
Risk Assessment and Personalized Treatment Options in Inherited Dilated Cardiomyopathies: A Narrative Review
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Defining Hereditary Dilated Cardiomyopathy (DCM)
3. Diagnosis of Hereditary DCM
3.1. Clinical Picture
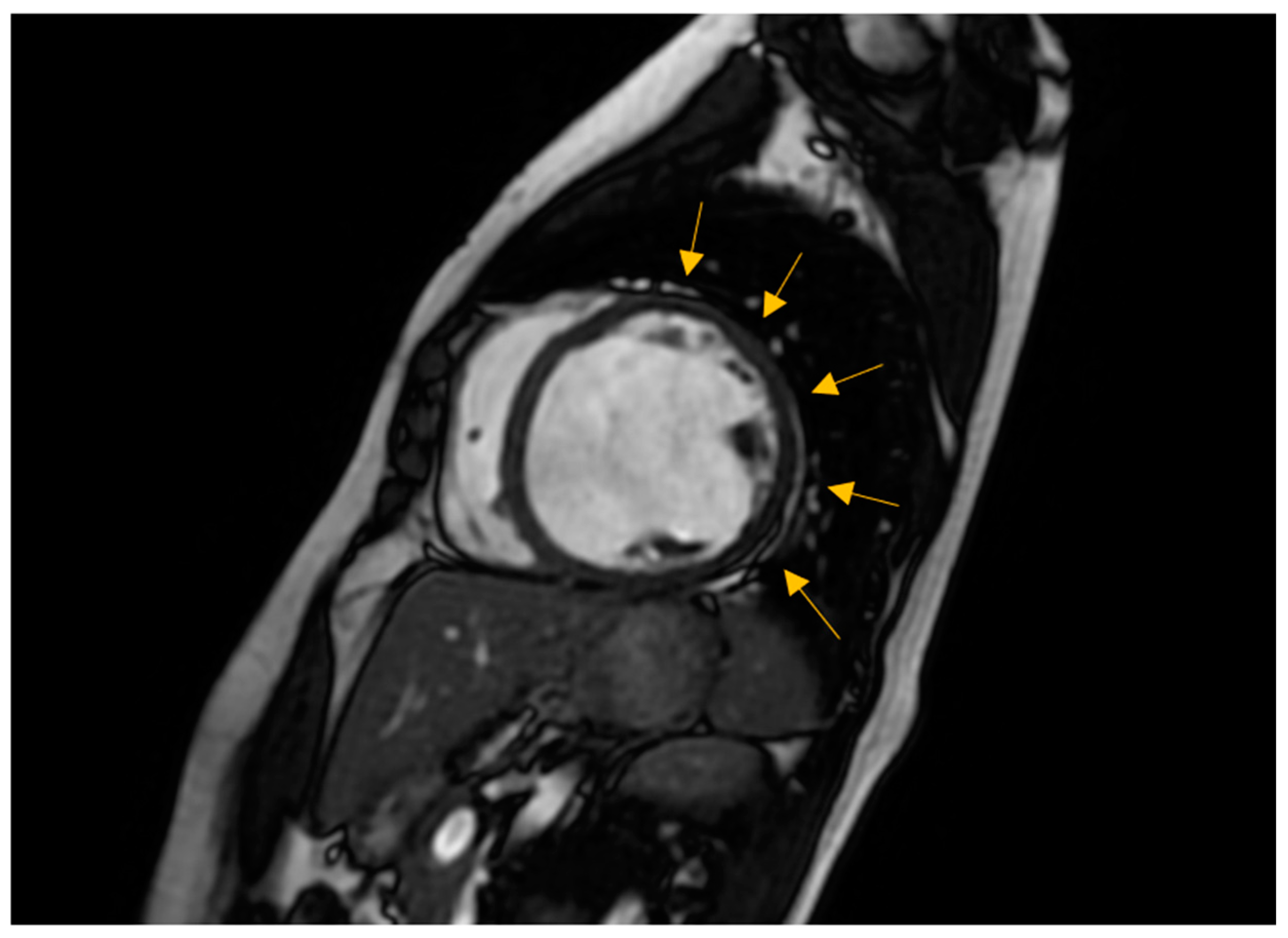
3.2. Non-Invasive Imaging Methods
3.3. Endomyocardial Biopsy
3.4. Laboratory Assessment
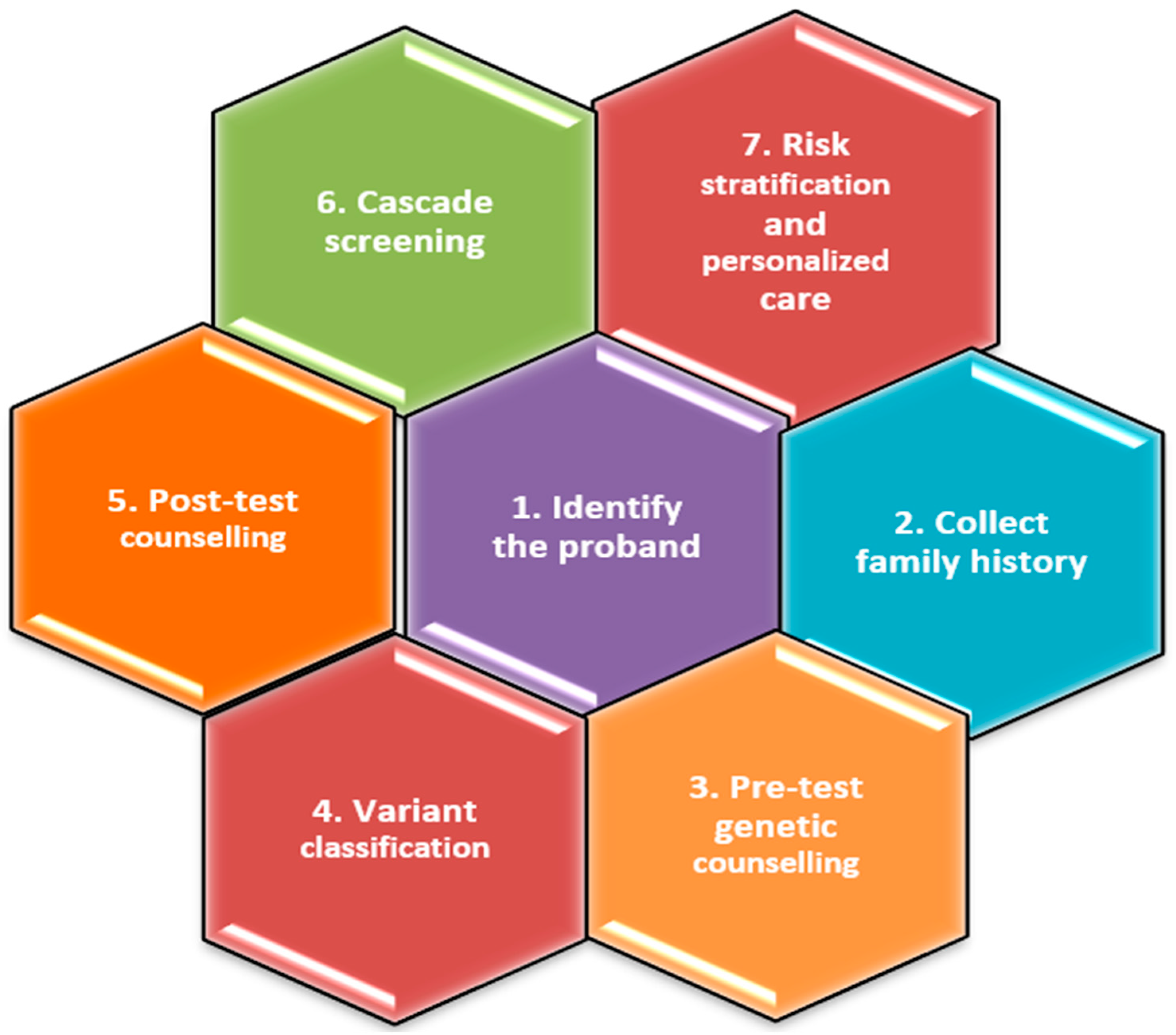
4. History Taking of the Initial Patient with Recently Diagnosed DCM and Family Screening
5. Genetic Testing
5.1. Benefits of Genetic Testing
5.2. Genetic Testing on the Proband
5.3. Prenatal Genetic Testing
5.4. Genetic Testing on the Relatives of the Proband
6. Genes Strongly Associated with DCM and Potential Therapeutic Implications
6.1. TTN
6.2. LMNA
6.3. MYH7
6.4. BAG3
6.5. RBM20
6.6. FLNC
6.7. SCN5A
6.8. TNNC1, TNNT2
6.9. DES
6.10. PLN
6.11. DSP
6.12. Mitofusin (MFN)
7. Genetic Risk Factors and Prognosis
8. Management
8.1. Non-Specific Therapy
8.2. Specific Therapy
9. Unresolved Issues and Future Directions
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Stehlik, J.; Edwards, L.B.; Kucheryavaya, A.Y.; Benden, C.; Christie, J.D.; Dobbels, F.; Kirk, R.; Rahmel, A.O.; Hetrz, M.I. The Registry of the International Society for Heart and Lung Transplantation: Twenty-eighth Adult Heart Transplant Report—2011. J. Heart Lung Transplant. 2011, 30, 1078–1094. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, P.A.; Albert, N.M.; Allen, L.A.; Bluemke, D.A.; Butler, J.; Fonarow, G.C.; Ikonomidis, J.S.; Khavjou, O.; Konstam, M.A.; Maddox, T.M.; et al. Forecasting the impact of heart failure in the United States a policy statement from the American Heart Association. Circ. Heart Fail. 2013, 6, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Paul, C.; Peters, S.; Perrin, M.; Fatkin, D.; Amerena, J. Non-ischemic dilated cardiomyopathy: Recognizing the genetic links. Intern. Med. J. 2023, 53, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Bui, Q.M.; Ding, J.; Hong, K.N.; Adler, E.A. The Genetic Evaluation of Dilated Cardiomyopathy. Struct. Heart 2023, 7, 100200. [Google Scholar] [CrossRef] [PubMed]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies: Developed by the task force on the management of cardiomyopathies of the European Society of Cardiology (ESC). Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Jordan, E. Dilated Cardiomyopathy Overview. In GeneReviews® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S., Bean, L., Gripp, K.W., Amamiya, A., Eds.; University of Washington: Seattle, WA, USA, 2022. [Google Scholar]
- Rosenbaum, A.N.; Agre, K.E.; Pereira, N.L. Genetics of dilated cardiomyopathy: Practical implications for heart failure management. Nat. Rev. Cardiol. 2020, 17, 286–297. [Google Scholar] [CrossRef]
- Sweet, M.E.; Taylor, M.R.; Mestroni, L. Diagnosis, prevalence, and screening of familial dilated cardiomyopathy. Expert. Opin. Orphan Drugs 2015, 3, 869–876. [Google Scholar] [CrossRef]
- Marian, A.J. Sporadic dilated cardiomyopathy is often familial. Cardiovasc. Res. 2022, 118, e69–e71. [Google Scholar] [CrossRef]
- Schmidt, M.A.; Michels, V.V.; Edwards, W.D.; Miller, F.A. Familial dilated cardiomyopathy. Am. J. Med. Genet. 1988, 31, 135–143. [Google Scholar] [CrossRef]
- Michels, V.V.; Moll, P.P.; Miller, F.A.; Tajik, A.J.; Chu, J.S.; Driscoll, D.J.; Burnett, J.C.; Rodeheffer, R.J.; Chesebro, J.H.; Tazelaar, H.D. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N. Engl. J. Med. 1992, 326, 77–82. [Google Scholar] [CrossRef]
- Huggins, G.S.; Kinnamon, D.D.; Haas, G.J.; Jordan, E.; Hofmeyer, M.; Kransdorf, E.; Ewald, G.A.; Morris, A.A.; Owens, A.; Lowes, B.; et al. DCM Precision Medicine Study of the DCM Consortium. Prevalence and cumulative risk of familial idiopathic dilated cardiomyopathy. JAMA 2022, 327, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Kumar, S.; Elliott, P.; Kalman, J.M.; Fatkin, D. Arrhythmic genotypes in familial dilated cardiomyopathy: Implications for genetic testing and clinical management. Heart Lung Circ. 2019, 28, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Ware, J.S.; Li, J.; Mazaika, E.; Yasso, C.M.; DeSouza, T.; Cappola, T.P.; Tsai, E.J.; Hilfiker-Kleiner, D.; Kamiya, C.A.; Mazzarotto, F.; et al. Shared genetic predisposition in peripartum and dilated cardiomyopathies. N. Engl. J. Med. 2016, 374, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Morales, A.; Siegfried, J.D. Clinical and genetic issues in dilated cardiomyopathy: A review for genetics pro-fessionals. Genet. Med. 2010, 12, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.; Vatta, M.; Ware, S.M. ACMG Professional Practice and Guidelines Committee. Genetic evaluation of cardiomyopathy: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 899. [Google Scholar]
