
CRISPR Base Editing to Create Potential Charcot–Marie–Tooth Disease Models with High Editing Efficiency: Human Induced Pluripotent Stem Cell Harboring SH3TC2 Variants
 , , , , , , , and
, , , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Editing Strategy: Plasmids and Oligonucleotides
2.2. Cell Culture and Transfection of HEK-293T
2.3. Genotyping of HEK-293T Cells
2.4. Culture and Nucleofection of hiPSCs
2.5. Flow Cytometry: hiPS Cells Sorting
2.6. hiPSC Clones Screening
2.7. Bystander Editing Analysis
2.8. Off-Target Site Selection and Amplicon Design
2.9. hiPSCs Characterization
3. Results
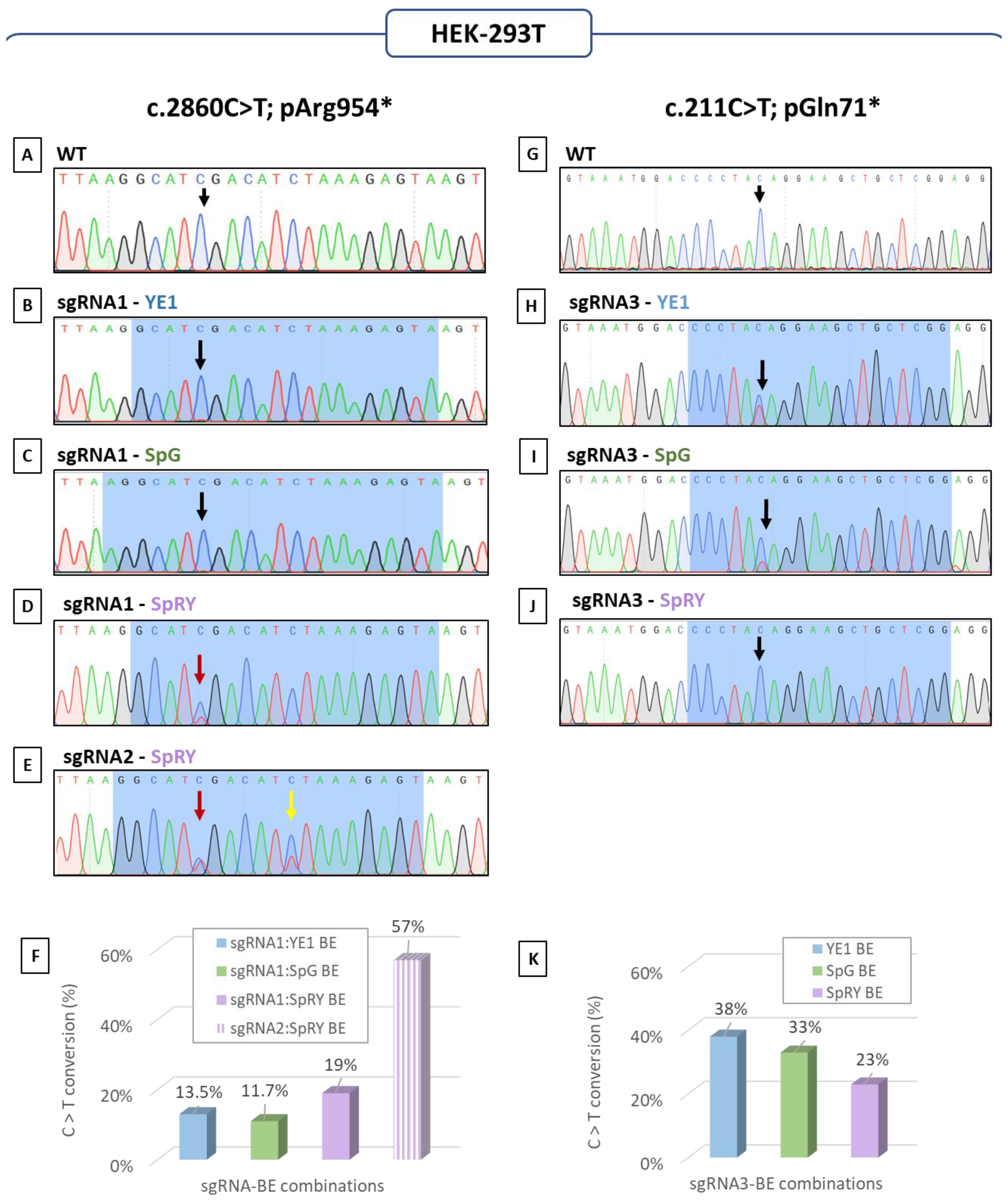
3.1. Base Editing: How to Quickly Determine the Best CRISPR Strategy Using HEK-293T Cells
3.2. Assessment of Base Editing Efficiency
3.3. Attempt for Generating SH3TC2 Alterations: From HEK-293T Cells to hiPSCs
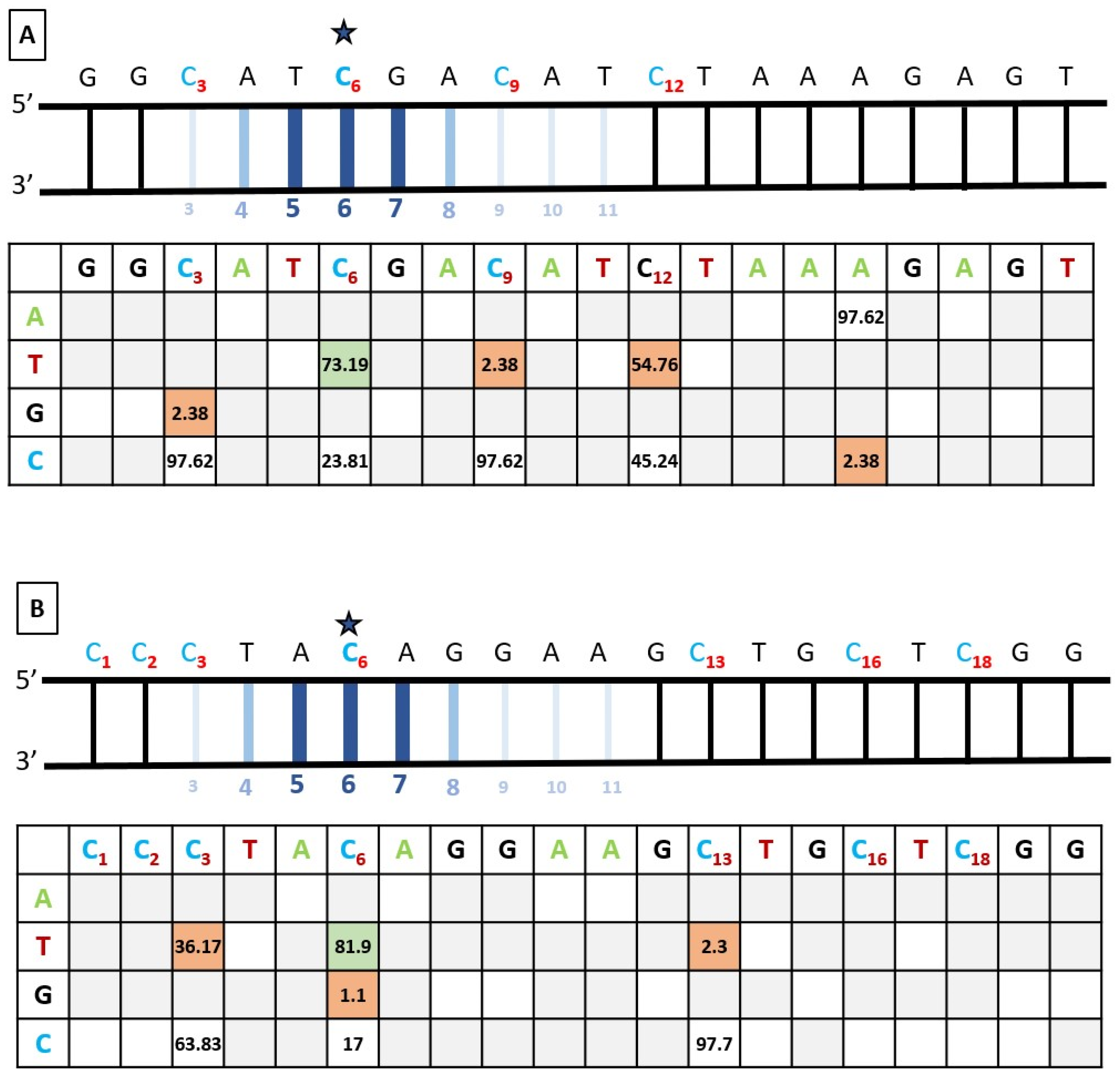
3.4. Bystander Editing
3.5. Off-Target Mutagenesis
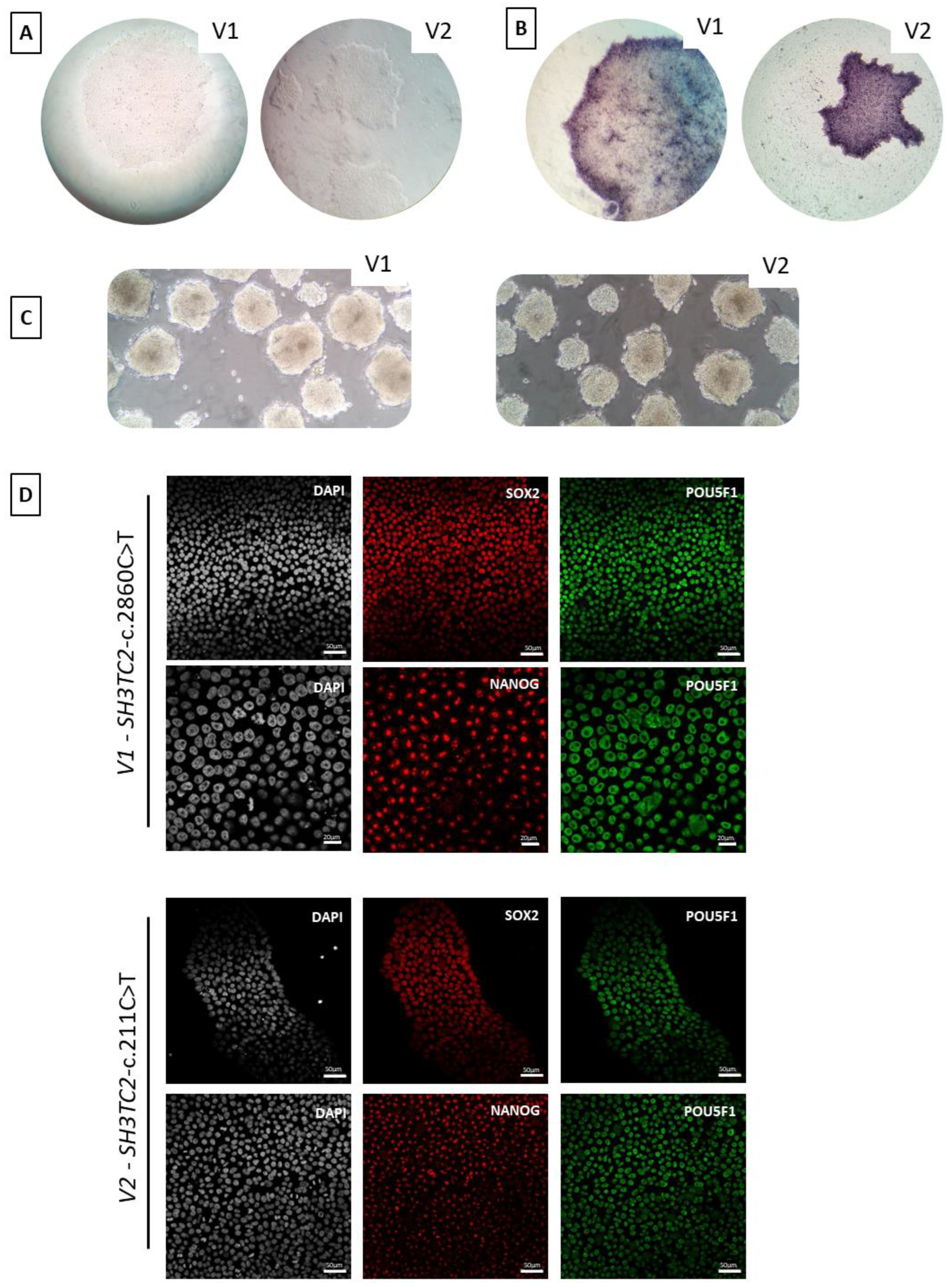
3.6. hiPSC Characterization
4. Discussion
4.1. SpRY Base Editor Combined with sgRNA2 Allowed an Unexpected High Editing Efficiency
4.2. Editing Efficiency Divergence between HEK-293T and hiPSCs
4.3. Base Editing outside the Activity Window and Consequences
4.4. Towards Improved Neural Models to Study Neurogenerative Disorders
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Faye, P.-A.; Vedrenne, N.; Miressi, F.; Rassat, M.; Romanenko, S.; Richard, L.; Bourthoumieu, S.; Funalot, B.; Sturtz, F.; Favreau, F.; et al. Optimized Protocol to Generate Spinal Motor Neuron Cells from Induced Pluripotent Stem Cells from Charcot Marie Tooth Patients. Brain Sci. 2020, 10, 407. [Google Scholar] [CrossRef] [PubMed]
- Sances, S.; Bruijn, L.I.; Chandran, S.; Eggan, K.; Ho, R.; Klim, J.R.; Livesey, M.R.; Lowry, E.; Macklis, J.D.; Rushton, D.; et al. Modeling ALS with Motor Neurons Derived from Human Induced Pluripotent Stem Cells. Nat. Neurosci. 2016, 19, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Hörner, S.J.; Couturier, N.; Bruch, R.; Koch, P.; Hafner, M.; Rudolf, R. hiPSC-Derived Schwann Cells Influence Myogenic Differentiation in Neuromuscular Cocultures. Cells 2021, 10, 3292. