
Diet and the Gut Microbiome as Determinants Modulating Metabolic Outcomes in Young Obese Adults
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
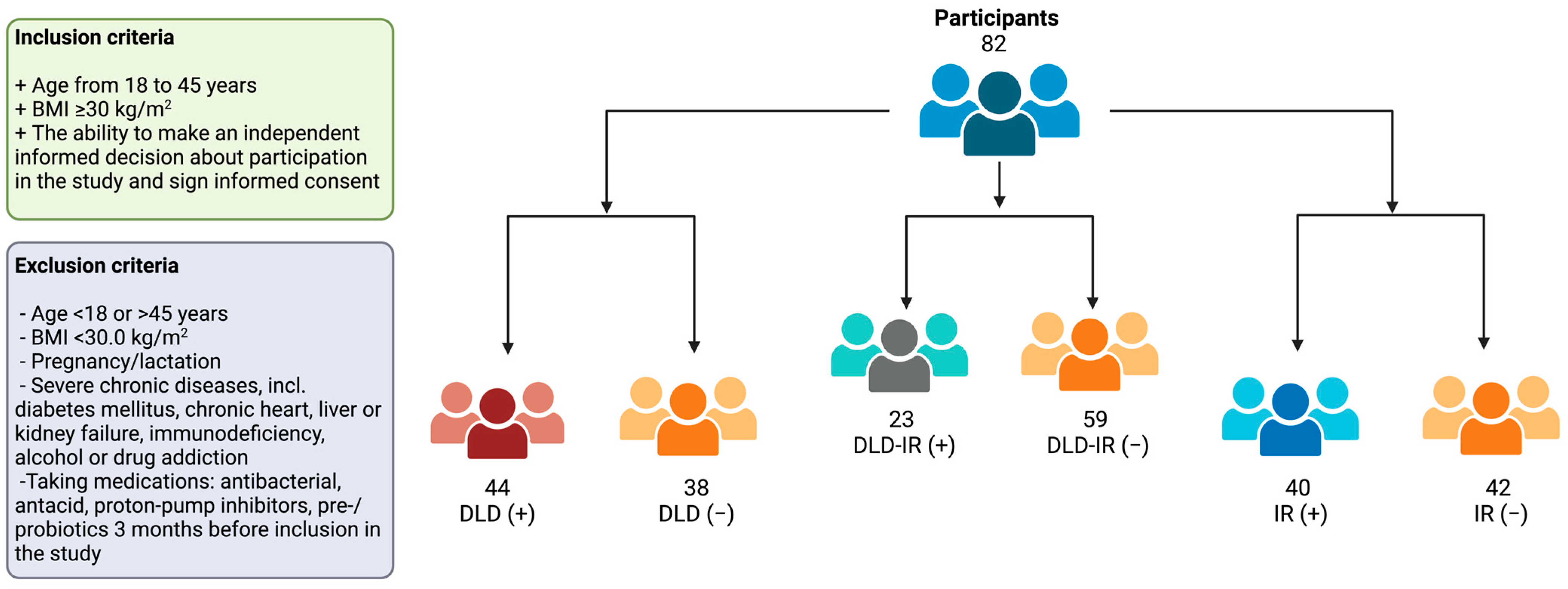
2.1. Subjects and Study Design
2.2. Diet Assessment
2.3. Anthropometry, Body Composition and Biochemical Indicators
2.4. Fecal Sample Collection
2.5. DNA Extraction, Library Construction and Sequencing
2.6. Gut Microbiota Composition Profiling
2.7. Statistical Analysis
3. Results
3.1. Characteristics of Carbohydrate and Lipid Metabolism
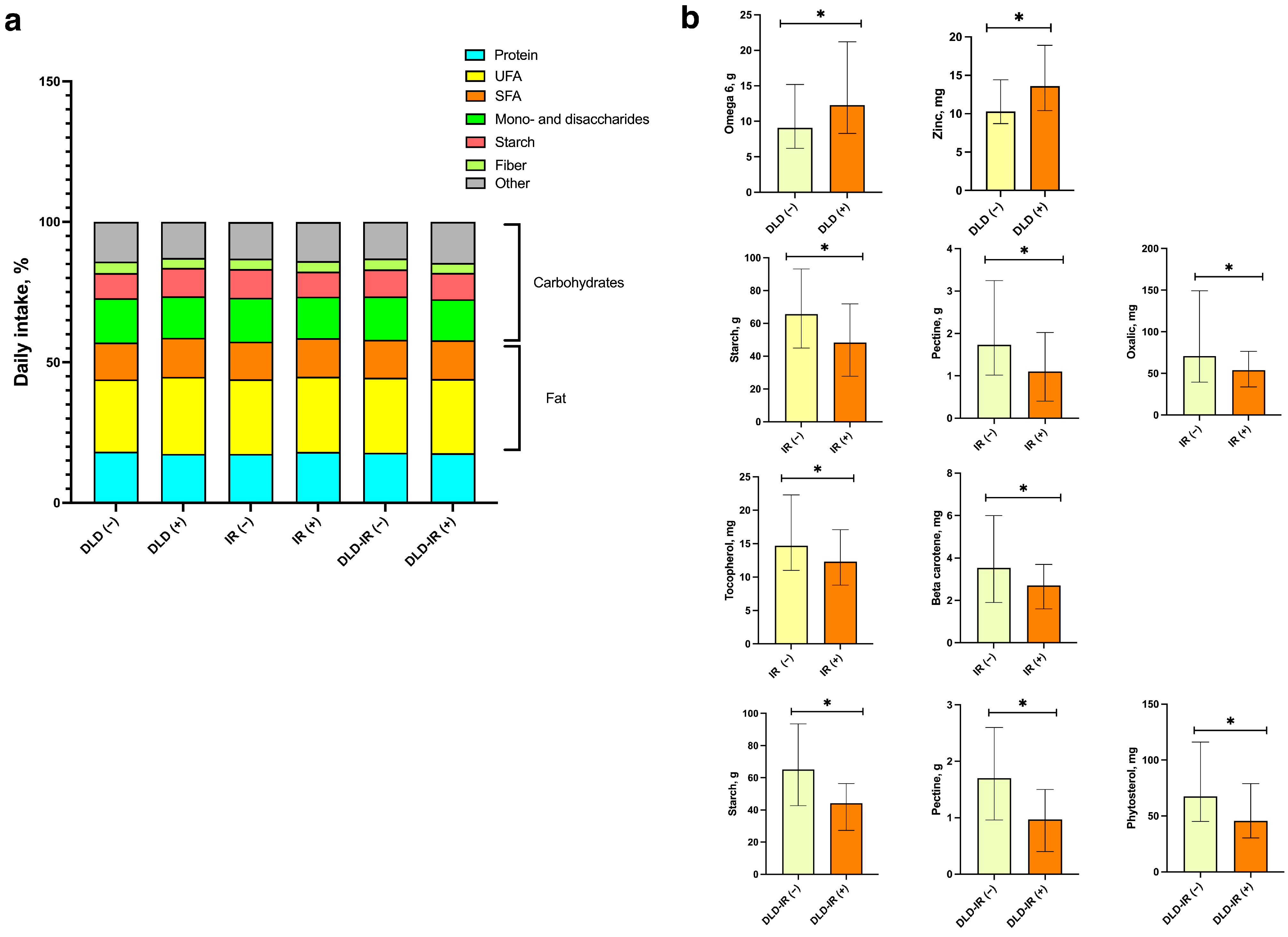
3.2. Diet
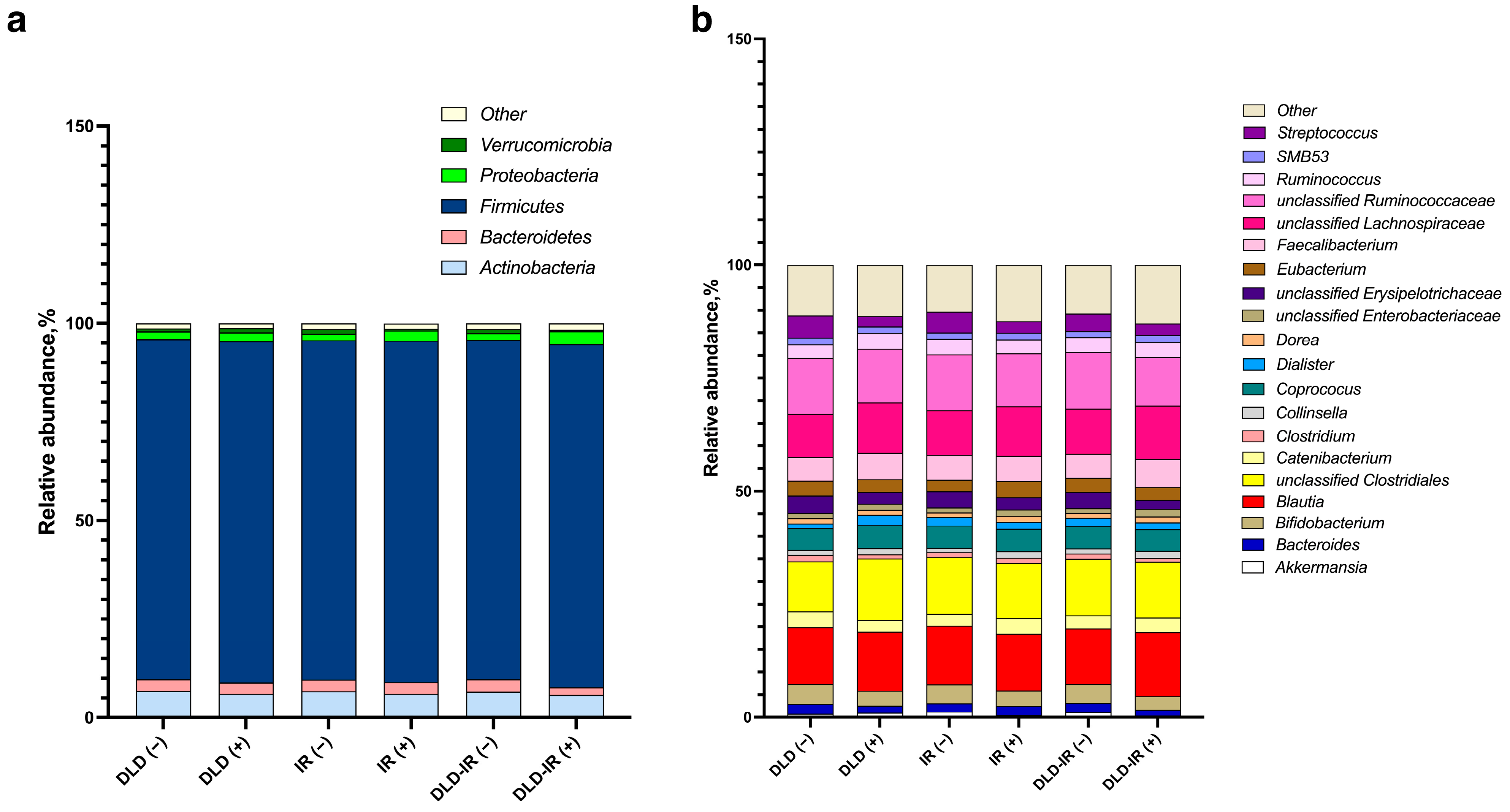
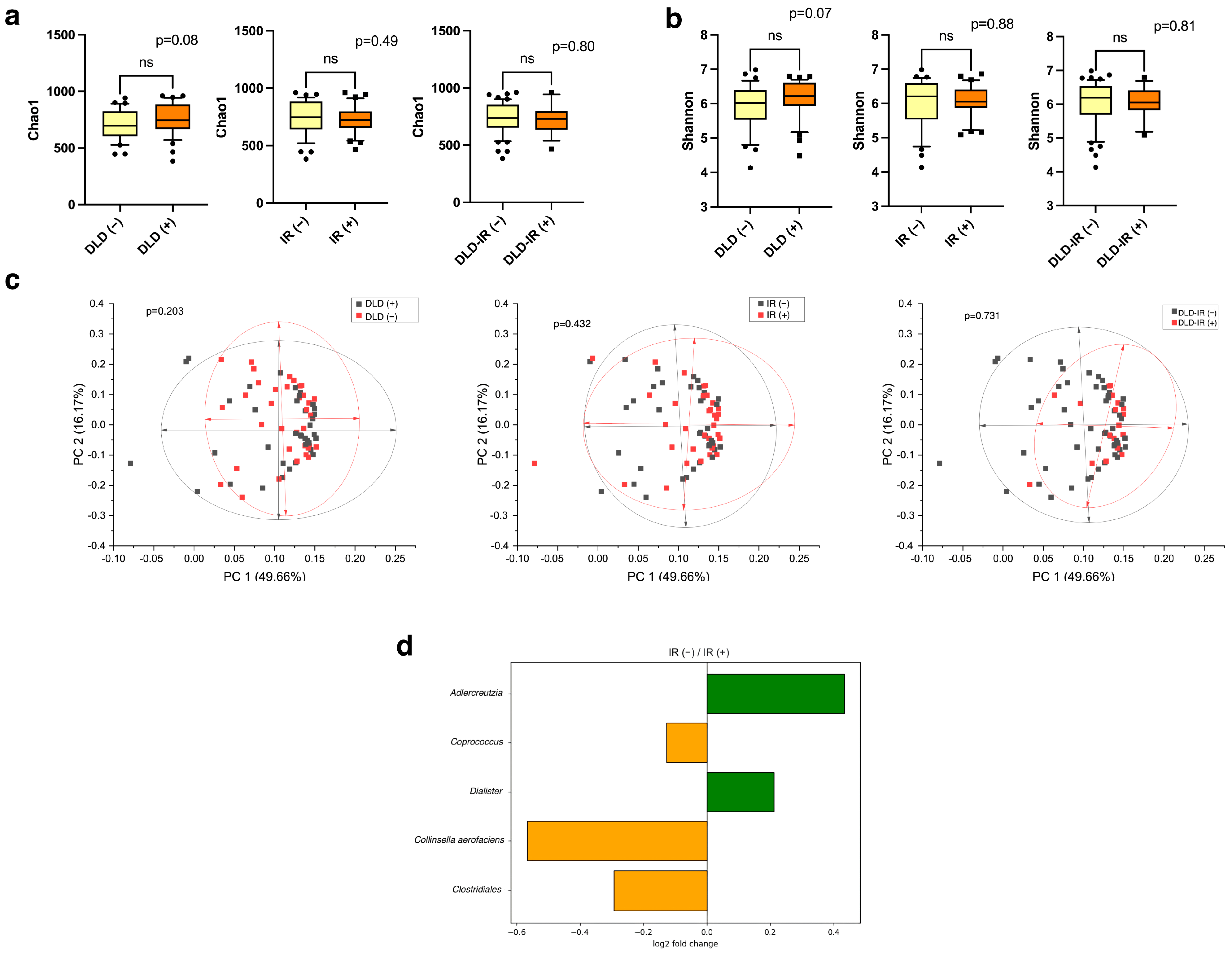
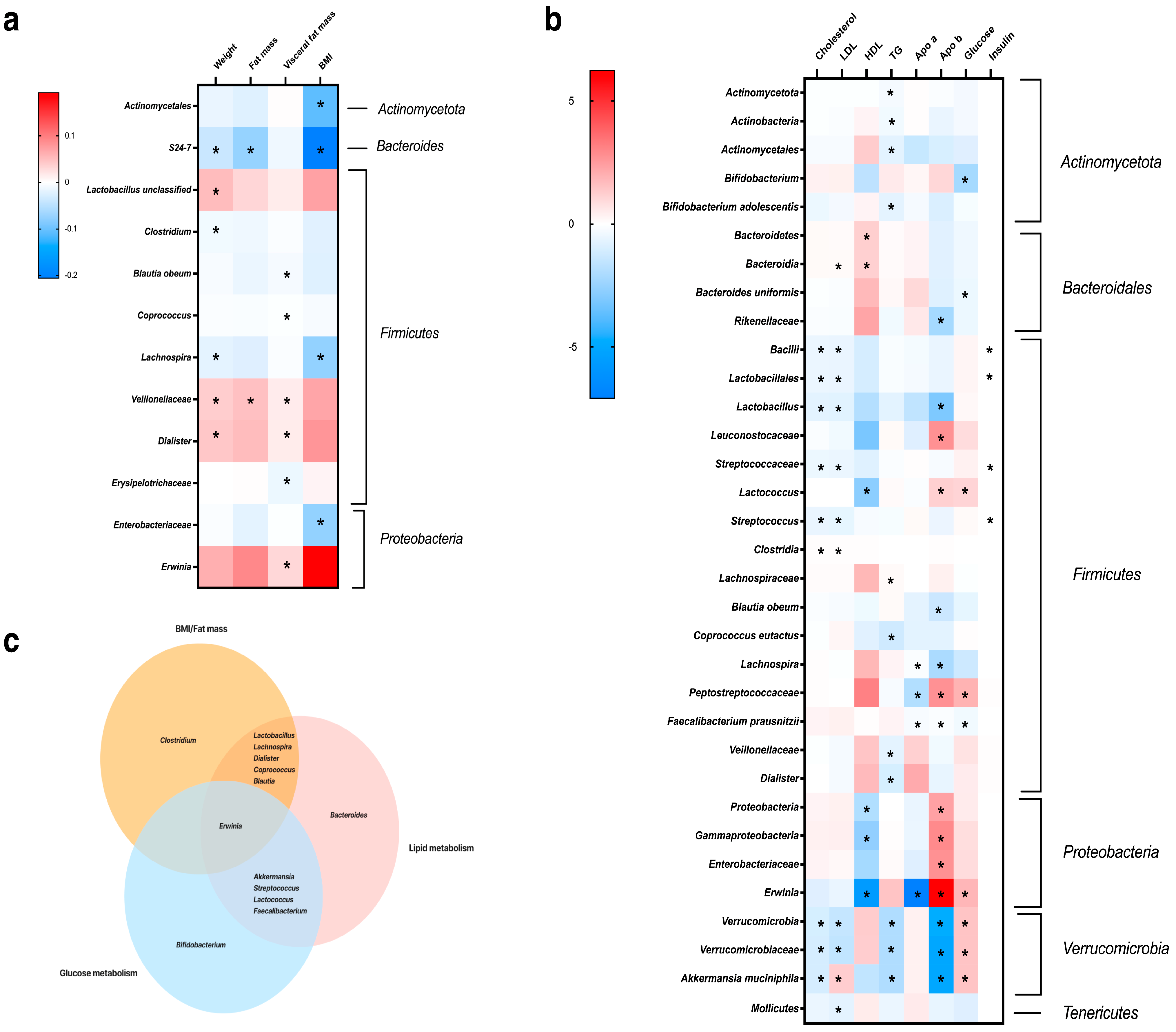
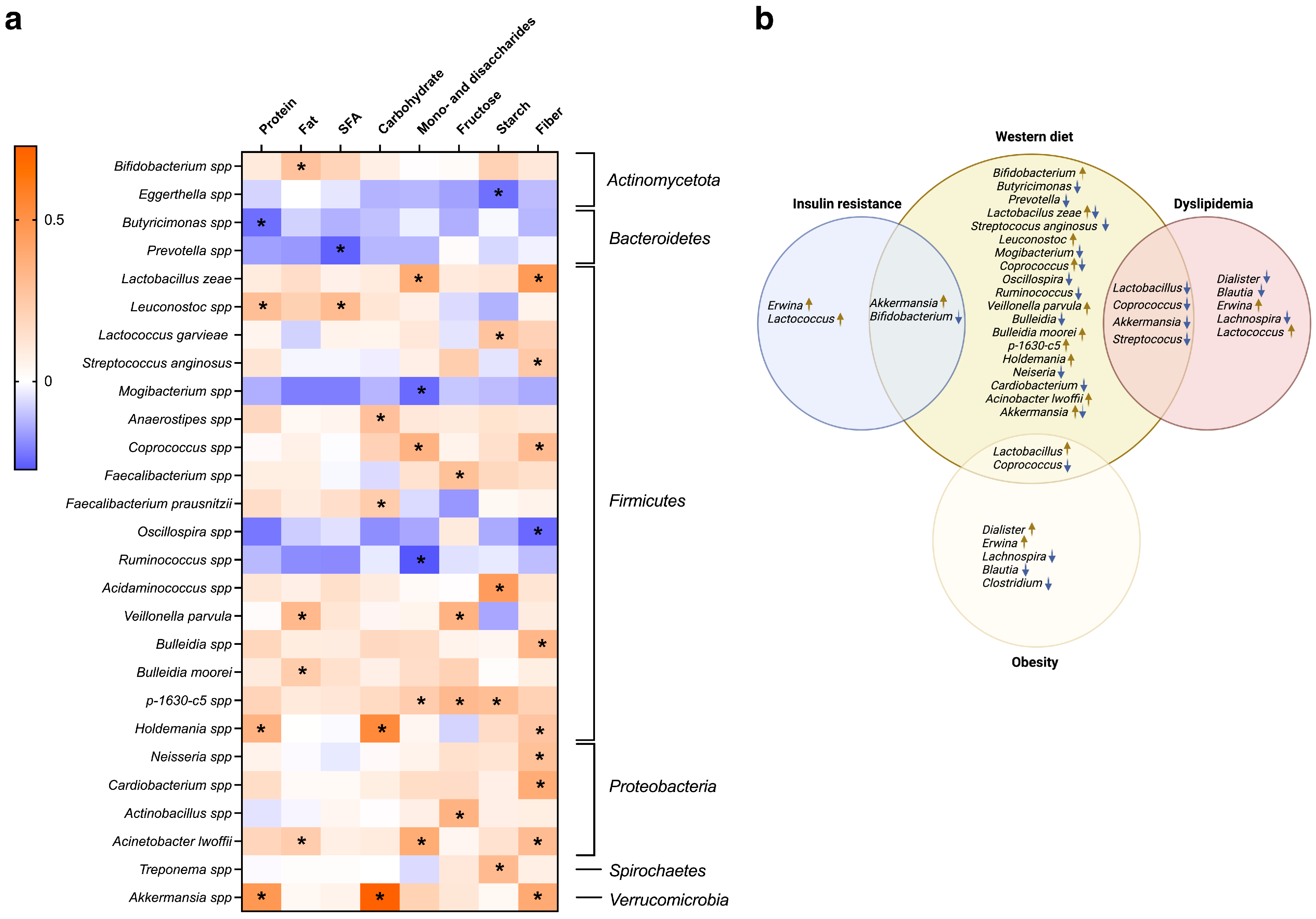
3.3. Association of Gut Microbiota Composition and Dietary Variation with Clinical Parameters and Metabolism
4. Discussion
Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fruh, S.M. Obesity: Risk Factors, Complications, and Strategies for Sustainable Long-term Weight Management. J. Am. Assoc. Nurse Pract. 2017, 29, S3. [Google Scholar] [CrossRef]
- Silva-Ochoa, A.D.; Velasteguí, E.; Falconí, I.B.; García-Solorzano, V.I.; Rendón-Riofrio, A.; Sanguña-Soliz, G.A.; Vanden Berghe, W.; Orellana-Manzano, A. Metabolic Syndrome: Nutri-Epigenetic Cause or Consequence? Heliyon 2023, 9, e21106. [Google Scholar] [CrossRef]
- Daniel, H. Diet and Gut Microbiome and the “Chicken or Egg” Problem. Front. Nutr. 2022, 8, 828630. [Google Scholar] [CrossRef]
- Wu, J.; Wang, K.; Wang, X.; Pang, Y.; Jiang, C. The Role of the Gut Microbiome and Its Metabolites in Metabolic Diseases. Protein Cell 2021, 12, 360–373. [Google Scholar] [CrossRef]
- Leonov, G.E.; Varaeva, Y.R.; Livantsova, E.N.; Starodubova, A.V. The Complicated Relationship of Short-Chain Fatty Acids and Oral Microbiome: A Narrative Review. Biomedicines 2023, 11, 2749. [Google Scholar] [CrossRef]
- Cuevas-Sierra, A.; Ramos-Lopez, O.; Riezu-Boj, J.I.; Milagro, F.I.; Martinez, J.A. Diet, Gut Microbiota, and Obesity: Links with Host Genetics and Epigenetics and Potential Applications. Adv. Nutr. 2019, 10, S17–S30. [Google Scholar] [CrossRef]