- Rapezzi, C.; Arbustini, E.; Caforio, A.L.; Charron, P.; Gimeno-Blanes, J.; Heliö, T.; Linhart, A.; Mogensen, J.; Pinto, Y.; Ristic, A.; et al. Diagnostic work-up in cardiomyopathies: Bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2013, 34, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; Hejtmancik, J.F.; Brink, P.; Gelb, B.; Zhu, X.M.; Chamberlain, J.S.; McCabe, E.R.; Swift, M. X-linked dilated cardiomyopathy-molecular-genetic evidence of linkage to the Duchenne muscular-dystrophy (dystrophin) gene at the Xp21 locus. Circulation 1993, 87, 1854–1865. [Google Scholar] [CrossRef] [PubMed]
- Bonne, G.; Di Barletta, M.R.; Varnous, S.; Bécane, H.M.; Hammouda, E.H.; Merlini, L.; Muntoni, F.; Greenberg, C.R.; Gary, F.; Urtizberea, J.A.; et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery- Dreifuss muscular dystrophy. Nat. Genet. 1999, 21, 285–288. [Google Scholar] [CrossRef]
- Fatkin, D.; Johnson, R.; McGaughran, J.; Weintraub, R.G.; Atherton, J.J. Position statement on the diagnosis and management of familial dilated cardiomyopathy. Heart Lung Circ. 2017, 26, 1127–1132. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Cowan, J.; Jordan, E.; Kinnamon, D.D. The complex and diverse genetic architecture of dilated cardiomyopathy. Circ. Res. 2021, 128, 1514–1532. [Google Scholar] [CrossRef]
- Peters, S.; Johnson, R.; Birch, S.; Zentner, D.; Hershberger, R.E.; Fatkin, D. Familial dilated cardiomyopathy. Heart Lung Circ. 2020, 29, 566–574. [Google Scholar] [CrossRef]
- Eldemire, R.; Mestroni, L.; Taylor, M.R.G. Genetics of Dilated Cardiomyopathy. Annu. Rev. Med. 2024, 7, 417–426. [Google Scholar] [CrossRef]
- Pinamonti, B.; Abate, E.; De Luca, A.; Finocchiaro, G.; Korcova, R. Role of cardiac imaging: Echocardiography. In Dilated Cardiomyopathy: From Genetics to Clinical Management; Sinagra, G., Merlo, M., Pinamonti, B., et al., Eds.; Springer: Cham, Switzerland, 2019; Chapter 7. Available online: https://www.ncbi.nlm.nih.gov/books/NBK553855/ (accessed on 23 February 2024).
- Hudson, L.; Morales, A.; Mauro, A.C.; Whellan, D.; Adams, K.F.; O’Connor, C.M.; Hershberger, R.E. Family history of dilated cardiomyopathy among patients with heart failure from the HF-ACTION genetic ancillary study. Clin. Transl. Sci. 2013, 6, 179–183. [Google Scholar] [CrossRef]
- Haugaa, K.H.; Hasselberg, N.E.; Edvardsen, T. Mechanical dispersion by strain echocardiography: A predictor of ventricular arrhythmias in subjects with lamin A/C mutations. JACC Cardiovasc. Imaging 2015, 8, 104–106. [Google Scholar] [CrossRef]
- Haugaa, K.H.; Goebel, B.; Dahlslett, T.; Meyer, K.; Jung, C.; Lauten, A.; Figulla, H.R.; Poerner, T.C.; Edvardsen, T. Risk assessment of ventricular arrhythmias in patients with nonischemic dilated cardiomyopathy by strain echocardiography. J. Am. Soc. Echocardiogr. 2012, 25, 667–673. [Google Scholar] [CrossRef]
- Leren, I.S.; Saberniak, J.; Haland, T.F.; Edvardsen, T.; Haugaa, K.H. Combination of ECG and echocardiography for identification of arrhythmic events in early ARVC. JACC Cardiovasc. Imaging 2017, 10, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Augusto, J.B.; Eiros, R.; Nakou, E.; MouraFerreira, S.; Treibel, T.A.; Captur, G.; Akhtar, M.M.; Protonotarios, A.; Gossios, T.D.; Savvatis, K.; et al. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: A comprehensive genotype-imaging phenotype study. Eur. Heart J. Cardiovasc. Imaging 2020, 21, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Amin, R.J.; Morris-Rosendahl, D.; Edwards, M.; Tayal, U.; Buchan, R.; Hammersley, D.J.; Jones, R.E.; Gati, S.; Khalique, Z.; Almogheer, B.; et al. The addition of genetic testing and cardiovascular magnetic resonance to routine clinical data for stratification of etiology in dilated cardiomyopathy. Front. Cardiovasc. Med. 2022, 9, 1017119. [Google Scholar] [CrossRef]
- Duboscq-Bidot, L.; Xu, P.; Charron, P.; Neyroud, N.; Dilanian, G.; Millaire, A.; Bors, V.; Komajda, M.; Villard, E. Mutations in the Z- band protein myopalladin gene and idiopathic dilated cardiomyopathy. Cardiovasc. Res. 2008, 77, 118–125. [Google Scholar] [CrossRef]
- Sam, D.; Feger, J.; Machang’a, K. Dilated Cardiomyopathy. Reference Article. Available online: https://radiopaedia.org/articles/dilated-cardiomyopathy?lang=us (accessed on 22 May 2024).
