, J.; et al. Identification of potential crucial genes associated with vasculogenic mimicry in human osteosarcoma based on gene expression profile. Neoplasma 2020, 67, 286–295. [Google Scholar] [CrossRef]
- Feleke, M.; Feng, W.; Song, D.; Li, H.; Rothzerg, E.; Wei, Q.; Kõks, S.; Wood, D.; Liu, Y.; Xu, J. Single-cell RNA sequencing reveals differential expression of EGFL7 and VEGF in giant-cell tumor of bone and osteosarcoma. Exp. Biol. Med. 2022, 247, 1214–1227. [Google Scholar] [CrossRef]
- Shao, H.; Ge, M.; Zhang, J.; Zhao, T.; Zhang, S. Osteoclasts differential-related prognostic biomarker for osteosarcoma based on single cell, bulk cell and gene expression datasets. BMC Cancer 2022, 22, 288. [Google Scholar] [CrossRef]
- Xu, F.; Yan, J.; Peng, Z.; Liu, J.; Li, Z. Comprehensive analysis of a glycolysis and cholesterol synthesis-related genes signature for predicting prognosis and immune landscape in osteosarcoma. Front. Immunol. 2022, 13, 1096009. [Google Scholar] [CrossRef] [PubMed]
- Dean, D.C.; Shen, S.; Hornicek, F.J.; Duan, Z. From genomics to metabolomics: Emerging metastatic biomarkers in osteosarcoma. Cancer Metastasis Rev. 2018, 37, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.; Wang, H.; Wang, H.; Mo, H.; Zhong, W.; Tang, J.; Lu, Y.; Zhou, W.; Tan, A.; Liu, Y.; et al. A three-gene signature based on tumour microenvironment predicts overall survival of osteosarcoma in adolescents and young adults. Aging (Albany NY) 2020, 13, 619–645. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, M.; Jing, Y.; Cheng, J.; Zhang, C.; Cheng, L.; Lu, H.; Cai, M.C.; Wu, J.; Wang, W.; et al. Baseline immunity and impact of chemotherapy on immune microenvironment in cervical cancer. Br. J. Cancer 2021, 124, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer 2020, 20, 485–503. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Jaillon, S.; Garlanda, C.; Allavena, P. Tumor-associated myeloid cells: Diversity and therapeutic targeting. Cell. Mol. Immunol. 2021, 18, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, L.; Li, X.; Zhuo, W. Neutrophil Extracellular Traps in Tumor Metastasis: Pathological Functions and Clinical Applications. Cancers 2021, 13, 2832. [Google Scholar] [CrossRef]
- Giese, M.A.; Hind, L.E.; Huttenlocher, A. Neutrophil plasticity in the tumor microenvironment. Blood 2019, 133, 2159–2167. [Google Scholar] [CrossRef]
- Gong, Q.; Chen, X.; Liu, F.; Cao, Y. Machine learning-based integration develops a neutrophil-derived signature for improving outcomes in hepatocellular carcinoma. Front. Immunol. 2023, 14, 1216585. [Google Scholar] [CrossRef]
- Yang, M.; Zhang, G.; Wang, Y.; He, M.; Xu, Q.; Lu, J.; Liu, H.; Xu, C. Tumour-associated neutrophils orchestrate intratumoural IL-8-driven immune evasion through Jagged2 activation in ovarian cancer. Br. J. Cancer 2020, 123, 1404–1416. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Dai, Y.; Tang, X.; Yin, T.; Wang, C.; Wang, T.; Dong, L.; Shi, M.; Qin, J.; et al. Single-cell RNA-seq analysis reveals BHLHE40-driven pro-tumour neutrophils with hyperactivated glycolysis in pancreatic tumour microenvironment. Gut 2023, 72, 958–971. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Feng, W.; Dai, Y.; Bao, M.; Yuan, Z.; He, M.; Qin, Z.; Liao, S.; He, J.; Huang, Q.; et al. Single-Cell Transcriptomics Reveals the Complexity of the Tumor Microenvironment of Treatment-Naive Osteosarcoma. Front. Oncol. 2021, 11, 709210. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Tong, C.; Xu, M.; He, H.; Chen, C. Bioinformatics Analysis Reveals an Association between Autophagy, Prognosis, Tumor Microenvironment, and Immunotherapy in Osteosarcoma. J. Oncol. 2022, 2022, 4220331. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A. A description of the Molecular Signatures Database (MSigDB) Web site. Methods Mol. Biol. 2014, 1150, 153–160. [Google Scholar] [CrossRef]