- Durazzi, F.; Sala, C.; Castellani, G.; Manfreda, G.; Remondini, D.; De Cesare, A. Comparison between 16S rRNA and Shotgun Sequencing Data for the Taxonomic Characterization of the Gut Microbiota. Sci. Rep. 2021, 11, 3030. [Google Scholar] [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What Is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The Firmicutes/Bacteroidetes Ratio: A Relevant Marker of Gut Dysbiosis in Obese Patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Bonfrate, L.; Khalil, M.; Garruti, G.; Portincasa, P. Contribution of the Microbiome for Better Phenotyping of People Living with Obesity. Rev. Endocr. Metab. Disord. 2023, 24, 839–870. [Google Scholar] [CrossRef]
- Geng, J.; Ni, Q.; Sun, W.; Li, L.; Feng, X. The Links between Gut Microbiota and Obesity and Obesity Related Diseases. Biomed. Pharmacother. 2022, 147, 112678. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, Z.; He, Y.; Li, P.; Zhou, H.; Zeng, N. Regional Distribution of Christensenellaceae and Its Associations with Metabolic Syndrome Based on a Population-Level Analysis. PeerJ 2020, 8, e9591. [Google Scholar] [CrossRef]
- Niu, H.; Zhou, M.; Zogona, D.; Xing, Z.; Wu, T.; Chen, R.; Cui, D.; Liang, F.; Xu, X. Akkermansia Muciniphila: A Potential Candidate for Ameliorating Metabolic Diseases. Front. Immunol. 2024, 15, 1370658. [Google Scholar] [CrossRef] [PubMed]
- Tsigalou, C.; Paraschaki, A.; Karvelas, A.; Kantartzi, K.; Gagali, K.; Tsairidis, D.; Bezirtzoglou, E. Gut Microbiome and Mediterranean Diet in the Context of Obesity. Current Knowledge, Perspectives and Potential Therapeutic Targets. Metab. Open 2021, 9, 100081. [Google Scholar] [CrossRef] [PubMed]
- Gundogdu, A.; Nalbantoglu, O.U. The Role of the Mediterranean Diet in Modulating the Gut Microbiome: A Review of Current Evidence. Nutrition 2023, 114, 112118. [Google Scholar] [CrossRef] [PubMed]
- Kohnert, E.; Kreutz, C.; Binder, N.; Hannibal, L.; Gorkiewicz, G.; Müller, A.; Storz, M.A.; Huber, R.; Lederer, A.-K. Changes in Gut Microbiota after a Four-Week Intervention with Vegan vs. Meat-Rich Diets in Healthy Participants: A Randomized Controlled Trial. Microorganisms 2021, 9, 727. [Google Scholar] [CrossRef] [PubMed]
