
CX-5461 Preferentially Induces Top2α-Dependent DNA Breaks at Ribosomal DNA Loci

 ,
, 

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell-Culture Conditions
2.2. In Vivo Experiments
2.3. Cloning and shRNA Transfection
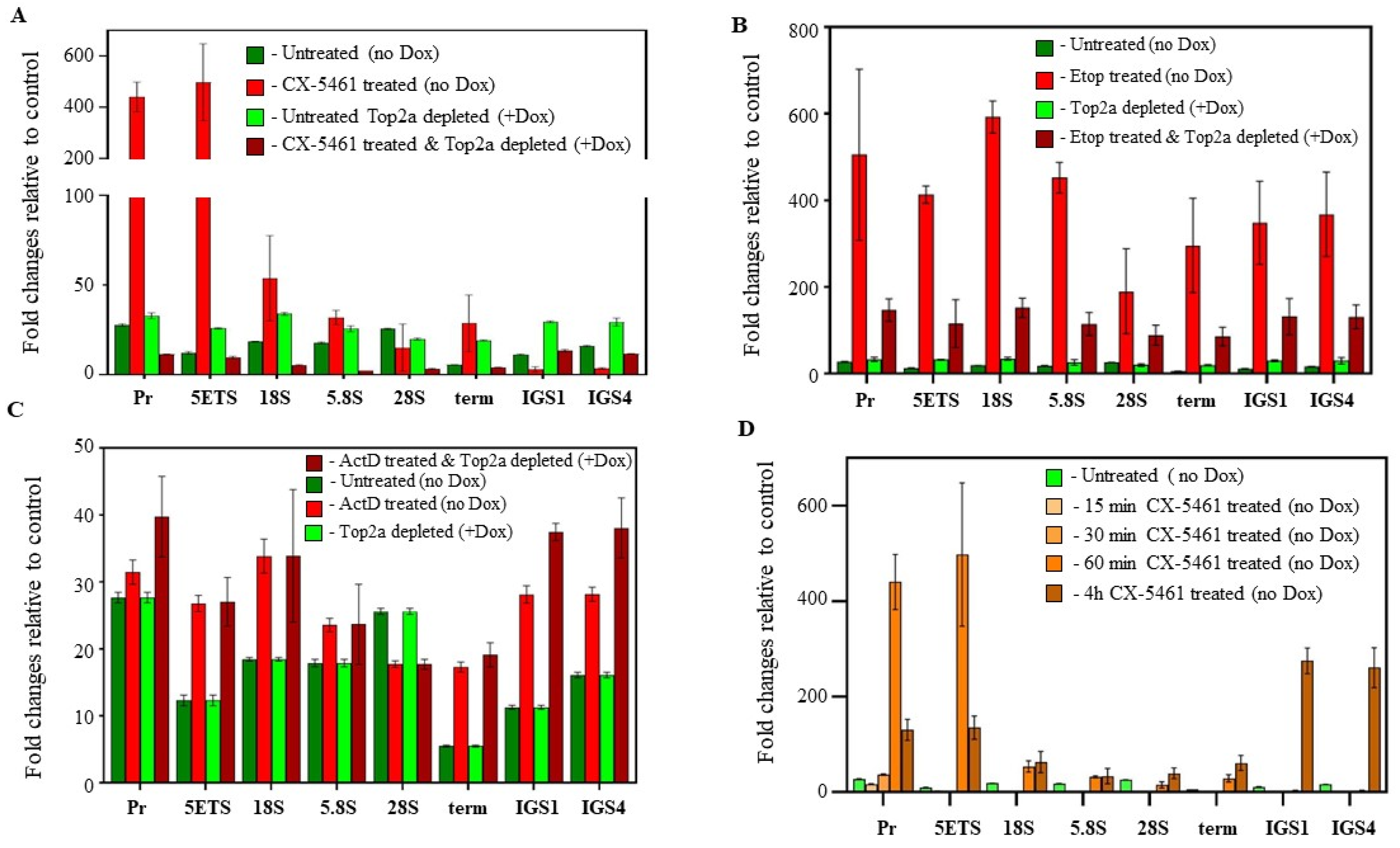
2.4. Quantitative Reverse Transcription PCR
2.5. Western Blot Assay
2.6. Dose–Response Assay
2.7. Immunofluorescence Imaging
2.8. Top2 Activity Assay
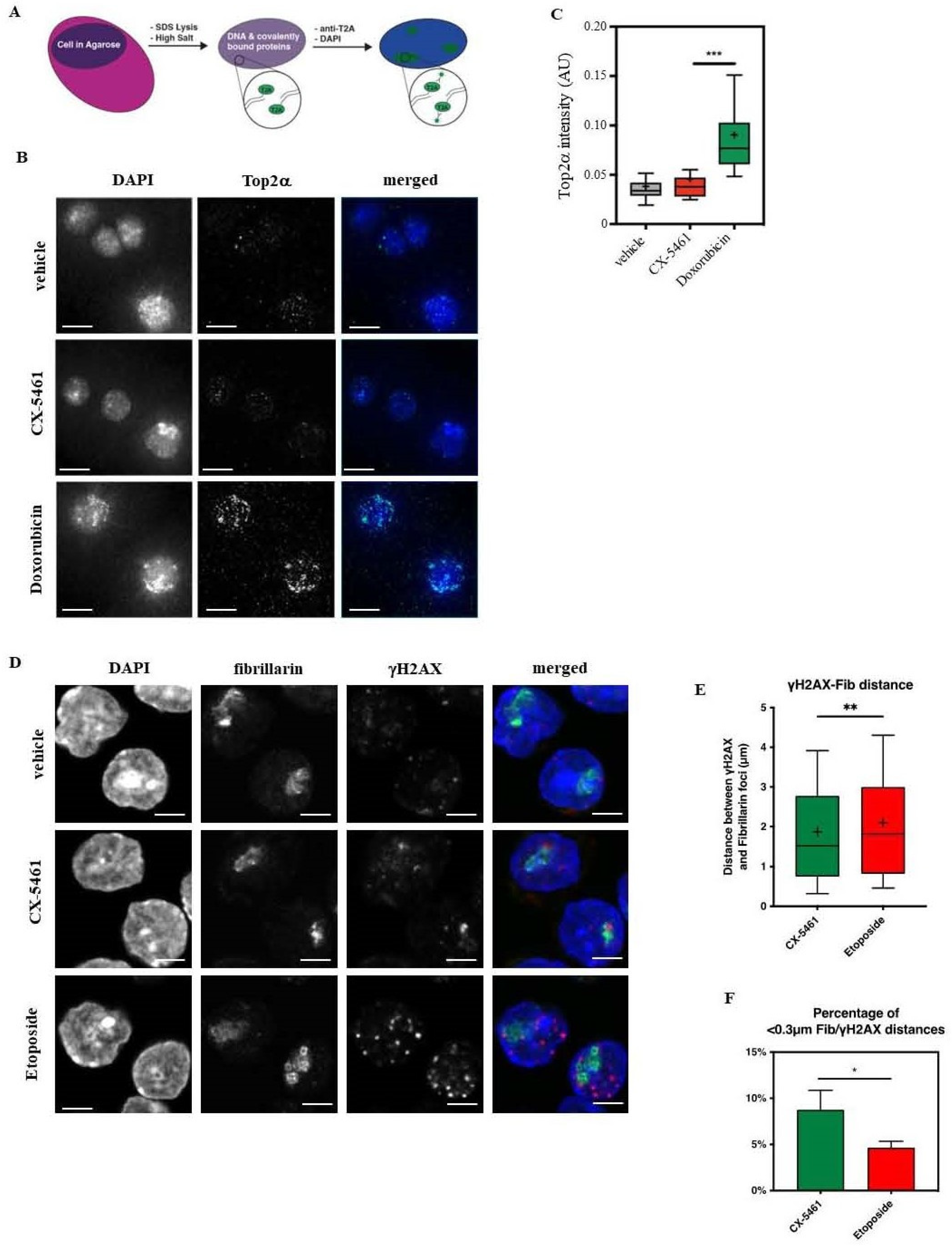
2.9. TARDIS Assay
2.10. DNA Double-Strand Break Labelling
2.11. Metabolic Labelling
3. Results
3.1. Topoisomerase 2α Mediates Resistance of Cancer Cells to CX-5461
3.1.1. Mutations in Top2α Mediate Resistance to CX-5461 in Eμ-Myc Lymphoma
3.1.2. Top2α Mediates Resistance to CX-5461 in Different Cellular Models
3.2. Alterations in Top2α Level/Activity Does Not Affect the Ability of CX-5461 to Inhibit rRNA Synthesis, but Modulates the Level of CX-5461-Induced DDR
3.2.1. The Expression Level of Top2α Does Not Affect the Pol I Inhibitory Activity of CX-5461
3.2.2. The Expression Level of Top2α Modulates the Level of DDR Induced by CX-5461 Treatment
3.3. Physiologically Relevant Concentrations of CX-5461 Do Not Cause Genome-Wide DNA Damage as an Early Response
3.3.1. CX-5461 Is a Top2α Inhibitor That Does Not Induce Genome-Wide Top2α-Mediated DNA Damage
3.3.2. CX-5461 Treatment Induces Targeted DNA Damage at the rDNA Loci
3.4. Induction/Stabilization of G4 Structures in the Eμ-Myc Lymphoma Cellular Model Is Not Sufficient to Induce Cell Death In Vitro
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pianese, G. Beitrag zur Histologie and Aetiologie des Karzinoms. Histologische und experimentelle untersuchungen. Beitr. Pathol. Anat. 1896, 142, 1–193. [Google Scholar]
- Ferreira, R.; Schneekloth, J.S., Jr.; Panov, K.I.; Hannan, K.M.; Hannan, R.D. Targeting the RNA Polymerase I Transcription for Cancer Therapy Comes of Age. Cells 2020, 9, 266. [Google Scholar] [CrossRef] [PubMed]
- Penzo, M.; Montanaro, L.; Trere, D.; Derenzini, M. The Ribosome Biogenesis-Cancer Connection. Cells 2019, 8, 55. [Google Scholar] [CrossRef]
- Hein, N.; Hannan, K.M.; George, A.J.; Sanij, E.; Hannan, R.D. The nucleolus: An emerging target for cancer therapy. Trends Mol. Med. 2013, 19, 643–654. [Google Scholar] [CrossRef]
- Drygin, D.; Lin, A.; Bliesath, J.; Ho, C.B.; O’Brien, S.E.; Proffitt, C.; Omori, M.; Haddach, M.; Schwaebe, M.K.; Siddiqui-Jain, A.; et al. Targeting RNA Polymerase I with an Oral Small Molecule CX-5461 Inhibits Ribosomal RNA Synthesis and Solid Tumor Growth. Cancer Res. 2011, 71, 1418–1430. [Google Scholar] [CrossRef]