- Castiglione, V.; Aimo, A.; Vergaro, G.; Saccaro, L.; Passino, C.; Emdin, M. Biomarkers for the diagnosis and management of heart failure. Heart Fail. Rev. 2022, 27, 625–643. [Google Scholar] [CrossRef]
- Shakur, R.; Ochoa, J.P.; Robinson, A.J.; Niroula, R.; Chandran, A.; Rahman, T.; Vihinen, M. Prognostic implications of troponin T variations in inherited cardiomyopathies using systems biology. npj Genom. Med. 2021, 6, 47. [Google Scholar] [CrossRef]
- Bonny, A.; Lellouche, N.; Ditah, I.; Hidden-Lucet, F.; Yitemben, M.T.; Granger, B.; Larrazet, F.; Frank, R.; Fontaine, G. C-reactive protein in arrhythmogenic right ventricular dysplasia/cardiomyopathy and relationship with ventricular tachycardia. Cardiol. Res. Pract. 2010, 2010, 919783. [Google Scholar] [CrossRef]
- Arany, Z. Peripartum Cardiomyopathy. N. Engl. J. Med. 2024, 390, 154–164. [Google Scholar] [CrossRef]
- Ni, H.; Jordan, E.; Kinnamon, D.D.; Cao, J.; Haas, G.J.; Hofmeyer, M.; Ewald, G.A.; Morris, A.A.; Owens, A.; Lowes, B.; et al. Screening for Dilated Cardiomyopathy in At-Risk First-Degree Relatives. J. Am. Coll. Cardiol. 2023, 81, 2059–2071. [Google Scholar] [CrossRef]
- Bozkcurt, B.; Colvin, M.; Cook, J.; Cooper, L.T.; Deswal, A.; Fonarow, G.C.; Francis, G.S.; Lenihan, D.; Lewis, E.F.; McNamara, D.M.; et al. Current diagnostic and treatment strategies for specific dilated cardiomyopathies: A scientific statement from the American Heart Association. Circulation 2016, 134, e579–e646. [Google Scholar] [CrossRef]
- Musunuru, K.; Hershberger, R.E.; Day, S.M.; Klinedinst, N.J.; Landstrom, A.P.; Parikh, V.N.; Prakash, S.; Semsarian, C.; Sturm, A.C.; American Heart Association Council on Genomic and Precision Medicine; et al. Genetic Testing for Inherited Cardiovascular Diseases: A Scientific Statement from the American Heart Association. Circ. Genom. Precis. Med. 2020, 13, e000067. [Google Scholar] [CrossRef]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: A report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2022, 79, e263–e421. [Google Scholar] [CrossRef] [PubMed]
- Paldino, A.; De Angelis, G.; Merlo, M.; Gigli, M.; Dal Ferro, M.; Severini, G.M.; Mestroni, L.; Sinagra, G. Genetics of Dilated Cardiomyopathy: Clinical Implications. Curr. Cardiol. Rep. 2018, 20, 83. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Zhao, Z.; Lan, H.; Li, X.; Yücel, G.; Lang, S.; Sattler, K.; Schünemann, J.D.; Zimmermann, W.H.; Cyganek, L.; et al. Ion Channel dysfunctions in dilated cardiomyopathy in limb-girdle muscular dystrophy. Circ. Genom. Precis. Med. 2018, 11, e001893. [Google Scholar] [CrossRef] [PubMed]
- Tayal, U.; Newsome, S.; Buchan, R.; Whiffin, N.; Halliday, B.; Lota, A.; Roberts, A.; Baksi, A.J.; Voges, I.; Midwinter, W.; et al. Phenotype and clinical outcomes of titin cardiomyopathy. J. Am. Coll. Cardiol. 2017, 70, 2264–2274. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Lopez, L.; Ochoa, J.P.; Royuela, A.; Verdonschot, J.A.J.; Dal Ferro, M.; Espinosa, M.A.; Sabater-Molina, M.; Gallego-Delgado, M.; Larrañaga-Moreira, J.M.; Garcia-Pinilla, J.M.; et al. Clinical risk score to predict pathogenic genotypes in patients with dilated cardiomyopathy. J. Am. Coll. Cardiol. 2022, 80, 1115–1126. [Google Scholar] [CrossRef]
- Mėlinytė-Ankudavičė, K.; Šukys, M.; Kasputytė, G.; Krikštolaitis, R.; Ereminienė, E.; Galnaitienė, G.; Mizarienė, V.; Šakalytė, G.; Krilavičius, T.; Jurkevičius, R. Association of uncertain significance genetic variants with myo-cardial mechanics and morphometrics in patients with nonischemic dilated cardiomyopathy. BMC Cardiovasc. Disord. 2024, 24, 224. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.T.; Lee, K.; Gordon, A.S.; Adelman, K.; Bale, S.J.; Chung, W.K.; Gollob, M.H.; Harrison, S.M.; Herman, G.E.; Hershberger, R.E.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2021 update: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 1391–1398. [Google Scholar] [CrossRef]
- Miller, D.T.; Lee, K.; Abul-Husn, N.S.; Brothers, K.; Chung, W.K.; Gollob, M.H.; Gordon, A.S.; Harrison, S.M.; Hershberger, R.E.; Klein, T.E.; et al. ACMG SF v3.1 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2022, 24, 1407–1414. [Google Scholar] [CrossRef]
- Wang, Y.; Dobreva, G. Epigenetics in LMNA-Related Cardiomyopathy. Cells 2023, 12, 783. [Google Scholar] [CrossRef]
- Rosario, K.F.; Karra, R.; Amos, K.; Landstrom, A.P.; Lakda, N.K. LMNA Cardiomyopathy: Important Considerations for the Heart Failure Clinician. J. Card. Fail. 2023, 29, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Zeppenfeld, K.; Tfelt-Hansen, J.; de Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; Charron, P.; Corrado, D.; Dagres, N.; de Chillou, C.; et al. 2022 ESC guidelines for the management of patients with ventricular ar-rhythmias and the prevention of sudden cardiac death. Eur. Heart J. 2022, 43, 3997–4126. [Google Scholar] [CrossRef] [PubMed]
- Mirelis, J.G.; Escobar-Lopez, L.; Ochoa, J.P.; Espinosa, M.Á.; Villacorta, E.; Navarro, M.; Casas, G.; Mora-Ayestarán, N.; Barriales-Villa, R.; Mogollón-Jiménez, M.V.; et al. Combination of late gadolinium enhancement and genotype improves prediction of prognosis in non-ischaemic dilated cardiomyopathy. Eur. J. Heart Fail. 2022, 24, 1183–1196. [Google Scholar] [CrossRef]
- Musunuru, K.; Hickey, K.T.; Al-Khatib, S.M.; Delles, C.; Fornage, M.; Fox, C.S.; Frazier, L.; Gelb, B.D.; Herrington, D.M.; Lanfear, D.E.; et al. American Heart Association Council on Functional Genomics and Translational Biology, Council on Clinical Cardiology, Council on Cardiovascular Disease in the Young, Council on Cardiovascular and Stroke Nursing, Council on Epidemiology and Prevention, Council on Hypertension, Council on Lifestyle and Cardiometabolic Health, Council on Quality of Care and Outcomes Research, and Stroke Council. Basic concepts and potential applications of genetics and genomics for cardi-ovascular and stroke clinicians: A scientific statement from the American Heart Association. Circ. Cardiovasc. Genet. 2015, 8, 216–242. [Google Scholar] [CrossRef]
- Verdonschot, J.A.J.; Hazebroek, M.R.; Wang, P.; Sanders-van Wijk, S.; Merken, J.J.; Adriaansen, Y.A.; Heymans, S.R. Clinical phenotype and genotype associations with improvement in left ventricular function in dilated cardiomyopathy. Circ. Heart Fail. 2018, 11, e005220. [Google Scholar] [CrossRef]
- Wilson, K.D.; Shen, P.; Fung, E.; Karakikes, I.; Zhang, A.; InanlooRahatloo, K.; Odegaard, J.; Sallam, K.; Davis, R.W.; Lui, G.K.; et al. A rapid, high-quality, cost-effective, comprehensive and expandable targeted next-generation sequencing assay for inherited heart diseases. Circ. Res. 2015, 117, 603–611. [Google Scholar] [CrossRef]
- Verdonschot, J.A.J.; Hazebroek, M.R.; Krapels, I.P.C.; Chen, H.; Haddad, F.; Kitani, T.; Wilson, K.D.; Tian, L.; Shrestha, R.; Wu, H.; et al. Implications of genetic testing in dilated cardiomyopathy. Circ. Genom. Precis. Med. 2020, 13, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Meynert, A.M.; Ansari, M.; FitzPatrick, D.R.; Taylor, M.S. Variant detection sensitivity and biases in whole genome and exome sequencing. BMC Bioinform. 2014, 15, 247. [Google Scholar] [CrossRef] [PubMed]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Seidman, C.E. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef]
- Bowles, N.E.; Bowles, K.R.; Towbin, J.A. The “final common pathway” hypothesis and inherited cardiovascular disease. The role of cytoskeletal proteins in dilated cardiomyopathy. Herz 2000, 25, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Heller, S.A.; Shih, R.; Kalra, R.; Kang, P.B. Emery-Dreifuss muscular dystrophy. Muscle Nerve. 2020, 61, 436–448. [Google Scholar] [CrossRef]