- Odermatt, A. The Western-Style Diet: A Major Risk Factor for Impaired Kidney Function and Chronic Kidney Disease. Am. J. Physiol. Ren. Physiol. 2011, 301, F919–F931. [Google Scholar] [CrossRef] [PubMed]
- Severino, A.; Tohumcu, E.; Tamai, L.; Dargenio, P.; Porcari, S.; Rondinella, D.; Venturini, I.; Maida, M.; Gasbarrini, A.; Cammarota, G.; et al. The Microbiome-Driven Impact of Western Diet in the Development of Noncommunicable Chronic Disorders. Best Pract. Res. Clin. Gastroenterol. 2024; in press. [Google Scholar] [CrossRef]
- Makki, K.; Deehan, E.C.; Walter, J.; Bäckhed, F. The Impact of Dietary Fiber on Gut Microbiota in Host Health and Disease. Cell Host Microbe 2018, 23, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Qin, C.; Wen, M. The Effects of Food Nutrients and Bioactive Compounds on the Gut Microbiota: A Comprehensive Review. Foods 2024, 13, 1345. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.S.; Seekatz, A.M.; Koropatkin, N.M.; Kamada, N.; Hickey, C.A.; Wolter, M.; Pudlo, N.A.; Kitamoto, S.; Terrapon, N.; Muller, A.; et al. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 2016, 167, 1339–1353.e21. [Google Scholar] [CrossRef]
- Chen, C.; Chen, W.; Ding, H.; Wu, P.; Zhang, G.; Xie, K.; Zhang, T. High-Fat Diet-Induced Gut Microbiota Alteration Promotes Lipogenesis by Butyric Acid/miR-204/ACSS2 Axis in Chickens. Poult. Sci. 2023, 102, 102856. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P. Influence of Foods and Nutrition on the Gut Microbiome and Implications for Intestinal Health. Int. J. Mol. Sci. 2022, 23, 9588. [Google Scholar] [CrossRef] [PubMed]
- Mu, C.; Yang, Y.; Luo, Z.; Zhu, W. Temporal Microbiota Changes of High-Protein Diet Intake in a Rat Model. Anaerobe 2017, 47, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Salvadori, M.; Rosso, G. Update on the Gut Microbiome in Health and Diseases. World J. Methodol. 2024, 14, 89196. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.; Zhang, K.; Paul, K.C.; Folle, A.D.; Del Rosario, I.; Jacobs, J.P.; Keener, A.M.; Bronstein, J.M.; Ritz, B. Diet and the Gut Microbiome in Patients with Parkinson’s Disease. NPJ Park. Dis. 2024, 10, 89. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, S.; Müller, A.; Kohnert, E.; Mehrbarzin, N.; Huber, R.; Häcker, G.; Kreutz, C.; Lederer, A.-K.; Badr, M.T. Longitudinal Dynamics of Gut Bacteriome and Mycobiome Interactions Pre- and Post-Visceral Surgery in Crohn’s Disease. Front. Cell. Infect. Microbiol. 2024, 13, 1275405. [Google Scholar] [CrossRef] [PubMed]