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. The New Frontier of Genome Engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Akram, F.; Sahreen, S.; Aamir, F.; ul Haq, I.; Malik, K.; Imtiaz, M.; Naseem, W.; Nasir, N.; Waheed, H.M. An Insight into Modern Targeted Genome-Editing Technologies with a Special Focus on CRISPR/Cas9 and Its Applications. Mol. Biotechnol. 2022, 65, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A. The promise and challenge of therapeutic genome editing. Nature 2020, 578, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Gersbach, C.A. Genome-Editing Technologies for Gene and Cell Therapy. Mol. Ther. 2016, 24, 430–446. [Google Scholar] [CrossRef]
- Cullot, G.; Boutin, J.; Toutain, J.; Prat, F.; Pennamen, P.; Rooryck, C.; Teichmann, M.; Rousseau, E.; Lamrissi-Garcia, I.; Guyonnet-Duperat, V.; et al. CRISPR-Cas9 Genome Editing Induces Megabase-Scale Chromosomal Truncations. Nat. Commun. 2019, 10, 1136. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable Editing of a Target Base in Genomic DNA without Double-Stranded DNA Cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef]
- Doman, J.L.; Raguram, A.; Newby, G.A.; Liu, D.R. Evaluation and Minimization of Cas9-Independent off-Target DNA Editing by Cytosine Base Editors. Nat. Biotechnol. 2020, 38, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Walton, R.T.; Christie, K.A.; Whittaker, M.N.; Kleinstiver, B.P. Unconstrained Genome Targeting with Near-PAMless Engineered CRISPR-Cas9 Variants. Science 2020, 368, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Ito, F.; Fu, Y.; Kao, S.-C.A.; Yang, H.; Chen, X.S. Family-Wide Comparative Analysis of Cytidine and Methylcytidine Deamination by Eleven Human APOBEC Proteins. J. Mol. Biol. 2017, 429, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Daer, R.M.; Cutts, J.P.; Brafman, D.A.; Haynes, K.A. The Impact of Chromatin Dynamics on Cas9-Mediated Genome Editing in Human Cells. ACS Synth. Biol. 2017, 6, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Skre, H. Genetic and Clinical Aspects of Charcot-Marie-Tooth’s Disease. Clin. Genet. 1974, 6, 98–118. [Google Scholar] [CrossRef]