- Bywater, M.J.; Poortinga, G.; Sanij, E.; Hein, N.; Peck, A.; Cullinane, C.; Wall, M.; Cluse, L.; Drygin, D.; Anderes, K.; et al. Inhibition of RNA Polymerase I as a Therapeutic Strategy to Promote Cancer-Specific Activation of p53. Cancer Cell 2012, 22, 51–65. [Google Scholar] [CrossRef]
- Lee, H.C.; Wang, H.; Baladandayuthapani, V.; Lin, H.; He, J.; Jones, R.J.; Kuiatse, I.; Gu, D.; Wang, Z.; Ma, W.; et al. RNA Polymerase I Inhibition with CX-5461 as a Novel Therapeutic Strategy to Target MYC in Multiple Myeloma. Br. J. Haematol. 2017, 177, 80–94. [Google Scholar] [CrossRef]
- Hein, N.; Cameron, D.P.; Hannan, K.M.; Nguyen, N.N.; Fong, C.Y.; Sornkom, J.; Wall, M.; Pavy, M.; Cullinane, C.; Diesch, J.; et al. Inhibition of Pol I transcription treats murine and human AML by targeting the leukemia-initiating cell population. Blood 2017, 129, 2882–2895. [Google Scholar] [CrossRef]
- Kim, D.W.; Wu, N.; Kim, Y.C.; Cheng, P.F.; Basom, R.; Kim, D.; Dunn, C.T.; Lee, A.Y.; Kim, K.; Lee, C.S.; et al. Genetic requirement for Mycl and efficacy of RNA Pol I inhibition in mouse models of small cell lung cancer. Genes Dev. 2016, 30, 1289–1299. [Google Scholar] [CrossRef]
- Lawrence, M.G.; Obinata, D.; Sandhu, S.; Selth, L.A.; Wong, S.Q.; Porter, L.H.; Lister, N.; Pook, D.; Pezaro, C.J.; Goode, D.L.; et al. Patient-derived Models of Abiraterone- and Enzalutamide-resistant Prostate Cancer Reveal Sensitivity to Ribosome-directed Therapy. Eur. Urol. 2018, 74, 562–572. [Google Scholar] [CrossRef]
- Cornelison, R.; Dobbin, Z.C.; Katre, A.A.; Jeong, D.H.; Zhang, Y.; Chen, D.; Petrova, Y.; Llaneza, D.C.; Steg, A.D.; Parsons, L.; et al. Targeting RNA-Polymerase I in Both Chemosensitive and Chemoresistant Populations in Epithelial Ovarian Cancer. Clin. Cancer Res. 2017, 23, 6529–6540. [Google Scholar] [CrossRef] [PubMed]
- Bruno, P.M.; Liu, Y.; Park, G.Y.; Murai, J.; Koch, C.E.; Eisen, T.J.; Pritchard, J.R.; Pommier, Y.; Lippard, S.J.; Hemann, M.T. A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat. Med. 2017, 23, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Burger, K.; Muhl, B.; Harasim, T.; Rohrmoser, M.; Malamoussi, A.; Orban, M.; Kellner, M.; Gruber-Eber, A.; Kremmer, E.; Holzel, M.; et al. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J. Biol. Chem. 2010, 285, 12416–12425. [Google Scholar] [CrossRef]
- Khot, A.; Brajanovski, N.; Cameron, D.P.; Hein, N.; Maclachlan, K.H.; Sanij, E.; Lim, J.; Soong, J.; Link, E.; Blombery, P.; et al. First-in-Human RNA Polymerase I Transcription Inhibitor CX-5461 in Patients with Advanced Hematologic Cancers: Results of a Phase I Dose-Escalation Study. Cancer Discov. 2019, 9, 1036–1049. [Google Scholar] [CrossRef]