- Mantri, A.; Klümpen, L.; Seel, W.; Krawitz, P.; Stehle, P.; Weber, B.; Koban, L.; Plassmann, H.; Simon, M.-C. Beneficial Effects of Synbiotics on the Gut Microbiome in Individuals with Low Fiber Intake: Secondary Analysis of a Double-Blind, Randomized Controlled Trial. Nutrients 2024, 16, 2082. [Google Scholar] [CrossRef] [PubMed]
- Horvath, A.; Zukauskaite, K.; Hazia, O.; Balazs, I.; Stadlbauer, V. Human Gut Microbiome: Therapeutic Opportunities for Metabolic Syndrome—Hype or Hope? Endocrinol. Diabetes Metab. 2024, 7, e436. [Google Scholar] [CrossRef]
- Popova, A.Y.; Tutelyan, V.A.; Nikityuk, D.V. On the new (2021) Norms of physiological requirements in energy and nutrients of various groups of the population of the Russian Federation. Vopr. Pitan. 2021, 90, 6–19. [Google Scholar] [CrossRef]
- Yu, Z.; Morrison, M. Improved Extraction of PCR-Quality Community DNA from Digesta and Fecal Samples. BioTechniques 2004, 36, 808–812. [Google Scholar] [CrossRef]
- Efimova, D.; Tyakht, A.; Popenko, A.; Vasilyev, A.; Altukhov, I.; Dovidchenko, N.; Odintsova, V.; Klimenko, N.; Loshkarev, R.; Pashkova, M.; et al. Knomics-Biota—A System for Exploratory Analysis of Human Gut Microbiota Data. BioData Min. 2018, 11, 25. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing Data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive Functional Profiling of Microbial Communities Using 16S rRNA Marker Gene Sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Brandt, B.W.; Bonder, M.J.; Huse, S.M.; Zaura, E. TaxMan: A Server to Trim rRNA Reference Databases and Inspect Taxonomic Coverage. Nucleic Acids Res. 2012, 40, W82–W87. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Chávez-Carbajal, A.; Nirmalkar, K.; Pérez-Lizaur, A.; Hernández-Quiroz, F.; Ramírez-del-Alto, S.; García-Mena, J.; Hernández-Guerrero, C. Gut Microbiota and Predicted Metabolic Pathways in a Sample of Mexican Women Affected by Obesity and Obesity Plus Metabolic Syndrome. Int. J. Mol. Sci. 2019, 20, 438. [Google Scholar] [CrossRef]