- Harding, A.E.; Thomas, P.K. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 1980, 103, 259–280. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, V.; Strickland, A.V.; Züchner, S. Genetics of Charcot-Marie-Tooth (CMT) Disease within the Frame of the Human Genome Project Success. Genes 2014, 5, 13–32. [Google Scholar] [CrossRef]
- Azzedine, H.; Salih, M.A. SH3TC2-Related Hereditary Motor and Sensory Neuropathy. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Fridman, V.; Bundy, B.; Reilly, M.M.; Pareyson, D.; Bacon, C.; Burns, J.; Day, J.; Feely, S.; Finkel, R.S.; Grider, T.; et al. CMT Subtypes and Disease Burden in Patients Enrolled in the Inherited Neuropathies Consortium Natural History Study: A Cross-Sectional Analysis. J. Neurol. Neurosurg. Psychiatry 2015, 86, 873–878. [Google Scholar] [CrossRef]
- Piscosquito, G.; Saveri, P.; Magri, S.; Ciano, C.; Gandioli, C.; Morbin, M.; Bella, D.D.; Moroni, I.; Taroni, F.F.; Pareyson, D. Screening for SH3TC2 Gene Mutations in a Series of Demyelinating Recessive Charcot-Marie-Tooth Disease (CMT4). J. Peripher. Nerv. Syst. 2016, 21, 142–149. [Google Scholar] [CrossRef]
- Lerat, J.; Magdelaine, C.; Lunati, A.; Dzugan, H.; Dejoie, C.; Rego, M.; Beze Beyrie, P.; Bieth, E.; Calvas, P.; Cintas, P.; et al. Implication of the SH3TC2 Gene in Charcot-Marie-Tooth Disease Associated with Deafness and/or Scoliosis: Illustration with Four New Pathogenic Variants. J. Neurol. Sci. 2019, 406, 116376. [Google Scholar] [CrossRef]
- Rehbein, T.; Wu, T.T.; Treidler, S.; Pareyson, D.; Lewis, R.; Yum, S.W.; McCray, B.A.; Ramchandren, S.; Burns, J.; Li, J.; et al. Neuropathy Due to Bi-Allelic SH3TC2 Variants: Genotype-Phenotype Correlation and Natural History. Brain 2023, 146, 3826–3835. [Google Scholar] [CrossRef] [PubMed]