- Hazebroek, M.R.; Krapels, I.; Verdonschot, J.; van den Wijngaard, A.; Vanhoutte, E.; Hoos, M.; Snijders, L.; van Montfort, L.; Witjens, M.; Dennert, R.; et al. Prevalence of pathogenic gene mutations and prognosis do not differ in isolated left ventricular dysfunction compared with dilated cardiomyopathy. Circ. Heart Fail. 2018, 11, e004682. [Google Scholar] [CrossRef]
- Gigli, M.; Stolfo, D.; Graw, S.L.; Merlo, M.; Gregorio, C.; Nee Chen, S.; Dal Ferro, M.; Paldino, M.D.A.; De Angelis, G.; Brun, F.; et al. Phenotypic expression, natural history, and risk stratification of cardiomyopathy caused by filamin C truncating variants. Circulation 2021, 144, 1600–1611. [Google Scholar] [CrossRef]
- Sylvius, N.; Bilinska, Z.T.; Veinot, J.P.; Fidzianska, A.; Bolongo, P.M.; Poon, S.; McKeown, P.; Davies, R.A.; Chan, K.L.; Tang, A.S.; et al. In vivo and in vitro examination of the functional significances of novel lamin gene mutations in heart failure patients. J. Med. Genet. 2005, 42, 639–647. [Google Scholar] [CrossRef]
- Shah, R.A.; Asatryan, B.; Sharaf Dabbagh, G.; Khanji, M.Y.; Lopes, L.R.; van Duijvenboden, S.; Holmes, A.; Muser, D.; Landstrom, A.P.; Lee, A.M.; et al. Frequency, penetrance, and variable expressivity of dilated cardiomyopa-thy-associated putative pathogenic gene variants in UK Biobank Participants. Circulation 2022, 146, 110–124. [Google Scholar] [CrossRef]
- Fatkin, D. Guidelines for the diagnosis and management of familial dilated cardiomyopathy. Heart Lung Circ. 2011, 20, 691–693. [Google Scholar] [CrossRef] [PubMed]
- Jordan, E.; Peterson, L.; Ai, T.; Asatryan, B.; Bronicki, L.; Brown, E.; Celeghin, R.; Edwards, M.; Fan, J.; Ingles, J.; et al. Evidence-based assessment of genes in dilated cardiomyopathy. Circulation 2021, 144, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Eldemire, R.; Tharp, C.A.; Taylor, M.R.G.; Sbaizero, O.; Mestroni, L. The sarcomerice spring protein titin: Biophysical properties, molecular mechanisms, and genetic mutations associated with heart failure and cardiomyopathy. Curr. Cardiol. Rep. 2021, 23, 121. [Google Scholar] [CrossRef] [PubMed]
- Gigli, M.; Begay, R.L.; Morea, G.; Graw, S.L.; Sinagra, G.; Taylor, M.R.; Granzier, H.; Mestroni, L. A review of the giant protein titin in clinical molecular diagnostics of cardiomyopathies. Front. Cardiovasc. Med. 2016, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.M.; Lorenzini, M.; Cicerchia, M.; Ochoa, J.P.; Hey, T.M.; Sabater Molina, M.; Restrepo-Cordoba, M.A.; Dal Ferro, M.; Stolfo, D.; Johnson, R.L.; et al. Clinical phenotypes and prognosis of dilated cardiomyopathy caused by truncating variants in the TTN gene. Circ. Heart Fail. 2020, 13, e006832. [Google Scholar] [CrossRef] [PubMed]
- Vissing, C.R.; Rasmussen, T.B.; Dybro, A.M.; Olesen, M.S.; Pedersen, L.N.; Jensen, M.; Bundgaard, H.; Christensen, A.H. Dilated cardiomyopathy caused by truncating titin variants: Long-term outcomes, arrhythmias, response to treatment and sex differences. J. Med. Genet. 2021, 58, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.M.; Ware, J.S.; Herman, D.S.; Schafer, S.; Baksi, J.; Bick, A.G.; Buchan, R.J.; Walsh, R.; John, S.; Wilkinson, S.; et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci. Transl. Med. 2015, 7, 270ra6. [Google Scholar] [CrossRef] [PubMed]
- Ware, J.S.; Cook, S.A. Role of titin in cardiomyopathy: From DNA variants to patient stratification. Nat. Rev. Cardiol. 2018, 15, 241–252. [Google Scholar] [CrossRef]
- McAfee, Q.; Chen, C.Y.; Yang, Y.; Caporizzo, M.A.; Morley, M.; Babu, A.; Jeong, S.; Brandimarto, J.; Bedi, K.C., Jr.; Flam, E.; et al. Truncated titin proteins in dilated cardiomyopathy. Sci. Transl. Med. 2021, 13, eabd7287. [Google Scholar] [CrossRef]
- Gramlich, M.; Pane, L.S.; Zhou, Q.; Chen, Z.; Murgia, M.; Schötterl, S.; Goedel, A.; Metzger, K.; Brade, T.; Parrotta, E.; et al. Antisense-mediated exon skipping: A therapeutic strategy for titin-based dilated cardiomyopathy. EMBO Mol. Med. 2015, 7, 562–576. [Google Scholar] [CrossRef]
- Romano, R.; Ghahremani, S.; Zimmerman, T.; Legere, N.; Thakar, K.; Ladha, F.A.; Pettinato, A.M.; Hinson, J.T. Reading frame repair of TTN truncation variants restores titin quantity and functions. Circulation 2022, 145, 194–205. [Google Scholar] [CrossRef]
- van Rijsingen, I.A.; Arbustini, E.; Elliott, P.M.; Mogensen, J.; Hermans-van Ast, J.F.; van der Kooi, A.J.; van Tintelen, J.P.; van den Berg, M.P.; Pilotto, A.; Pasotti, M.; et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J. Am. Coll. Cardiol. 2012, 59, 493–500. [Google Scholar] [CrossRef]
- Parks, S.B.; Kushner, J.D.; Nauman, D.; Burgess, D.; Ludwigsen, S.; Peterson, A.; Li, D.; Jakobs, P.; Litt, M.; Porter, C.B.; et al. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am. Heart J. 2008, 156, 161–169. [Google Scholar] [CrossRef]
- Yamada, S.; Ko, T.; Ito, M.; Sassa, T.; Nomura, S.; Okuma, H.; Sato, M.; Imasaki, T.; Kikkawa, S.; Zhang, B.; et al. TEAD1 trapping by the Q353R-lamin A/C causes dilated cardiomyopathy. Sci. Adv. 2023, 9, eade7047. [Google Scholar] [CrossRef] [PubMed]
- Quarta, G.; Syrris, P.; Ashworth, M.; Jenkins, S.; Zuborne Alapi, K.; Morgan, J.; Muir, A.; Pantazis, A.; McKenna, W.J.; Elliott, P.M. Mutations in the lamin A/C gene mimic ar-rhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2012, 33, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Chatzifrangkeskou, M.; Le Dour, C.; Wu, W.; Morrow, J.P.; Joseph, L.C.; Beuvin, M.; Sera, F.; Homma, S.; Vignier, N.; Mougenot, N.; et al. ERK1/2 Directly acts on CTGF/CCN2 expression to mediate myocardial fibrosis in cardiomyopathy caused by mutations in the lamin A/C gene. Hum. Mol. Genet. 2016, 25, 2220–2233. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Jordan, E. LMNA-Related Dilated Cardiomyopathy. In GeneReviews® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2022. [Google Scholar] [PubMed]
- Chai, R.J.; Werner, H.; Li, P.Y.; Lee, Y.L.; Nyein, K.T.; Solovei, I.; Luu, T.D.A.; Sharma, B.; Navasankari, R.; Maric, M.; et al. Disrupting the LINC complex by AAV mediated gene transduction prevents progression of lamin induced cardiomyopathy. Nat. Commun. 2021, 2, 4722. [Google Scholar] [CrossRef]
- Ramos, F.J.; Chen, S.C.; Garelick, M.G.; Dai, D.F.; Liao, C.Y.; Schreiber, K.H.; MacKay, V.L.; An, E.H.; Strong, R.; Ladiges, W.C.; et al. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci. Transl. Med. 2012, 4, 144ra03. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Muchir, A.; Shan, J.; Bonne, G.; Worman, H.J. Mitogen-activated protein kinase inhibitors improve heart function and prevent fibrosis in cardiomyopathy caused by mutation in lamin A/C gene. Circulation 2011, 123, 53–61. [Google Scholar] [CrossRef]
- Auguste, G.; Rouhi, L.; Matkovich, S.J.; Coarfa, C.; Robertson, M.J.; Czernuszewicz, G.; Gurha, P.; Marian, A.J. BET Bromodomain inhibition attenuates cardiac phenotype in myocyte-specific lamin A/C-deficient mice. J. Clin. Investig. 2020, 130, 4740–4758. [Google Scholar] [CrossRef]
- Zheng, K.; Liu, L.; Zhang, Y.Q. Recent research on childhood hypertrophic cardiomyopathy caused by MYH7 gene mutations. Zhongguo Dang Dai Er Ke Za Zhi 2023, 25, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Peng, L.; Zhao, C. MYH7 in cardiomyopathy and skeletal muscle myopathy. Mol. Cell. Biochem. 2023, 479, 393–417. [Google Scholar] [CrossRef] [PubMed]