- Thingholm, L.B.; Rühlemann, M.C.; Koch, M.; Fuqua, B.; Laucke, G.; Boehm, R.; Bang, C.; Franzosa, E.A.; Hübenthal, M.; Rahnavard, A.; et al. Obese Individuals with and without Type 2 Diabetes Show Different Gut Microbial Functional Capacity and Composition. Cell Host Microbe 2019, 26, 252–264.e10. [Google Scholar] [CrossRef]
- Sroka-Oleksiak, A.; Młodzińska, A.; Bulanda, M.; Salamon, D.; Major, P.; Stanek, M.; Gosiewski, T. Metagenomic Analysis of Duodenal Microbiota Reveals a Potential Biomarker of Dysbiosis in the Course of Obesity and Type 2 Diabetes: A Pilot Study. J. Clin. Med. 2020, 9, 369. [Google Scholar] [CrossRef]
- Naderpoor, N.; Mousa, A.; Gomez-Arango, L.F.; Barrett, H.L.; Dekker Nitert, M.; de Courten, B. Faecal Microbiota Are Related to Insulin Sensitivity and Secretion in Overweight or Obese Adults. J. Clin. Med. 2019, 8, 452. [Google Scholar] [CrossRef]
- Ciubotaru, I.; Green, S.J.; Kukreja, S.; Barengolts, E. Significant Differences in Fecal Microbiota Are Associated with Various Stages of Glucose Tolerance in African American Male Veterans. Transl. Res. J. Lab. Clin. Med. 2015, 166, 401–411. [Google Scholar] [CrossRef]
- Yan, H.; Qin, Q.; Chen, J.; Yan, S.; Li, T.; Gao, X.; Yang, Y.; Li, A.; Ding, S. Gut Microbiome Alterations in Patients with Visceral Obesity Based on Quantitative Computed Tomography. Front. Cell. Infect. Microbiol. 2022, 11, 823262. [Google Scholar] [CrossRef]
- Deng, K.; Shuai, M.; Zhang, Z.; Jiang, Z.; Fu, Y.; Shen, L.; Zheng, J.-S.; Chen, Y.-M. Temporal Relationship among Adiposity, Gut Microbiota, and Insulin Resistance in a Longitudinal Human Cohort. BMC Med. 2022, 20, 171. [Google Scholar] [CrossRef]
- Choi, B.S.-Y.; Daniel, N.; Houde, V.P.; Ouellette, A.; Marcotte, B.; Varin, T.V.; Vors, C.; Feutry, P.; Ilkayeva, O.; Ståhlman, M.; et al. Feeding Diversified Protein Sources Exacerbates Hepatic Insulin Resistance via Increased Gut Microbial Branched-Chain Fatty Acids and mTORC1 Signaling in Obese Mice. Nat. Commun. 2021, 12, 3377. [Google Scholar] [CrossRef]