- Hilton, J.; Cescon, D.W.; Bedard, P.; Ritter, H.; Tu, D.; Soong, J.; Gelmon, K.; Aparicio, S.; Seymour, L. CCTG IND. 231: A phase 1 trial evaluating CX-5461 in patients with advanced solid tumors. Ann. Oncol. 2018, 29, iii8. [Google Scholar] [CrossRef]
- Quin, J.; Chan, K.T.; Devlin, J.R.; Cameron, D.P.; Diesch, J.; Cullinane, C.; Ahern, J.; Khot, A.; Hein, N.; George, A.J.; et al. Inhibition of RNA polymerase I transcription initiation by CX-5461 activates non-canonical ATM/ATR signaling. Oncotarget 2016, 7, 49800–49818. [Google Scholar] [CrossRef]
- Negi, S.S.; Brown, P. rRNA synthesis inhibitor, CX-5461, activates ATM/ATR pathway in acute lymphoblastic leukemia, arrests cells in G2 phase and induces apoptosis. Oncotarget 2015, 6, 18094–18104. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Santos, N.D.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef]
- Li, G.; Shen, J.; Cao, J.; Zhou, G.; Lei, T.; Sun, Y.; Gao, H.; Ding, Y.; Xu, W.; Zhan, Z.; et al. Alternative splicing of human telomerase reverse transcriptase in gliomas and its modulation mediated by CX-5461. J. Exp. Clin. Cancer Res. 2018, 37, 78. [Google Scholar] [CrossRef]
- Yan, S.; Xuan, J.; Brajanovski, N.; Tancock, M.R.C.; Madhamshettiwar, P.B.; Simpson, K.J.; Ellis, S.; Kang, J.; Cullinane, C.; Sheppard, K.E.; et al. The RNA polymerase I transcription inhibitor CX-5461 cooperates with topoisomerase 1 inhibition by enhancing the DNA damage response in homologous recombination-proficient high-grade serous ovarian cancer. Br. J. Cancer 2021, 124, 616–627. [Google Scholar] [CrossRef]
- Bruno, P.M.; Lu, M.; Dennis, K.A.; Inam, H.; Moore, C.J.; Sheehe, J.; Elledge, S.J.; Hemann, M.T.; Pritchard, J.R. The primary mechanism of cytotoxicity of the chemotherapeutic agent CX-5461 is topoisomerase II poisoning. Proc. Natl. Acad. Sci. USA 2020, 117, 4053–4060. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.; Wright, W.C.; Chapple, R.H.; Zubair, A.; Sandhu, M.; Batchelder, J.E.; Huddle, B.C.; Low, J.; Blankenship, K.B.; Wang, Y.; et al. The chemotherapeutic CX-5461 primarily targets TOP2B and exhibits selective activity in high-risk neuroblastoma. Nat. Commun. 2021, 12, 6468. [Google Scholar] [CrossRef]
- Bossaert, M.; Pipier, A.; Riou, J.F.; Noirot, C.; Nguyen, L.T.; Serre, R.F.; Bouchez, O.; Defrancq, E.; Calsou, P.; Britton, S.; et al. Transcription-associated topoisomerase 2alpha (TOP2A) activity is a major effector of cytotoxicity induced by G-quadruplex ligands. eLife 2021, 10, e65184. [Google Scholar] [CrossRef] [PubMed]