- de Frutos, F.; Ochoa, J.P.; Navarro-Peñalver, M.; Baas, A.; Bjerre, J.V.; Zorio, E.; Méndez, I.; Lorca, R.; Verdonschot, J.A.J.; García-Granja, P.E.; et al. Natural history of MYH7-related dilated cardiomyopathy. J. Am. Coll. Cardiol. 2022, 80, 1447–1461. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.S.; Pahl, E.; Dellefave-Castillo, L.; Rychlik, K.; Ing, A.; Yap, K.L.; Brew, C.; Johnston, J.R.; McNally, E.M.; Webster, G. Genotype and cardiac outcomes in pediatric dilated cardiomyopathy. J. Am. Heart Assoc. 2022, 11, e022854. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wilson, J.; Madani, M.; Feld, G.; Greenberg, B. Atrial arrhythmias and extensive left atrial fibrosis as the initial presentation of MYH7 gene mutation. JACC Clin. Electrophysiol. 2018, 4, 1488–1490. [Google Scholar] [CrossRef]
- Petropoulou, E.; Soltani, M.; Firoozabadi, A.D.; Namayandeh, S.M.; Crockford, J.; Maroofian, R.; Jamshidi, Y. Digenic inheritance of mutations in the cardiac troponin (TNNT2) and cardiac beta myosin heavy chain (MYH7) as the cause of severe dilated cardiomyopathy. Eur. J. Med. Genet. 2017, 60, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, A.M.; Carlus, S.J.; Al-Mazroea, A.H.; Alluqmani, M.; Almohammadi, Y.; Bhuiyan, Z.A.; Al-Harbi, K.M. Digenic inheritance of LAMA4 and MYH7 mutations in patient with infantile dilated cardiomyopathy. Medicina 2019, 55, 17. [Google Scholar] [CrossRef] [PubMed]
- Selvi Rani, D.; Nallari, P.; Dhandapany, P.S.; Rani, J.; Meraj, K.; Ganesan, M.; Narasimhan, C.; Thangaraj, K. Coexistence of digenic mutations in both thin (TPM1) and thick (MYH7) filaments of sarcomeric genes leads to severe hypertrophic cardiomyopathy in a south Indian FHCM. DNA Cell Biol. 2015, 34, 350–359. [Google Scholar] [CrossRef]
- Chang, A.C.Y.; Chang, A.C.H.; Kirillova, A.; Sasagawa, K.; Su, W.; Weber, G.; Termglinchan, V.; Karakikes, I.; Seeger, T.; Dainis, A.M.; et al. Telomere shortening is a hallmark of genetic cardiomyopathies. Proc. Natl. Acad. Sci. USA 2018, 115, 9276–9281. [Google Scholar] [CrossRef]
- Chang, A.C.Y.; Blau, H.M. Short telomeres—A hallmark of heritable cardiomyopathies. Differentiation 2018, 100, 31–36. [Google Scholar] [CrossRef]
- Eguchi, A.; Gonzalez, A.; Torres-Bigio, S.I.; Koleckar, K.; Birnbaum, F.; Zhang, J.Z.; Wang, V.Y.; Wu, J.C.; Artandi, S.E.; Blau, H.M. TRF2 rescues telomere attrition and prolongs cell survival in duchenne muscular dystrophy cardiomyocytes derived from human iPSCs. Proc. Natl. Acad. Sci. USA 2023, 120, e2209967120. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C.M.; Choudhary, M.; McCormick, M.G.; Cheung, D.; Landesberg, G.P.; Wang, J.F.; Song, J.; Martin, T.G.; Cheung, J.Y.; Qu, H.Q.; et al. BAG3: Nature’s quintessential multifunctional protein functions as a ubiquitous intra-cellular glue. Cells 2023, 12, 937. [Google Scholar] [CrossRef] [PubMed]
- Kirk, J.A.; Cheung, J.Y.; Feldman, A.M. Therapeutic targeting of BAG3: Considering its complexity in cancer and heart disease. J. Clin. Investig. 2021, 131, e149415. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Sun, K.; Zhang, X.; Tang, Y.; Xu, D. Advances in the role and mechanism of BAG3 in dilated cardiomyopathy. Heart Fail. Rev. 2021, 26, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.Q.; Feldman, A.M.; Hakonarson, H. Genetics of BAG3: A paradigm for developing precision therapies for dilated cardio-myopathies. J. Am. Heart Assoc. 2022, 11, e027373. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, W.; Sadoshima, J. BAG3 plays a central role in proteostasis in the heart. J. Clin. Investig. 2017, 127, 2900–2903. [Google Scholar] [CrossRef] [PubMed]
- Domínguez, F.; Cuenca, S.; Bilińska, Z.; Toro, R.; Villard, E.; Barriales-Villa, R.; Domínguez, F.; Cuenca, S.; Bilińska, Z.; Toro, R.; et al. Dilated cardiomyopathy due to BLC2-associated athanogene 3 (BAG3) mutations. J. Am. Coll. Cardiol. 2018, 72, 2471–2481. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Bogomolovas, J.; Wu, T.; Zhang, W.; Liu, C.; Veevers, J.; Stroud, M.J.; Zhang, Z.; Ma, X.; Mu, Y.; et al. Loss-of function mutations in co-chaperone BAG3 destabilize small HSPs and cause cardiomyopathy. J. Clin. Investig. 2017, 127, 3189–3200. [Google Scholar] [CrossRef]
- Gamerdinger, M.; Hajieva, P.; Kaya, A.M.; Wolfrum, U.; Hartl, F.U.; Behl, C. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. 2009, 28, 889–901. [Google Scholar] [CrossRef]
- Small, E.M.; Olson, E.N. Pervasive roles of microRNAs in cardiovascular biology. Nature 2011, 469, 336–342. [Google Scholar] [CrossRef]
- Kalayinia, S.; Arjmand, F.; Maleki, M.; Malakootian, M.; Singh, C.P. MicroRNAs: Roles in cardiovascular development and disease. Cardiovasc. Pathol. 2021, 50, 107296. [Google Scholar] [CrossRef] [PubMed]
- Zaragoza, C.; Saura, M.; Hernández, I.; Ramirez-Carracedo, R.; García-García, F.; Zamorano, J.L.; Mangas, A.; Toro, R. Differential expression of circulating miRNAs as a novel tool to assess BAG3-associated familial dilated cardiomyopathy. Biosci. Rep. 2019, 39, BSR20180934. [Google Scholar] [CrossRef] [PubMed]
- Diofano, F.; Weinmann, K.; Schneider, I.; Thiessen, K.D.; Rottbauer, W.; Just, S. Genetic compensation prevents myopathy and heart failure in an in vivo model of Bag3 deficiency. PLoS Genet. 2020, 16, e1009088. [Google Scholar] [CrossRef] [PubMed]
- Refaat, M.M.; Lubitz, S.A.; Makino, S.; Islam, Z.; Frangiskakis, J.M.; Mehdi, H.; Gutmann, R.; Zhang, M.L.; Bloom, H.L.; MacRae, C.A.; et al. Genetic variation in the alternative splicing regulator RBM20 is associated with dilated cardiomyopathy. Heart Rhythm. 2012, 9, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Kimura, A.; Kuroyanagi, H. Alternative splicing regulator RBM20 and cardiomyopathy. Front. Mol. Biosci. 2018, 5, 105. [Google Scholar] [CrossRef] [PubMed]
- Brauch, K.M.; Karst, M.L.; Herron, K.J.; de Andrade, M.; Pellikka, P.A.; Rodeheffer, R.J.; Michels, V.V.; Olson, T.M. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 2009, 54, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, T.; Zhang, Y.; Cui, M.; Li, H.; Sanchez-Ortiz, E.; McAnally, J.R.; Tan, W.; Kim, J.; Chen, K.; Xu, L.; et al. Precise genomic editing of pathogenic mutations in RBM20 rescues dilated cardiomyopathy. Sci. Transl. Med. 2022, 14, eade1633. [Google Scholar] [CrossRef]
- LeWinter, M.M.; Granzier, H.L. Titin is a major human disease gene. Circulation 2013, 127, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Schafer, S.; Greaser, M.L.; Radke, M.H.; Liss, M.; Govindarajan, T.; Maatz, H.; Schulz, H.; Li, S.; Parrish, A.M.; et al. RBM20, A gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012, 18, 766–773. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, C.; Sun, M.; Jin, Y.; Braz, C.U.; Khatib, H.; Hacker, T.A.; Liss, M.; Gotthardt, M.; Granzier, H.; et al. RBM20 Phosphorylation and its role in nucleocytoplasmic transport and cardiac pathogenesis. FASEB J. 2022, 36, e22302. [Google Scholar] [CrossRef]
- Ihara, K.; Sasano, T.; Hiraoka, Y.; Togo-Ohno, M.; Soejima, Y.; Sawabe, M.; Tsuchiya, M.; Ogawa, H.; Furukawa, T.; Kuroyanagi, H. A missense mutation in the RSRSP stretch of Rbm20 causes dilated cardiomyopathy and atrial fibrillation in mice. Sci. Rep. 2020, 10, 17894. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Shi, A.; Lian, H.; Hu, S.; Nie, Y. Filamin C in cardiomyopathy: From physiological roles to DNA variants. Heart Fail. Rev. 2022, 27, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Nakamura, F. Structure and function of filamin C in the muscle Z-disc. Int. J. Mol. Sci. 2020, 21, 2696. [Google Scholar] [CrossRef] [PubMed]