- Oñate, F.P.; Chamignon, C.; Burz, S.D.; Lapaque, N.; Monnoye, M.; Philippe, C.; Bredel, M.; Chêne, L.; Farin, W.; Paillarse, J.-M.; et al. Adlercreutzia Equolifaciens Is an Anti-Inflammatory Commensal Bacterium with Decreased Abundance in Gut Microbiota of Patients with Metabolic Liver Disease. Int. J. Mol. Sci. 2023, 24, 12232. [Google Scholar] [CrossRef]
- Gomez-Arango, L.F.; Barrett, H.L.; Wilkinson, S.A.; Callaway, L.K.; McIntyre, H.D.; Morrison, M.; Dekker Nitert, M. Low Dietary Fiber Intake Increases Collinsella Abundance in the Gut Microbiota of Overweight and Obese Pregnant Women. Gut Microbes 2018, 9, 189–201. [Google Scholar] [CrossRef]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The Impact of the Gut Microbiota on Human Health: An Integrative View. Cell 2012, 148, 1258. [Google Scholar] [CrossRef]
- Wang, H.; Lv, X.; Zhao, S.; Yuan, W.; Zhou, Q.; Sadiq, F.A.; Zhao, J.; Lu, W.; Wu, W. Weight Loss Promotion in Individuals with Obesity through Gut Microbiota Alterations with a Multiphase Modified Ketogenic Diet. Nutrients 2023, 15, 4163. [Google Scholar] [CrossRef]
- Hur, H.J.; Wu, X.; Yang, H.J.; Kim, M.J.; Lee, K.-H.; Hong, M.; Park, S.; Kim, M.-S. Beneficial Effects of a Low-Glycemic Diet on Serum Metabolites and Gut Microbiota in Obese Women with Prevotella and Bacteriodes Enterotypes: A Randomized Clinical Trial. Front. Nutr. 2022, 9, 861880. [Google Scholar] [CrossRef]
- Bolte, L.A.; Vich Vila, A.; Imhann, F.; Collij, V.; Gacesa, R.; Peters, V.; Wijmenga, C.; Kurilshikov, A.; Campmans-Kuijpers, M.J.E.; Fu, J.; et al. Long-Term Dietary Patterns Are Associated with Pro-Inflammatory and Anti-Inflammatory Features of the Gut Microbiome. Gut 2021, 70, 1287–1298. [Google Scholar] [CrossRef]
- Cui, J.; Ramesh, G.; Wu, M.; Jensen, E.T.; Crago, O.; Bertoni, A.G.; Gao, C.; Hoffman, K.L.; Sheridan, P.A.; Wong, K.E.; et al. Butyrate-Producing Bacteria and Insulin Homeostasis: The Microbiome and Insulin Longitudinal Evaluation Study (MILES). Diabetes 2022, 71, 2438–2446. [Google Scholar] [CrossRef]