- Diesch, J.; Bywater, M.J.; Sanij, E.; Cameron, D.P.; Schierding, W.; Brajanovski, N.; Son, J.; Sornkom, J.; Hein, N.; Evers, M.; et al. Changes in long-range rDNA-genomic interactions associate with altered RNA polymerase II gene programs during malignant transformation. Commun. Biol. 2019, 2, 39. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, A.J.; Porter, A.C. Construction, characterization, and complementation of a conditional-lethal DNA topoisomerase IIalpha mutant human cell line. Mol. Biol. Cell 2004, 15, 5700–5711. [Google Scholar] [CrossRef]
- Devlin, J.R.; Hannan, K.M.; Hein, N.; Cullinane, C.; Kusnadi, E.; Ng, P.Y.; George, A.J.; Shortt, J.; Bywater, M.J.; Poortinga, G.; et al. Combination Therapy Targeting Ribosome Biogenesis and mRNA Translation Synergistically Extends Survival in MYC-Driven Lymphoma. Cancer Discov. 2016, 6, 59–70. [Google Scholar] [CrossRef]
- Fellmann, C.; Hoffmann, T.; Sridhar, V.; Hopfgartner, B.; Muhar, M.; Roth, M.; Lai, D.Y.; Barbosa, I.A.; Kwon, J.S.; Guan, Y.; et al. An optimized microRNA backbone for effective single-copy RNAi. Cell Rep. 2013, 5, 1704–1713. [Google Scholar] [CrossRef]
- Zuber, J.; McJunkin, K.; Fellmann, C.; Dow, L.E.; Taylor, M.J.; Hannon, G.J.; Lowe, S.W. Toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi. Nat. Biotechnol. 2011, 29, 79–83. [Google Scholar] [CrossRef]
- Pear, W.S.; Nolan, G.P.; Scott, M.L.; Baltimore, D. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. USA 1993, 90, 8392–8396. [Google Scholar] [CrossRef]
- Kamentsky, L.; Jones, T.R.; Fraser, A.; Bray, M.A.; Logan, D.J.; Madden, K.L.; Ljosa, V.; Rueden, C.; Eliceiri, K.W.; Carpenter, A.E. Improved structure, function and compatibility for CellProfiler: Modular high-throughput image analysis software. Bioinformatics 2011, 27, 1179–1180. [Google Scholar] [CrossRef]
- Gilles, J.F.; Dos Santos, M.; Boudier, T.; Bolte, S.; Heck, N. DiAna, an ImageJ tool for object-based 3D co-localization and distance analysis. Methods 2017, 115, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L.; Soans, E.; Rogojina, A.; Seth, A.; Mishina, M. Topoisomerase assays. Curr. Protoc. Pharmacol. 2012, 57, 3. [Google Scholar] [CrossRef] [PubMed]
- Willmore, E.; Frank, A.J.; Padget, K.; Tilby, M.J.; Austin, C.A. Etoposide targets topoisomerase IIalpha and IIbeta in leukemic cells: Isoform-specific cleavable complexes visualized and quantified in situ by a novel immunofluorescence technique. Mol. Pharmacol. 