- Bagaev, A.; Kotlov, N.; Nomie, K.; Svekolkin, V.; Gafurov, A.; Isaeva, O.; Osokin, N.; Kozlov, I.; Frenkel, F.; Gancharova, O.; et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell 2021, 39, 845–865.e7. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef]
- Liang, J.Y.; Wang, D.S.; Lin, H.C.; Chen, X.X.; Yang, H.; Zheng, Y.; Li, Y.H. A Novel Ferroptosis-related Gene Signature for Overall Survival Prediction in Patients with Hepatocellular Carcinoma. Int. J. Biol. Sci. 2020, 16, 2430–2441. [Google Scholar] [CrossRef]
- Khanna, C.; Prehn, J.; Yeung, C.; Caylor, J.; Tsokos, M.; Helman, L. An orthotopic model of murine osteosarcoma with clonally related variants differing in pulmonary metastatic potential. Clin. Exp. Metastasis 2000, 18, 261–271. [Google Scholar] [CrossRef]
- Zhu, T.; Han, J.; Yang, L.; Cai, Z.; Sun, W.; Hua, Y.; Xu, J. Immune Microenvironment in Osteosarcoma: Components, Therapeutic Strategies and Clinical Applications. Front. Immunol. 2022, 13, 907550. [Google Scholar] [CrossRef]
- Wu, X.; Yan, L.; Liu, Y.; Shang, L. Circ_0000527 promotes osteosarcoma cell progression through modulating miR-646/ARL2 axis. Aging 2021, 13, 6091–6102. [Google Scholar] [CrossRef]
- Wu, Z.; Zhang, X.; Chen, D.; Li, Z.; Wu, X.; Wang, J.; Deng, Y. N6-Methyladenosine-Related LncRNAs Are Potential Remodeling Indicators in the Tumor Microenvironment and Prognostic Markers in Osteosarcoma. Front. Immunol. 2022, 12, 806189. [Google Scholar] [CrossRef]
- Lim, B.; Lin, Y.; Navin, N. Advancing Cancer Research and Medicine with Single-Cell Genomics. Cancer Cell 2020, 37, 456–470. [Google Scholar] [CrossRef]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Zhang, Y.; He, R.; Lei, X.; Mao, L.; Jiang, P.; Ni, C.; Yin, Z.; Zhong, X.; Chen, C.; Zheng, Q.; et al. A Novel Pyroptosis-Related Signature for Predicting Prognosis and Indicating Immune Microenvironment Features in Osteosarcoma. Front. Genet. 2021, 12, 780780. [Google Scholar] [CrossRef]
- Fu, Y.; Jin, Z.; Shen, Y.; Zhang, Z.; Li, M.; Liu, Z.; He, G.; Wu, J.; Wen, J.; Bao, Q.; et al. Construction and validation of a novel apoptosis-associated prognostic signature related to osteosarcoma metastasis and immune infiltration. Transl. Oncol. 2022, 22, 101452. [Google Scholar] [CrossRef]
- Qian, H.; Lei, T.; Lei, P.; Hu, Y. Construction and Validation of an Autophagy-Related Prognostic Model for Osteosarcoma Patients. J. Oncol. 2021, 2021, 9943465. [Google Scholar] [CrossRef]
- Coulthard, L.G.; Woodruff, T.M. Is the complement activation product C3a a proinflammatory molecule? Re-evaluating the evidence and the myth. J. Immunol. 2015, 194, 3542–3548. [Google Scholar] [CrossRef]
- Veerhuis, R.; Nielsen, H.M.; Tenner, A.J. Complement in the brain. Mol. Immunol. 2011, 48, 1592–1603. [Google Scholar] [CrossRef]
- Tian, H. Identification of candidate genes for myeloma-induced osteocyte death based on microarray data. J. Orthop. Surg. Res. 2016, 11, 81. [Google Scholar] [CrossRef]
- Xing, S.; Wang, Y.; Hu, K.; Wang, F.; Sun, T.; Li, Q. WGCNA reveals key gene modules regulated by the combined treatment of colon cancer with PHY906 and CPT11. Biosci. Rep. 2020, 40, BSR20200935. [Google Scholar] [CrossRef]
- Xia, Z.; Shang, H.; Cholewa, J.; Wang, Q.; Ding, X.; Su, Q.; Zhao, Y.; Zanchi, N.E. The Effect of Exercise on Gene Expression and Signaling in Mouse Melanoma Tumors. Med. Sci. Sports Exerc. 2020, 52, 1485–1494. [Google Scholar] [CrossRef]
- Xu, C.; Xu, J.; Lu, L.; Tian, W.; Ma, J.; Wu, M. Identification of key genes and novel immune infiltration-associated biomarkers of sepsis. Innate Immun. 2020, 26, 666–682. [Google Scholar] [CrossRef]
- Litvinchuk, A.; Wan, Y.W.; Swartzlander, D.B.; Chen, F.; Cole, A.; Propson, N.E.; Wang, Q.; Zhang, B.; Liu, Z.; Zheng, H. Complement C3aR Inactivation Attenuates Tau Pathology and Reverses an Immune Network Deregulated in Tauopathy Models and Alzheimer’s Disease. Neuron 2018, 100, 1337–1353.e5. [Google Scholar] [CrossRef]