- Bacquet, J.; Stojkovic, T.; Boyer, A.; Martini, N.; Audic, F.; Chabrol, B.; Salort-Campana, E.; Delmont, E.; Desvignes, J.-P.; Verschueren, A.; et al. Molecular Diagnosis of Inherited Peripheral Neuropathies by Targeted Next-Generation Sequencing: Molecular Spectrum Delineation. BMJ Open 2018, 8, e021632. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Ren, Y.; Xu, X.; Wang, H.; Wei, C. Application of Allele Specific PCR in Identifying Offspring Genotypes of Bi-Allelic SbeIIb Mutant Lines in Rice. Plants 2022, 11, 524. [Google Scholar] [CrossRef] [PubMed]
- Miressi, F.; Benslimane, N.; Favreau, F.; Rassat, M.; Richard, L.; Bourthoumieu, S.; Laroche, C.; Magy, L.; Magdelaine, C.; Sturtz, F.; et al. GDAP1 Involvement in Mitochondrial Function and Oxidative Stress, Investigated in a Charcot-Marie-Tooth Model of hiPSCs-Derived Motor Neurons. Biomedicines 2021, 9, 945. [Google Scholar] [CrossRef] [PubMed]
- van der Burg, M.; Kreyenberg, H.; Willasch, A.; Barendregt, B.H.; Preuner, S.; Watzinger, F.; Lion, T.; Roosnek, E.; Harvey, J.; Alcoceba, M.; et al. Standardization of DNA Isolation from Low Cell Numbers for Chimerism Analysis by PCR of Short Tandem Repeats. Leukemia 2011, 25, 1467–1470. [Google Scholar] [CrossRef] [PubMed]
- Concordet, J.P.; Haeussler, M. CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018, 46, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Hallmann, A.-L.; Araúzo-Bravo, M.J.; Mavrommatis, L.; Ehrlich, M.; Röpke, A.; Brockhaus, J.; Missler, M.; Sterneckert, J.; Schöler, H.R.; Kuhlmann, T.; et al. Astrocyte Pathology in a Human Neural Stem Cell Model of Frontotemporal Dementia Caused by Mutant TAU Protein. Sci. Rep. 2017, 7, 42991. [Google Scholar] [CrossRef]
- Allende, M.L.; Cook, E.K.; Larman, B.C.; Nugent, A.; Brady, J.M.; Golebiowski, D.; Sena-Esteves, M.; Tifft, C.J.; Proia, R.L. Cerebral Organoids Derived from Sandhoff Disease-Induced Pluripotent Stem Cells Exhibit Impaired Neurodifferentiation. J. Lipid Res. 2018, 59, 550–563. [Google Scholar] [CrossRef]
- Cui, L.; Zheng, J.; Zhao, Q.; Chen, J.-R.; Liu, H.; Peng, G.; Wu, Y.; Chen, C.; He, Q.; Shi, H.; et al. Mutations of MAP1B Encoding a Microtubule-Associated Phosphoprotein Cause Sensorineural Hearing Loss. JCI Insight 2020, 5, e136046. [Google Scholar] [CrossRef]