- Caputo, M.; Pigni, S.; Antoniotti, V.; Agosti, E.; Caramaschi, A.; Antonioli, A.; Aimaretti, G.; Manfredi, M.; Bona, E.; Prodam, F. Targeting Microbiota in Dietary Obesity Management: A Systematic Review on Randomized Control Trials in Adults. Crit. Rev. Food Sci. Nutr. 2023, 63, 11449–11481. [Google Scholar] [CrossRef]
- Lakshmanan, A.P.; Al Zaidan, S.; Bangarusamy, D.K.; Al-Shamari, S.; Elhag, W.; Terranegra, A. Increased Relative Abundance of Ruminoccocus Is Associated with Reduced Cardiovascular Risk in an Obese Population. Front. Nutr. 2022, 9, 849005. [Google Scholar] [CrossRef]
- Cai, Y.; Kang, Y. Gut Microbiota and Metabolites in Diabetic Retinopathy: Insights into Pathogenesis for Novel Therapeutic Strategies. Biomed. Pharmacother. 2023, 164, 114994. [Google Scholar] [CrossRef]
- Naqvi, S.; Asar, T.O.; Kumar, V.; Al-Abbasi, F.A.; Alhayyani, S.; Kamal, M.A.; Anwar, F. A Cross-Talk between Gut Microbiome, Salt and Hypertension—ScienceDirect. Biomed. Pharmacother. 2021, 134, 111156. [Google Scholar] [CrossRef]
- Gong, H.; Gao, H.; Ren, Q.; He, J. The Abundance of Bifidobacterium in Relation to Visceral Obesity and Serum Uric Acid. Sci. Rep. 2022, 12, 13073. [Google Scholar] [CrossRef]
- Yu, D.; Nguyen, S.M.; Yang, Y.; Xu, W.; Cai, H.; Wu, J.; Cai, Q.; Long, J.; Zheng, W.; Shu, X.-O. Long-Term Diet Quality Is Associated with Gut Microbiome Diversity and Composition among Urban Chinese Adults. Am. J. Clin. Nutr. 2021, 113, 684–694. [Google Scholar] [CrossRef]
- Dao, M.C.; Everard, A.; Aron-Wisnewsky, J.; Sokolovska, N.; Prifti, E.; Verger, E.O.; Kayser, B.D.; Levenez, F.; Chilloux, J.; Hoyles, L.; et al. Akkermansia Muciniphila and Improved Metabolic Health during a Dietary Intervention in Obesity: Relationship with Gut Microbiome Richness and Ecology. Gut 2016, 65, 426–436. [Google Scholar] [CrossRef]
- Chen, Y.; Lin, H.; Cole, M.; Morris, A.; Martinson, J.; Mckay, H.; Mimiaga, M.; Margolick, J.; Fitch, A.; Methe, B.; et al. Signature Changes in Gut Microbiome Are Associated with Increased Susceptibility to HIV-1 Infection in MSM. Microbiome 2021, 9, 237. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Mean (95% CI)/n (%) |
|---|---|
| Age | 34.0 (32.4; 35.9) |
| Gender | |
| male | 28 (34.1) |
| female | 54 (65.9) |
| Obesity | |
| Class I | 41 (50.0) |
| Class II | 25 (30.5) |
| Class III | 16 (19.5) |