- Noureddine, M.; Gehmlich, K. Structural and signaling proteins in the Z-disk and their role in cardiomyopathies. Front. Physiol. 2023, 14, 1143858. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.D.; Kirkland, N.J.; Liu, C.; Razu, S.S.; Fang, X.; Engler, A.J.; Chen, J.; McCulloch, A.D. Subcellular remodeling in filamin C deficient mouse hearts impairs myocyte tension development during progression of dilated cardiomyopathy. Int. J. Mol. Sci. 2022, 23, 871. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, Z.; Zhang, L.; Zhu, M.; Tan, C.; Zhou, X.; Evans, S.M.; Fang, X.; Feng, W.; Chen, J. Loss of filamin C is catastrophic for heart function. Circulation 2020, 141, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Begay, R.L.; Graw, S.L.; Sinagra, G.; Asimaki, A.; Rowland, T.J.; Slavov, D.B. Filamin C truncation mutations are associated with arrhythmogenic dilated cardiomyopathy and changes in the cell-cell adhesion structures. JACC Clin. Electrophysiol. 2018, 4, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Genga, M.F.; Cuenca, S.; Dal Ferro, M.; Zorio, E.; Salgado-Aranda, R.; Climent, V.; Padrón-Barthe, L.; Duro-Aguado, I.; Jiménez-Jáimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC mutations are asso-ciated with high-risk dilated and arrhythmogenic cardiomyopathies. J. Am. Coll. Cardiol. 2016, 68, 2440–2451. [Google Scholar] [CrossRef]
- Celeghin, R.; Cipriani, A.; Bariani, R.; Bueno Marinas, M.; Cason, M.; Bevilacqua, M. Filamin-C variant-associated cardiomyopathy: A pooled analysis of individualpatient data to evaluate the clinical profile and risk of sudden cardiac death. Heart Rhythm. 2022, 19, 235–243. [Google Scholar] [CrossRef]
- Chen, S.N.; Lam, C.K.; Wan, Y.W.; Gao, S.; Malak, O.A.; Zhao, S.R.; Lombardi, R.; Ambardekar, A.V.; Bristow, M.R.; Cleveland, J.; et al. Activation of PDGFRA signaling contributes to filamin C-related arrhythmogenic cardiomyopathy. Sci. Adv. 2022, 8, eabk0052. [Google Scholar] [CrossRef]
- Remme, C.A. SCN5A Channelopathy: Arrhythmia, cardiomyopathy, epilepsy and beyond. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2023, 378, 20220164. [Google Scholar] [CrossRef] [PubMed]
- Zegkos, T.; Panagiotidis, T.; Parcharidou, D.; Efthimiadis, G. Emerging concepts in arrhythmogenic dilated cardiomyopathy. Heart Fail. Rev. 2021, 26, 1219–1229. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yin, L.; Shen, C.; Hu, K.; Ge, J.; Sun, A. SCN5A variants: Association with cardiac disorders. Front. Physiol. 2018, 9, 1372. [Google Scholar] [CrossRef] [PubMed]
- Moreau, A.; Gosselin-Badaroudine, P.; Delemotte, L.; Klein, M.L.; Chahine, M. Gating pore currents are defects in common with two Nav1.5 mutations in patients with mixed arrhythmias and dilated cardiomyopathy. J. Gen. Physiol. 2015, 145, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Poller, W.; Escher, F.; Haas, J.; Heidecker, B.; Schultheiss, H.P.; Attanasio, P.; Skurk, C.; Haghikia, A.; Meder, B.; Klaassen, S. Missense variant E1295K of sodium channel SCN5A associated with recurrent ventricular fibrillation and myocardial inflammation. JACC Case Rep. 2022, 4, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.M.; Amin, A.S. Clinical Spectrum of SCN5A mutations: Long QT syndrome, brugada syndrome, and cardiomyopathy. JACC Clin. Electrophysiol. 2018, 4, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Thompson, B.A.; Perrin, M.; James, P.; Zentner, D.; Kalman, J.M.; Vandenberg, J.I.; Fatkin, D. Arrhythmic phenotypes are a defining feature of dilated cardiomyopathy-associated SCN5A variants: A systematic review. Circ. Genom. Precis. Med. 2022, 15, e003432. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Xie, A.; Kim, T.Y.; Liu, H.; Shi, G.; Kang, G.J.; Jiang, N.; Liu, M.; Jeong, E.M.; Choi, B.R.; et al. HuR-mediated SCN5A messenger RNA stability reduces arrhythmic risk in heart failure. Heart Rhythm. 2018, 15, 1072–1080. [Google Scholar] [CrossRef]
- Li, M.X.; Hwang, P.M. Structure and function of cardiac troponin C (TNNC1): Implications for heart failure, cardiomyopathies, and troponin modulating drugs. Gene 2015, 571, 153–166. [Google Scholar] [CrossRef]
- Keyt, L.K.; Duran, J.M.; Bui, Q.M.; Chen, C.; Miyamoto, M.I.; Silva Enciso, J.; Tardiff, J.C.; Adler, E.D. Thin filament cardio-myopathies: A review of genetics, disease mechanisms, and emerging therapeutics. Front. Cardiovasc. Med. 2022, 9, 972301. [Google Scholar] [CrossRef]
- Kalyva, A.; Parthenakis, F.I.; Marketou, M.E.; Kontaraki, J.E.; Vardas, P.E. Biochemical characterisation of troponin C mutations causing hypertrophic and dilated cardiomyopathies. J. Muscle Res. Cell Motil. 2014, 35, 161–178. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, J.; Murphy, R.T.; Shaw, T.; Bahl, A.; Redwood, C.; Watkins, H.; Burke, M.; Elliott, P.M.; McKenna, W.J. Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 2004, 44, 2033–2040. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Norton, N.; Morales, A.; Li, D.; Siegfried, J.D.; Gonzalez-Quintana, J. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2010, 3, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Parks, S.B.; Kushner, J.D.; Li, D.; Ludwigsen, S.; Jakobs, P.; Nauman, D.; Burgess, D.; Partain, J.; Litt, M. Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3,and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin. Transl. Sci. 2008, 1, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Tadros, H.J.; Life, C.S.; Garcia, G.; Pirozzi, E.; Jones, E.G.; Datta, S.; Parvatiyar, M.S.; Chase, P.B.; Allen, H.D.; Kim, J.J.; et al. Meta-analysis of cardiomyopathy-associated variants in troponin genes identifies loci and intragenic hot spots that are associated with worse clinical outcomes. J. Mol. Cell Cardiol. 2020, 142, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Alves, M.L.; Warren, C.M.; Simon, J.N.; Gaffin, R.D.; Montminy, E.M.; Wieczorek, D.F.; Solaro, R.J.; Wolska, B.M. Early sensitization of myofilaments to Ca2+ prevents genetically linked dilated cardiomyopathy in mice. Cardiovasc. Res. 2017, 113, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Guo, Y.; Zhan, Y.; Zhou, X.; Li, Y.; Zhao, C.; Sun, N.; Xu, C.; Liang, Q. Cardiac overexpression of XIN prevents dilated cardiomyopathy caused by TNNT2 ΔK210 mutation. Front. Cell Dev. Biol. 2021, 9, 691749. [Google Scholar] [CrossRef] [PubMed]
- Gomes, G.; Seixas, M.R.; Azevedo, S.; Audi, K.; Jurberg, A.D.; Mermelstein, C.; Costa, M.L. What does desmin do: A bibliometric assessment of the functions of the muscle intermediate filament. Exp. Biol. Med. 2022, 247, 538–550. [Google Scholar] [CrossRef]
- Schröder, R.; Vrabie, A.; Goebel, H.H. Primary desminopathies. J. Cell Mol. Med. 2007, 11, 416–426. [Google Scholar] [CrossRef]
- Tsikitis, M.; Galata, Z.; Mavroidis, M.; Psarras, S.; Capetanaki, Y. Intermediate filaments in cardiomyopathy. Biophys. Rev. 2018, 10, 1007–1031. [Google Scholar] [CrossRef]
- Tesson, F.; Sylvius, N.; Pilotto, A.; Dubosq-Bidot, L.; Peuchmaurd, M.; Bouchier, C.; Benaiche, A.; Mangin, L.; Charron, P.; Gavazzi, A.; et al. Epidemiology of desmin and cardiac actin gene mutations in a European population of dilated cardiomyopathy. Eur. Heart J. 2000, 21, 1872–1876. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, I.; Dieding, M.; Klauke, B.; Brodehl, A.; Gaertner-Rommel, A.; Walhorn, V.; Gummert, J.; Schulz, U.; Paluszkiewicz, L.; Anselmetti, D.; et al. A novel desmin (DES) indel mutation causes severe atypical cardiomyopathy in combination with atrio-ventricular block and skeletal myopathy. Mol. Genet. Genom. Med. 2018, 6, 288–293. [Google Scholar] [CrossRef] [PubMed]