1998, 54, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Cowell, I.G.; Tilby, M.J.; Austin, C.A. An overview of the visualisation and quantitation of low and high MW DNA adducts using the trapped in agarose DNA immunostaining (TARDIS) assay. Mutagenesis 2011, 26, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Panova, T.; Miller, G.; Volkov, A.; Porter, A.C.; Russell, J.; Panov, K.I.; Zomerdijk, J.C. Topoisomerase IIalpha promotes activation of RNA polymerase I transcription by facilitating pre-initiation complex formation. Nat. Commun. 2013, 4, 1598. [Google Scholar] [CrossRef]
- Ju, B.G.; Lunyak, V.V.; Perissi, V.; Garcia-Bassets, I.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 2006, 312, 1798–1802. [Google Scholar] [CrossRef] [PubMed]
- Dahl, J.A.; Collas, P. A rapid micro chromatin immunoprecipitation assay (microChIP). Nat. Protoc. 2008, 3, 1032–1045. [Google Scholar] [CrossRef]
- Nelson, J.D.; Denisenko, O.; Bomsztyk, K. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat. Protoc. 2006, 1, 179–185. [Google Scholar] [CrossRef]
- Lefebure, M.; Tothill, R.W.; Kruse, E.; Hawkins, E.D.; Shortt, J.; Matthews, G.M.; Gregory, G.P.; Martin, B.P.; Kelly, M.J.; Todorovski, I.; et al. Genomic characterisation of Emu-Myc mouse lymphomas identifies Bcor as a Myc co-operative tumour-suppressor gene. Nat. Commun. 2017, 8, 14581. [Google Scholar] [CrossRef]
- Wendorff, T.J.; Schmidt, B.H.; Heslop, P.; Austin, C.A.; Berger, J.M. The structure of DNA-bound human topoisomerase II alpha: Conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J. Mol. Biol. 2012, 424, 109–124. [Google Scholar] [CrossRef]
- Negi, S.S.; Brown, P. Transient rRNA synthesis inhibition with CX-5461 is sufficient to elicit growth arrest and cell death in acute lymphoblastic leukemia cells. Oncotarget 2015, 6, 34846–34858. [Google Scholar] [CrossRef]
- So, S.; Davis, A.J.; Chen, D.J. Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. J. Cell Biol. 2009, 187, 977–990. [Google Scholar] [CrossRef]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Canman, C.E.; Lim, D.S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar] [CrossRef] [PubMed]
- Banin, S.; Moyal, L.; Shieh, S.; Taya, Y.; Anderson, C.W.; Chessa, L.; Smorodinsky, N.I.; Prives, C.; Reiss, Y.; Shiloh, Y.; et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998, 281, 1674–1677. [Google Scholar] [CrossRef]
- Hickson, I.; Zhao, Y.; Richardson, C.J.; Green, S.J.; Martin, N.M.; Orr, A.I.; Reaper, P.M.; Jackson, S.P.; Curtin, N.J.; Smith, G.C. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004, 64, 9152–9159. [Google Scholar] [CrossRef] [PubMed]
- Tamaichi, H.; Sato, M.; Porter, A.C.; Shimizu, T.; Mizutani, S.; Takagi, M. Ataxia telangiectasia mutated-dependent regulation of topoisomerase II alpha expression and sensitivity to topoisomerase II inhibitor. Cancer Sci. 2013, 104, 178–184. [Google Scholar] [CrossRef]