| Smoking status | |
| Current smokers | 22 (26.8) |
| Non-smokers | 60 (73.2) |
| Dyslipidemia | 44 (53.7) |
| Insulin resistance | 40 (48.8) |
| Hyperuricemia | 38 (46.3) |
| Hypertension | 24 (29.3) |
| Parameter | Mean (95% CI) |
|---|---|
| Body weight, kg | 105.02 (100.57; 109.48) |
| BMI, kg/m2 | 35.80 (34.71; 36.97) |
| Waist circumference, cm | 109.7 (107.1; 112.3) |
| Fat mass, kg | 45.61 (43.02; 48.20) |
| Visceral adipose tissue area, cm2 | 204.72 (195.45; 213.98) |
| Muscle mass, kg | 33.53 (31.72; 35.34) |
| Total body water, L | 43.55 (41.36; 45.74) |
| Extracellular fluid, L | 16,46 (15.64; 17.28) |
| Parameter | Mean (95% CI) | Reference |
|---|---|---|
| Glucose, mmol/L | 5.05 (4.94; 5.15) | 3.9–5.6 |
| AST, U/L | 22.65 (20.40; 24.90) | 8–33 |
| ALT, U/L | 29.45 (24.72; 34.18) | 7–56 |
| Uric acid, μmol/L | 356.61 (338.28; 374.94) | M 110–420; W 110–360 |
| TC, mmol/L | 5.19 (4.98; 5.41) | 3.5–5.2 |
| TG, mmol/L | 1.49 (1.31; 1.68) | 0.68–1.69 |
| LDL cholesterol, mmol/L | 3.43 (3.24; 3.62) | <3.2 |
| HDL cholesterol, mmol/L | 1.17 (1.13; 1.22) | 0.9–1.2 |
| Insulin, μIU/mL | 14.35 (12.43; 16.26) | 2.6–24.9 |
| HOMA index | 3.26 (2.79; 3.73) | ≤2.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Livantsova, E.N.; Leonov, G.E.; Starodubova, A.V.; Varaeva, Y.R.; Vatlin, A.A.; Koshechkin, S.I.; Korotkova, T.N.; Nikityuk, D.B. Diet and the Gut Microbiome as Determinants Modulating Metabolic Outcomes in Young Obese Adults. Biomedicines 2024, 12, 1601. https://doi.org/10.3390/biomedicines12071601
Livantsova EN, Leonov GE, Starodubova AV, Varaeva YR, Vatlin AA, Koshechkin SI, Korotkova TN, Nikityuk DB. Diet and the Gut Microbiome as Determinants Modulating Metabolic Outcomes in Young Obese Adults. Biomedicines. 2024; 12(7):1601. https://doi.org/10.3390/biomedicines12071601
Chicago/Turabian StyleLivantsova, Elena N., Georgy E. Leonov, Antonina V. Starodubova, Yurgita R. Varaeva, Aleksey A. Vatlin, Stanislav I. Koshechkin, Tatyana N. Korotkova, and Dmitry B. Nikityuk. 2024. "Diet and the Gut Microbiome as Determinants Modulating Metabolic Outcomes in Young Obese Adults" Biomedicines 12, no. 7: 1601. https://doi.org/10.3390/biomedicines12071601