- Tsuji, M.; Ezumi, Y.; Arai, M.; Takayama, H. A novel association of Fc receptor gamma-chain with glycoprotein VI and their co-expression as a collagen receptor in human platelets. J. Biol. Chem. 1997, 272, 23528–23531. [Google Scholar] [CrossRef]
- Xu, H.; Zhu, Q.; Tang, L.; Jiang, J.; Yuan, H.; Zhang, A.; Lou, M. Prognostic and predictive value of FCER1G in glioma outcomes and response to immunotherapy. Cancer Cell Int. 2021, 21, 103. [Google Scholar] [CrossRef]
- Liu, S.; Wang, C.; Yang, H.; Zhu, T.; Jiang, H.; Chen, J. Weighted gene co-expression network analysis identifies FCER1G as a key gene associated with diabetic kidney disease. Ann. Transl. Med. 2020, 8, 1427. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, S.; Lang, J.H. Development and validation of nomogram with tumor microenvironment-related genes and clinical factors for predicting overall survival of endometrial cancer. J. Cancer 2021, 12, 3530–3538. [Google Scholar] [CrossRef]
- Zou, M.; Su, X.; Wang, L.; Yi, X.; Qiu, Y.; Yin, X.; Zhou, X.; Niu, X.; Wang, L.; Su, M. The Molecular Mechanism of Multiple Organ Dysfunction and Targeted Intervention of COVID-19 Based on Time-Order Transcriptomic Analysis. Front. Immunol. 2021, 12, 729776. [Google Scholar] [CrossRef]
- Li, J.; Shi, H.; Yuan, Z.; Wu, Z.; Li, H.; Liu, Y.; Lu, M.; Lu, M. The role of SPI1-TYROBP-FCER1G network in oncogenesis and prognosis of osteosarcoma, and its association with immune infiltration. BMC Cancer 2022, 22, 108. [Google Scholar] [CrossRef]
- Nguyen, V.C.; Ray, D.; Gross, M.S.; de Tand, M.F.; Frézal, J.; Moreau-Gachelin, F. Localization of the human oncogene SPI1 on chromosome 11, region p11.22. Hum. Genet. 1990, 84, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Wang, X.; Zhou, Q. Long noncoding RNA SNHG16 promotes the tumorigenicity of cervical cancer cells by recruiting transcriptional factor SPI1 to upregulate PARP9. Cell Biol. Int. 2020, 44, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Qiu, X.; Gao, J.; Gong, Z.; Zhou, X.; Luo, H.; Geng, X. SPI1 involvement in malignant melanoma pathogenesis by regulation of HK2 through the AKT1/mTOR pathway. J. Cell. Mol. Med. 2023, 27, 2675–2683. [Google Scholar] [CrossRef]
- Yang, J.; Liu, C.; Guan, J.; Wang, Y.; Su, J.; Wang, Y.; Liu, S. SPI1 mediates transcriptional activation of TPX2 and RNF2 to regulate the radiosensitivity of lung squamous cell carcinoma. Arch. Biochem. Biophys. 2022, 730, 109425. [Google Scholar] [CrossRef]
- Xu, Y.; Gu, S.; Bi, Y.; Qi, X.; Yan, Y.; Lou, M. Transcription factor PU.1 is involved in the progression of glioma. Oncol. Lett. 2018, 15, 3753–3759. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, J.; Rui, J.; Xu, X.-Y.; Shen, J.-K. Exploring the Role of Neutrophil-Related Genes in Osteosarcoma via an Integrative Analysis of Single-Cell and Bulk Transcriptome. Biomedicines 2024, 12, 1513. https://doi.org/10.3390/biomedicines12071513
Lu J, Rui J, Xu X-Y, Shen J-K. Exploring the Role of Neutrophil-Related Genes in Osteosarcoma via an Integrative Analysis of Single-Cell and Bulk Transcriptome. Biomedicines. 2024; 12(7):1513. https://doi.org/10.3390/biomedicines12071513
Chicago/Turabian StyleLu, Jing, Jiang Rui, Xiao-Yu Xu, and Jun-Kang Shen. 2024. "Exploring the Role of Neutrophil-Related Genes in Osteosarcoma via an Integrative Analysis of Single-Cell and Bulk Transcriptome" Biomedicines 12, no. 7: 1513. https://doi.org/10.3390/biomedicines12071513
APA StyleLu, J., Rui, J., Xu, X.-Y., & Shen, J.-K. (2024). Exploring the Role of Neutrophil-Related Genes in Osteosarcoma via an Integrative Analysis of Single-Cell and Bulk Transcriptome. Biomedicines, 12(7), 1513. https://doi.org/10.3390/biomedicines12071513