- van Spaendonck-Zwarts, K.Y.; van Hessem, L.; Jongbloed, J.D.; de Walle, H.E.; Capetanaki, Y.; van der Kooi, A.J.; van Langen, I.M.; van den Berg, M.P.; van Tintelen, J.P. Desmin-related myopathy. Clin. Genet. 2011, 80, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Elsnicova, B.; Hornikova, D.; Tibenska, V.; Kolar, D.; Tlapakova, T.; Schmid, B.; Mallek, M.; Eggers, B.; Schlötzer-Schrehardt, U.; Peeva, V.; et al. Desmin knock-out cardiomyopathy: A heart on the verge of metabolic crisis. Int. J. Mol. Sci. 2022, 23, 12020. [Google Scholar] [CrossRef] [PubMed]
- Diermeier, S.; Iberl, J.; Vetter, K.; Haug, M.; Pollmann, C.; Reischl, B.; Buttgereit, A.; Schürmann, S.; Spörrer, M.; Goldmann, W.H.; et al. Early signs of architectural and biomechanical failure in isolated myofibers and immortalized myoblasts from desmin-mutant knock-in mice. Sci. Rep. 2017, 7, 1391. [Google Scholar] [CrossRef] [PubMed]
- Koopman, W.J.; Willems, P.H.; Smeitink, J.A. Monogenic mitochondrial disorders. N. Engl. J. Med. 2012, 366, 1132–1141. [Google Scholar] [CrossRef]
- Diokmetzidou, A.; Soumaka, E.; Kloukina, I.; Tsikitis, M.; Makridakis, M.; Varela, A.; Davos, C.H.; Georgopoulos, S.; Anesti, V.; Vlahou, A.; et al. Desmin and αB-crystallin interplay in the maintenance of mitochondrial homeostasis and cardiomyocyte survival. J. Cell Sci. 2016, 129, 3705–3720. [Google Scholar] [CrossRef] [PubMed]
- Feyen, D.A.M.; Perea-Gil, I.; Maas, R.G.C.; Harakalova, M.; Gavidia, A.A.; Arthur Ataam, J.; Wu, T.H.; Vink, A.; Pei, J.; Vadgama, N.; et al. Unfolded protein response as a compensatory mechanism and potential therapeutic target in PLN R14del cardiomyopathy. Circulation 2021, 144, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Hof, I.E.; van der Heijden, J.F.; Kranias, E.G.; Sanoudou, D.; de Boer, R.A.; van Tintelen, J.P.; van der Zwaag, P.A.; Doevendans, P.A. Prevalence and cardiac phenotype of patients with a phospholamban mutation. Neth. Heart J. 2019, 27, 64–69. [Google Scholar] [CrossRef]
- van der Zwaag, P.A.; van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef]
- Medin, M.; Hermida-Prieto, M.; Monserrat, L.; Laredo, R.; Rodriguez-Rey, J.C.; Fernandez, X.; Castro-Beiras, A. Mutational screening of phos-pholamban gene in hypertrophic and idiopathic dilated cardiomyopathy and functional study of the PLN-42 C>G mutation. Eur. J. Heart Fail. 2007, 9, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Villard, E.; Duboscq-Bidot, L.; Charron, P.; Benaiche, A.; Conraads, V.; Sylvius, N.; Komajda, M. Mutation screening in dilated cardiomyo-pathy: Prominent role of the beta myosin heavy chain gene. Eur. Heart J. 2005, 26, 794–803. [Google Scholar] [CrossRef]
- van Rijsingen, I.A.; van der Zwaag, P.A.; Groeneweg, J.A.; Nannenberg, E.A.; Jongbloed, J.D.; Zwinderman, A.H.; Pinto, Y.M.; Dit Deprez, R.H.; Post, J.G.; Tan, H.L.; et al. Outcome in phospholamban R14del carriers: Results of a large multicentre cohort study. Circ. Cardiovasc. Genet. 2014, 7, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Deiman, F.E.; Bomer, N.; van der Meer, P.; Grote Beverborg, N. Review: Precision medicine approaches for genetic cardiomyo-pathy: Targeting phospholamban R14del. Curr. Heart Fail. Rep. 2022, 19, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Doevendans, P.A.; Glijnis, P.C.; Kranias, E.G. Leducq transatlantic network of excellence to cure phospholamban-induced car-diomyopathy (CURE-PLaN). Circ. Res. 2019, 125, 720–724. [Google Scholar] [CrossRef] [PubMed]
- de Brouwer, R.; Meems, L.M.G.; Verstraelen, T.E.; Mahmoud, B.; Proost, V.; Wilde, A.A.M.; Bosman, L.P.; van Drie, E.; van der Zwaag, P.A.; van Tintelen, J.P.; et al. Sex-specific aspects of phospholamban cardiomyopathy: The importance and prognostic value of low-voltage electrocardiograms. Heart Rhythm. 2022, 19, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Te Rijdt, W.P.; Hoorntje, E.T.; de Brouwer, R.; Oomen, A.; Amin, A.; van der Heijden, J.F.; Karper, J.C.; Westenbrink, B.D.; Silljé, H.H.W.; Te Riele, S.J.M.; et al. Rationale and design of the PHOs-pholamban RElated Cardiomyopathy intervention STudy (i-PHORECAST). Neth. Heart J. 2022, 30, 84–95. [Google Scholar] [CrossRef]
- Grote Beverborg, N.; Später, D.; Knöll, R.; Hidalgo, A.; Yeh, S.T.; Elbeck, Z.; Silljé, H.H.W.; Eijgenraam, T.R.; Siga, H.; Zurek, M.; et al. Phospholamban antisense oligonucleotides improve cardiac function in murine cardiomyopathy. Nat. Commun. 2021, 12, 5180. [Google Scholar] [CrossRef] [PubMed]
- Dave, J.; Raad, N.; Mittal, N.; Zhang, L.; Fargnoli, A.; Oh, J.G.; Savoia, M.E.; Hansen, J.; Fava, M.; Yin, X.; et al. Gene editing reverses arrhythmia susceptibility in humanized PLN-R14del mice: Modelling a European cardiomyopathy with global impact. Cardiovasc. Res. 2022, 118, 3140–3150. [Google Scholar] [CrossRef]
- Vafiadaki, E.; Glijnis, P.C.; Doevendans, P.A.; Kranias, E.G.; Sanoudou, D. Phospholamban R14del disease: The past, the present and the future. Front. Cardiovasc. Med. 2023, 10, 1162205. [Google Scholar] [CrossRef]
- Pugh, T.J.; Kelly, M.A.; Gowrisankar, S.; Hynes, E.; Seidman, M.A.; Baxter, S.M.; Bowser, M.; Harrison, B.; Aaron, D.; Mahanta, L.M.; et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet. Med. 2014, 16, 601–608. [Google Scholar] [CrossRef]
- Karvonen, V.; Harjama, L.; Heliö, K.; Kettunen, K.; Elomaa, O.; Koskenvuo, J.W.; Kere, J.; Weckström, S.; Holmström, M.; Saarela, J.; et al. A novel desmoplakin mutation causes dilated cardiomyopathy with palmoplantar keratoderma as an early clinical sign. J. Eur. Acad. Dermatol. Venereol. 2022, 36, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Gigli, M.; Merlo, M.; Graw, S.L.; Barbati, G.; Rowland, T.J.; Slavov, D.B.; Stolfo, D.; Haywood, M.E.; Dal Ferro, M.; Altinier, A.; et al. Genetic risk of arrhythmic phenotypes in patients with dilated cardiomyopathy. J. Am. Coll. Cardiol. 2019, 74, 1480–1490. [Google Scholar] [CrossRef] [PubMed]
- Brandão, M.; Bariani, R.; Rigato, I.; Bauce, B. Desmoplakin cardiomyopathy: Comprehensive review of an increasingly recognized entity. J. Clin. Med. 2023, 12, 2660. [Google Scholar] [CrossRef]
- Smith, E.D.; Lakdawala, N.K.; Papoutsidakis, N.; Aubert, G.; Mazzanti, A.; McCanta, A.C.; Agarwal, P.P.; Arscott, P.; Dellefave-Castillo, L.M.; Vorovich, E.E.; et al. Desmoplakin cardiomyopathy, a fibrotic and inflammatory form of cardiomyopathy distinct from typical dilated or arrhythmogenic right ventricular cardiomyopathy. Circulation 2020, 141, 1872–1884. [Google Scholar] [CrossRef] [PubMed]
- Bariani, R.; Cason, M.; Rigato, I.; Cipriani, A.; Celeghin, R.; De Gaspari, M.; Bueno Marinas, M.; Mattesi, G.; Pergola, V.; Rizzo, S.; et al. Clinical profile and long-term follow-up of a cohort of patients with desmoplakin cardiomyopathy. Heart Rhythm. 2022, 19, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Chelko, S.P.; Asimaki, A.; Lowenthal, J.; Bueno-Beti, C.; Bedja, D.; Scalco, A.; Amat-Alarcon, N.; Andersen, P.; Judge, D.P.; Tung, L.; et al. Therapeutic modulation of the immune response in arrhythmogenic cardiomyopathy. Circulation 2019, 140, 1491–1505. [Google Scholar] [CrossRef] [PubMed]
- Bauce, B.; Basso, C.; Rampazzo, A.; Beffagna, G.; Daliento, L.; Frigo, G.; Malagrida, S.; Settimo, L.; Danieli, G.A.; Thiene, G.; et al. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur. Heart J. 2005, 26, 1666–1675. [Google Scholar] [CrossRef]