- Molla, K.A.; Yang, Y. CRISPR/Cas-Mediated Base Editing: Technical Considerations and Practical Applications. Trends Biotechnol. 2019, 37, 1121–1142. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.; Zheng, Z.; Joung, J.K. High-Fidelity CRISPR-Cas9 Variants with Undetectable Genome-Wide off-Targets. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Hwang, G.-H.; Park, J.; Lim, K.; Kim, S.; Yu, J.; Yu, E.; Kim, S.-T.; Eils, R.; Kim, J.-S.; Bae, S. Web-Based Design and Analysis Tools for CRISPR Base Editing. BMC Bioinform. 2018, 19, 542. [Google Scholar] [CrossRef] [PubMed]
- Arbab, M.; Shen, M.W.; Mok, B.; Wilson, C.; Matuszek, Ż.; Cassa, C.A.; Liu, D.R. Determinants of Base Editing Outcomes from Target Library Analysis and Machine Learning. Cell 2020, 182, 463–480.e30. [Google Scholar] [CrossRef] [PubMed]
- Pallaseni, A.; Peets, E.M.; Koeppel, J.; Weller, J.; Vanderstichele, T.; Ho, U.L.; Crepaldi, L.; van Leeuwen, J.; Allen, F.; Parts, L. Predicting Base Editing Outcomes Using Position-Specific Sequence Determinants. Nucleic Acids Res. 2022, 50, 3551–3564. [Google Scholar] [CrossRef] [PubMed]
- Vicencio, J.; Sánchez-Bolaños, C.; Moreno-Sánchez, I.; Brena, D.; Vejnar, C.E.; Kukhtar, D.; Ruiz-López, M.; Cots-Ponjoan, M.; Rubio, A.; Melero, N.R.; et al. Genome Editing in Animals with Minimal PAM CRISPR-Cas9 Enzymes. Nat. Commun. 2022, 13, 2601. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Zhang, Y.; Li, L.; Yang, Y.; Fei, J.-F.; Liu, Y.; Qin, W. SpG and SpRY Variants Expand the CRISPR Toolbox for Genome Editing in Zebrafish. Nat. Commun. 2022, 13, 3421. [Google Scholar] [CrossRef]
- Rosello, M.; Serafini, M.; Mignani, L.; Finazzi, D.; Giovannangeli, C.; Mione, M.C.; Concordet, J.-P.; Del Bene, F. Disease Modeling by Efficient Genome Editing Using a near PAM-Less Base Editor In Vivo. Nat. Commun. 2022, 13, 3435. [Google Scholar] [CrossRef]