- Korwek, Z.; Sewastianik, T.; Bielak-Zmijewska, A.; Mosieniak, G.; Alster, O.; Moreno-Villanueva, M.; Burkle, A.; Sikora, E. Inhibition of ATM blocks the etoposide-induced DNA damage response and apoptosis of resting human T cells. DNA Repair 2012, 11, 864–873. [Google Scholar] [CrossRef]
- Sordet, O.; Nakamura, A.J.; Redon, C.E.; Pommier, Y. DNA double-strand breaks and ATM activation by transcription-blocking DNA lesions. Cell Cycle 2010, 9, 274–278. [Google Scholar] [CrossRef]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. 2009, 9, 327–337. [Google Scholar] [CrossRef]
- Gao, R.; Schellenberg, M.J.; Huang, S.Y.; Abdelmalak, M.; Marchand, C.; Nitiss, K.C.; Nitiss, J.L.; Williams, R.S.; Pommier, Y. Proteolytic degradation of topoisomerase II (Top2) enables the processing of Top2.DNA and Top2.RNA covalent complexes by tyrosyl-DNA-phosphodiesterase 2 (TDP2). J. Biol. Chem. 2014, 289, 17960–17969. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Lyu, Y.L.; Lin, C.P.; Zhou, N.; Azarova, A.M.; Wood, L.M.; Liu, L.F. A protease pathway for the repair of topoisomerase II-DNA covalent complexes. J. Biol. Chem. 2006, 281, 35997–36003. [Google Scholar] [CrossRef] [PubMed]
- Tewey, K.M.; Rowe, T.C.; Yang, L.; Halligan, B.D.; Liu, L.F. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science 1984, 226, 466–468. [Google Scholar] [CrossRef] [PubMed]
- Kinner, A.; Wu, W.; Staudt, C.; Iliakis, G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008, 36, 5678–5694. [Google Scholar] [CrossRef]
- Bassing, C.H.; Alt, F.W. H2AX may function as an anchor to hold broken chromosomal DNA ends in close proximity. Cell Cycle 2004, 3, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.A.; Matsunaga, S.; Ma, N.; Takata, H.; Yokoyama, M.; Uchiyama, S.; Fukui, K. Fibrillarin, a nucleolar protein, is required for normal nuclear morphology and cellular growth in HeLa cells. Biochem. Biophys. Res. Commun. 2007, 360, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Tollervey, D.; Lehtonen, H.; Jansen, R.; Kern, H.; Hurt, E.C. Temperature-sensitive mutations demonstrate roles for yeast fibrillarin in pre-rRNA processing, pre-rRNA methylation, and ribosome assembly. Cell 1993, 72, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef]
- Izbicka, E.; Wheelhouse, R.T.; Raymond, E.; Davidson, K.K.; Lawrence, R.A.; Sun, D.; Windle, B.E.; Hurley, L.H.; Von Hoff, D.D. Effects of cationic porphyrins as G-quadruplex interactive agents in human tumor cells. Cancer Res. 1999, 59, 639–644. [Google Scholar]