- Franco, A.; Li, J.; Kelly, D.P.; Hershberger, R.E.; Marian, A.J.; Lewis, R.M.; Song, M.; Dang, X.; Schmidt, A.D.; Mathyer, M.E.; et al. A human mitofusin 2 mutation can cause mitophagic cardiomyopathy. eLife 2023, 12, e84235. [Google Scholar] [CrossRef]
- Paldino, A.; Dal Ferro, M.; Stolfo, D.; Gandin, I.; Medo, K.; Graw, S.; Gigli, M.; Gagno, G.; Zaffalon, D.; Castrichini, M.; et al. Prognostic prediction of genotype vs phenotype in genetic cardiomyopathies. J. Am. Coll. Cardiol. 2022, 80, 1981–1994. [Google Scholar] [CrossRef]
- Meune, C.; Van Berlo, J.H.; Anselme, F.; Bonne, G.; Pinto, Y.M.; Duboc, D. Primary prevention of sudden death in patients with lamin A/C gene mutations. N. Engl. J. Med. 2006, 354, 209–210. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019, 16, e301–e372. [Google Scholar] [CrossRef] [PubMed]
- Ichida, F.; Tsubata, S.; Bowles, K.R.; Haneda, N.; Uese, K.; Miyawaki, T.; Dreyer, W.J.; Messina, J.; Li, H.; Bowles, N.E.; et al. Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation 2001, 103, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.G.; Felix, J.F.; Morrison, A.C.; Kalogeropoulos, A.; Trompet, S.; Wilk, J.B.; Gidlöf, O.; Wang, X.; Morley, M.; Mendelson, M.; et al. Discovery of genetic variation on chromosome 5q22 associated with mortality in heart failure. PLOS Genet. 2016, 12, e1006034. [Google Scholar] [CrossRef]
- Merlo, M.; Cannatà, A.; Pio Loco, C.; Stolfo, D.; Barbati, G.; Artico, J.; Gentile, P.; De Paris, V.; Ramani, F.; Zecchin, M.; et al. Contemporary survival trends and aetiological characterization in non-ischaemic dilated cardiomyopathy. Eur. J. Heart Fail. 2020, 22, 1111–1121. [Google Scholar] [CrossRef]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure. J. Card. Fail. 2022, 28, e1–e167. [Google Scholar] [CrossRef]
- Cresci, S.; Kelly, R.J.; Cappola, T.P.; Diwan, A.; Dries, D.; Kardia, S.L.; Dorn, G.W., 2nd. Clinical and genetic modifiers of long-term survival in heart failure. J. Am. Coll. Cardiol. 2009, 54, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Gaertner, A.; Bloebaum, J.; Brodehl, A.; Klauke, B.; Sielemann, K.; Kassner, A.; Fox, H.; Morshuis, M.; Tiesmeier, J.; Schulz, U.; et al. The Combined Human Genotype of Truncating TTN and RBM20 Mutations Is Associated with Severe and Early Onset of Dilated Cardiomyopathy. Genes 2021, 12, 883. [Google Scholar] [CrossRef]
- Owens, A.T.; Day, S.M. Reappraising Genes for Dilated Cardiomyopathy: Stepping Back to Move Forward. Circulation 2021, 144, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.M.; Semsarian, C.; Márquez, M.F.; Shamloo, A.S.; Ackerman, M.J.; Ashley, E.A.; Sternick, E.B.; Barajas-Martinez, H.; Behr, E.R.; Bezzina, C.R.; et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the State of Genetic Testing for Cardiac Diseases. Heart Rhythm. 2022, 19, 623–629. [Google Scholar] [CrossRef]
- White, H.; Maqbool, A.; McMahon, A.D.; Yates, L.; Ball, S.G.; Hall, A.S.; Balmforth, A.J. An evaluation of the β1 adrenergic receptor Arg389Gly polymorphism in individuals at risk of coronary events: A WOSCOPS substudy. Eur. Heart J. 2002, 23, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- White, H.L.; de Boer, R.A.; Maqbool, A.; Greenwood, D.; van Veldhuisen, D.J.; Cuthbert, R.; Ball, S.G.; Hall, A.S.; Balmforth, A.J. An evaluation of the β1 adrenergic receptor Arg389Gly polymorphism in individuals with heart failure: A MERIT-HF sub-study. Eur. J. Heart Fail. 2003, 5, 463–468. [Google Scholar] [PubMed]
- Pare, G.; Kubo, M.; Byrd, J.B.; McCarty, C.A.; Woodard-Grice, A.; Teo, K.K.; Anand, S.S.; Zuvich, R.L.; Bradford, Y.; Ross, S.; et al. Genetic variants associated with angiotensin- converting enzyme inhibitor- associated angioedema. Pharmacogenet. Genomics 2013, 23, 470–478. [Google Scholar]
- Nelveg-Kristensen, K.E.; Busk Madsen, M.; Torp-Pedersen, C.; Køber, L.; Egfjord, M.; Berg Rasmussen, H.; Riis Hansen, P. Pharmacogenetic risk stratification in angiotensin-converting enzyme inhibitor- treated patients with congestive heart failure: A retrospective cohort study. PLoS ONE 2015, 10, e0144195. [Google Scholar] [CrossRef] [PubMed]
- Van Spaendonck-Zwarts, K.Y.; van Tintelen, J.P.; van Veldhuisen, D.J.; van der Werf, R.; Jongbloed, J.D.; Paulus, W.J.; Dooijes, D.; van den Berg, M.P. Peripartum cardiomyopathy as a part of familial dilated cardiomyopathy. Circulation 2010, 121, 2169–2175. [Google Scholar] [CrossRef] [PubMed]
- Scott, E.; Hasbullah, J.S.; Ross, C.J.; Carleton, B.C. Reducing anthracycline-induced cardiotoxicity through pharmacogenetics. Pharmacogenomics 2018, 19, 1147–1150. [Google Scholar] [CrossRef] [PubMed]
- Hazebroek, M.R.; Moors, S.; Dennert, R.; van den Wijngaard, A.; Krapels, I.; Hoos, M.; Verdonschot, J.; Merken, J.J.; de Vries, B.; Wolffs, P.F.; et al. Prognostic relevance of gene-environment interactions in patients with dilated cardiomyopathy: Applying the MOGE(S) classification. J. Am. Coll. Cardiol. 2015, 66, 1313–1323. [Google Scholar] [CrossRef]
- Mann, S.A.; Castro, M.L.; Ohanian, M.; Guo, G.; Zodgekar, P.; Sheu, A.; Stockhammer, K.; Thompson, T.; Playford, D.; Subbiah, R.; et al. R222Q SCN5A mutation is associated with reversible ventricular ectopy and dilated cardiomyopathy. J. Am. Coll. Cardiol. 2012, 60, 1566–1573. [Google Scholar] [CrossRef] [PubMed]
- Skjølsvik, E.T.; Hasselberg, N.E.; Dejgaard, L.A.; Lie, Ø.H.; Andersen, K.; Holm, T.; Edvardsen, T.; Haugaa, K.H. Exercise is associated with impaired left ventricular systolic function in patients with Lamin A/C genotype. J. Am. Heart Assoc. 2020, 9, e012937. [Google Scholar] [CrossRef]
- Regitz-Zagrosek, V.; Roos-Hesselink, J.W.; Bauersachs, J.; Blomström-Lundqvist, C.; Cífkova, R.; De Bonis, M.; Iung, B.; Johnson, M.R.; Kintscher, U.; Kranke, P.; et al. 2018 ESC guidelines for the management of cardiovascular diseases during pregnancy: The Task Force for the Management of Cardi-ovascular Diseases during pregnancy of the European Society of Cardiology (ESC). Eur. Heart J. 2018, 39, 3165–3241. [Google Scholar] [CrossRef]
- De Boer, R.A.; Heymans, S.; Backs, J.; Carrier, L.; Coats, A.J.S.; Dimmeler, S.; Eschenhagen, T.; Filippatos, G.; Gepstein, L.; Hulot, J.S.; et al. Targeted therapies in genetic dilated and hypertrophic cardiomyopathies: From molecular mechanisms to therapeutic targets. A position paper from the Heart Failure Association (HFA) and the Working Group on Myocardial Function of the European Society of Cardiology (ESC). Eur. J. Heart Fail. 2022, 24, 406–420. [Google Scholar] [PubMed]
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Böhm, M.; Duboc, D.; Gimeno, J.; de Groote, P.; Imazio, M.; et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnautu, D.-A.; Cozma, D.; Lala, I.-R.; Arnautu, S.-F.; Tomescu, M.-C.; Andor, M. Risk Assessment and Personalized Treatment Options in Inherited Dilated Cardiomyopathies: A Narrative Review. Biomedicines 2024, 12, 1643. https://doi.org/10.3390/biomedicines12081643
Arnautu D-A, Cozma D, Lala I-R, Arnautu S-F, Tomescu M-C, Andor M. Risk Assessment and Personalized Treatment Options in Inherited Dilated Cardiomyopathies: A Narrative Review. Biomedicines. 2024; 12(8):1643. https://doi.org/10.3390/biomedicines12081643
Chicago/Turabian StyleArnautu, Diana-Aurora, Dragos Cozma, Ioan-Radu Lala, Sergiu-Florin Arnautu, Mirela-Cleopatra Tomescu, and Minodora Andor. 2024. "Risk Assessment and Personalized Treatment Options in Inherited Dilated Cardiomyopathies: A Narrative Review" Biomedicines 12, no. 8: 1643. https://doi.org/10.3390/biomedicines12081643