- Moreno-Mateos, M.A.; Vejnar, C.E.; Beaudoin, J.-D.; Fernandez, J.P.; Mis, E.K.; Khokha, M.K.; Giraldez, A.J. CRISPRscan: Designing Highly Efficient sgRNAs for CRISPR-Cas9 Targeting In Vivo. Nat. Methods 2015, 12, 982–988. [Google Scholar] [CrossRef]
- Sürün, D.; Schneider, A.; Mircetic, J.; Neumann, K.; Lansing, F.; Paszkowski-Rogacz, M.; Hänchen, V.; Lee-Kirsch, M.A.; Buchholz, F. Efficient Generation and Correction of Mutations in Human iPS Cells Utilizing mRNAs of CRISPR Base Editors and Prime Editors. Genes 2020, 11, 511. [Google Scholar] [CrossRef]
- Yu, C.; Chen, J.; Ma, J.; Zang, L.; Dong, F.; Sun, J.; Zheng, M. Identification of Key Genes and Signaling Pathways Associated with the Progression of Gastric Cancer. Pathol. Oncol. Res. 2019, 26, 1903–1919. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Sretenovic, S.; Liu, S.; Tang, X.; Huang, L.; He, Y.; Liu, L.; Guo, Y.; Zhong, Z.; Liu, G.; et al. PAM-Less Plant Genome Editing Using a CRISPR-SpRY Toolbox. Nat. Plants 2021, 7, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Beale, R.C.L.; Petersen-Mahrt, S.K.; Watt, I.N.; Harris, R.S.; Rada, C.; Neuberger, M.S. Comparison of the Differential Context-Dependence of DNA Deamination by APOBEC Enzymes: Correlation with Mutation Spectra In Vivo. J. Mol. Biol. 2004, 337, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Saraconi, G.; Severi, F.; Sala, C.; Mattiuz, G.; Conticello, S.G. The RNA Editing Enzyme APOBEC1 Induces Somatic Mutations and a Compatible Mutational Signature Is Present in Esophageal Adenocarcinomas. Genome Biol. 2014, 15, 417. [Google Scholar] [CrossRef]
- Sharma, J.; Du, M.; Wong, E.; Mutyam, V.; Li, Y.; Chen, J.; Wangen, J.; Thrasher, K.; Fu, L.; Peng, N.; et al. A Small Molecule That Induces Translational Readthrough of CFTR Nonsense Mutations by eRF1 Depletion. Nat. Commun. 2021, 12, 4358. [Google Scholar] [CrossRef] [PubMed]
- Saporta, M.A.; Dang, V.; Volfson, D.; Zou, B.; Xie, X.; Adebola, A.; Liem, R.K.; Shy, M.; Dimos, J.T. Axonal Charcot-Marie-Tooth Disease Patient-Derived Motor Neurons Demonstrate Disease-Specific Phenotypes Including Abnormal Electrophysiological Properties. Exp. Neurol. 2015, 263, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Kropf, E.; Hou, Y.-M.; Iacovitti, L. A Stress-Free Strategy to Correct Point Mutations in Patient iPS Cells. Stem Cell Res. 2021, 53, 102332. [Google Scholar] [CrossRef] [PubMed]