- Olivieri, M.; Cho, T.; Alvarez-Quilon, A.; Li, K.; Schellenberg, M.J.; Zimmermann, M.; Hustedt, N.; Rossi, S.E.; Adam, S.; Melo, H.; et al. A Genetic Map of the Response to DNA Damage in Human Cells. Cell 2020, 182, 481–496.e21. [Google Scholar] [CrossRef]
- Bywater, M.J.; Pearson, R.B.; McArthur, G.A.; Hannan, R.D. Dysregulation of the basal RNA polymerase transcription apparatus in cancer. Nat. Rev. 2013, 13, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Johnson-Arbor, K.; Dubey, R. StatPearls: Doxorubicin. In Doxorubicin; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Reyhanoglu, G.; Tadi, P. StatPearls: Etoposide. In Etoposide; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Advani, R.H.; Hurwitz, H.I.; Gordon, M.S.; Ebbinghaus, S.W.; Mendelson, D.S.; Wakelee, H.A.; Hoch, U.; Silverman, J.A.; Havrilla, N.A.; Berman, C.J.; et al. Voreloxin, a first-in-class anticancer quinolone derivative, in relapsed/refractory solid tumors: A report on two dosing schedules. Clin. Cancer Res. 2010, 16, 2167–2175. [Google Scholar] [CrossRef] [PubMed]
- Do, P.M.; Varanasi, L.; Fan, S.; Li, C.; Kubacka, I.; Newman, V.; Chauhan, K.; Daniels, S.R.; Boccetta, M.; Garrett, M.R.; et al. Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes. Dev. 2012, 26, 830–845. [Google Scholar] [CrossRef] [PubMed]
- Aas, T.; Borresen, A.L.; Geisler, S.; Smith-Sorensen, B.; Johnsen, H.; Varhaug, J.E.; Akslen, L.A.; Lonning, P.E. Specific P53 mutations are associated with de novo resistance to doxorubicin in breast cancer patients. Nat. Med. 1996, 2, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Yasuda, H. Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J. 1999, 18, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Deisenroth, C.; Zhang, Y. Ribosome biogenesis surveillance: Probing the ribosomal protein-Mdm2-p53 pathway. Oncogene 2010, 29, 4253–4260. [Google Scholar] [CrossRef]
- Miliani de Marval, P.L.; Zhang, Y. The RP-Mdm2-p53 pathway and tumorigenesis. Oncotarget 2011, 2, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Hannan, K.M.; Soo, P.; Wong, M.S.; Lee, J.K.; Hein, N.; Poh, P.; Wysoke, K.D.; Williams, T.D.; Montellese, C.; Smith, L.K.; et al. Nuclear stabilization of p53 requires a functional nucleolar surveillance pathway. Cell Rep. 2022, 41, 111571. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. 2009, 9, 338–350. [Google Scholar] [CrossRef]
- Das, S.K.; Kuzin, V.; Cameron, D.P.; Sanford, S.; Jha, R.K.; Nie, Z.; Rosello, M.T.; Holewinski, R.; Andresson, T.; Wisniewski, J.; et al. MYC assembles and stimulates topoisomerases 1 and 2 in a “topoisome”. Mol. Cell 2022, 82, 140–158.e112. [Google Scholar] [CrossRef]