- Feliciano, C.M.; Wu, K.; Watry, H.L.; Marley, C.B.E.; Ramadoss, G.N.; Ghanim, H.Y.; Liu, A.Z.; Zholudeva, L.V.; McDevitt, T.C.; Saporta, M.A.; et al. Allele-Specific Gene Editing Rescues Pathology in a Human Model of Charcot-Marie-Tooth Disease Type 2E. Front. Cell Dev. Biol. 2021, 9, 723023. [Google Scholar] [CrossRef]
- Van Lent, J.; Verstraelen, P.; Asselbergh, B.; Adriaenssens, E.; Mateiu, L.; Verbist, C.; De Winter, V.; Eggermont, K.; Van Den Bosch, L.; De Vos, W.H.; et al. Induced Pluripotent Stem Cell-Derived Motor Neurons of CMT Type 2 Patients Reveal Progressive Mitochondrial Dysfunction. Brain 2021, 144, 2471–2485. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loret, C.; Pauset, A.; Faye, P.-A.; Prouzet-Mauleon, V.; Pyromali, I.; Nizou, A.; Miressi, F.; Sturtz, F.; Favreau, F.; Turcq, B.; et al. CRISPR Base Editing to Create Potential Charcot–Marie–Tooth Disease Models with High Editing Efficiency: Human Induced Pluripotent Stem Cell Harboring SH3TC2 Variants. Biomedicines 2024, 12, 1550. https://doi.org/10.3390/biomedicines12071550
Loret C, Pauset A, Faye P-A, Prouzet-Mauleon V, Pyromali I, Nizou A, Miressi F, Sturtz F, Favreau F, Turcq B, et al. CRISPR Base Editing to Create Potential Charcot–Marie–Tooth Disease Models with High Editing Efficiency: Human Induced Pluripotent Stem Cell Harboring SH3TC2 Variants. Biomedicines. 2024; 12(7):1550. https://doi.org/10.3390/biomedicines12071550
Chicago/Turabian StyleLoret, Camille, Amandine Pauset, Pierre-Antoine Faye, Valérie Prouzet-Mauleon, Ioanna Pyromali, Angélique Nizou, Federica Miressi, Franck Sturtz, Frédéric Favreau, Béatrice Turcq, and et al. 2024. "CRISPR Base Editing to Create Potential Charcot–Marie–Tooth Disease Models with High Editing Efficiency: Human Induced Pluripotent Stem Cell Harboring SH3TC2 Variants" Biomedicines 12, no. 7: 1550. https://doi.org/10.3390/biomedicines12071550