- Rubbi, C.P.; Milner, J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. Embo J. 2003, 22, 6068–6077. [Google Scholar] [CrossRef]
- Woods, S.J.; Hannan, K.M.; Pearson, R.B.; Hannan, R.D. The nucleolus as a fundamental regulator of the p53 response and a new target for cancer therapy. Biochim. Biophys. Acta 2015, 1849, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.; D’Costa, Z.; Ray, S.; Gorski, J.; Harkin, D.P.; Mullan, P.; Panov, K.I. The identification of a novel role for BRCA1 in regulating RNA polymerase I transcription. Oncotarget 2016, 7, 68097. [Google Scholar] [CrossRef] [PubMed]
- van Sluis, M.; McStay, B. Nucleolar reorganization in response to rDNA damage. Curr. Opin. Cell Biol. 2017, 46, 81–86. [Google Scholar] [CrossRef]
- Koh, G.C.C.; Boushaki, S.; Zhao, S.J.; Pregnall, A.M.; Sadiyah, F.; Badja, C.; Memari, Y.; Georgakopoulos-Soares, I.; Nik-Zainal, S. The chemotherapeutic drug CX-5461 is a potent mutagen in cultured human cells. Nat. Genet. 2024, 56, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Haddach, M.; Schwaebe, M.K.; Michaux, J.; Nagasawa, J.; O’Brien, S.E.; Whitten, J.P.; Pierre, F.; Kerdoncuff, P.; Darjania, L.; Stansfield, R.; et al. Discovery of CX-5461, the First Direct and Selective Inhibitor of RNA Polymerase I, for Cancer Therapeutics. ACS Med. Chem. Lett. 2012, 3, 602–606. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cameron, D.P.; Sornkom, J.; Alsahafi, S.; Drygin, D.; Poortinga, G.; McArthur, G.A.; Hein, N.; Hannan, R.; Panov, K.I. CX-5461 Preferentially Induces Top2α-Dependent DNA Breaks at Ribosomal DNA Loci. Biomedicines 2024, 12, 1514. https://doi.org/10.3390/biomedicines12071514
Cameron DP, Sornkom J, Alsahafi S, Drygin D, Poortinga G, McArthur GA, Hein N, Hannan R, Panov KI. CX-5461 Preferentially Induces Top2α-Dependent DNA Breaks at Ribosomal DNA Loci. Biomedicines. 2024; 12(7):1514. https://doi.org/10.3390/biomedicines12071514
Chicago/Turabian StyleCameron, Donald P., Jirawas Sornkom, Sameerh Alsahafi, Denis Drygin, Gretchen Poortinga, Grant A. McArthur, Nadine Hein, Ross Hannan, and Konstantin I. Panov. 2024. "CX-5461 Preferentially Induces Top2α-Dependent DNA Breaks at Ribosomal DNA Loci" Biomedicines 12, no. 7: 1514. https://doi.org/10.3390/biomedicines12071514
APA StyleCameron, D. P., Sornkom, J., Alsahafi, S., Drygin, D., Poortinga, G., McArthur, G. A., Hein, N., Hannan, R., & Panov, K. I. (2024). CX-5461 Preferentially Induces Top2α-Dependent DNA Breaks at Ribosomal DNA Loci. Biomedicines, 12(7), 1514. https://doi.org/10.3390/biomedicines12071514