
The Biallelic Inheritance of Two Novel SCN1A Variants Results in Developmental and Epileptic Encephalopathy Responsive to Levetiracetam


 ,
,  , , , , , , ,
, , , , , , ,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical and Genetic Analysis
2.2. Mutagenesis and Nav1.1 Channel Expression
2.3. Electrophysiology
2.4. Statistical Analysis
3. Results
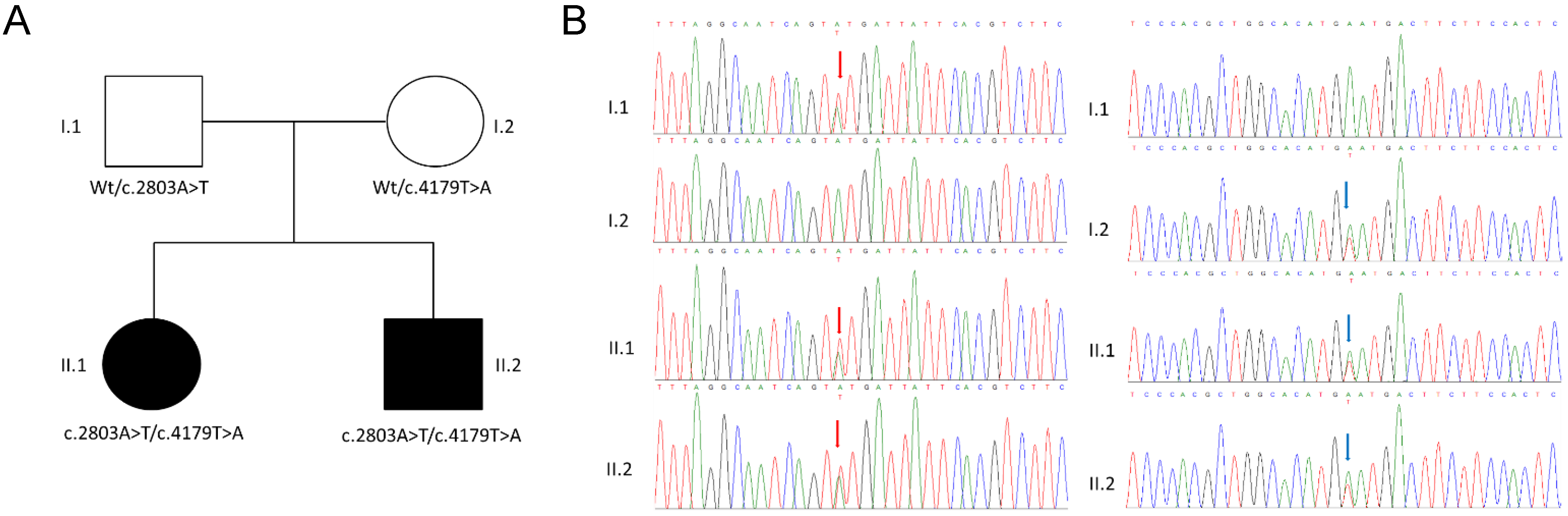
3.1. Clinical Description
3.2. Genetic Analysis
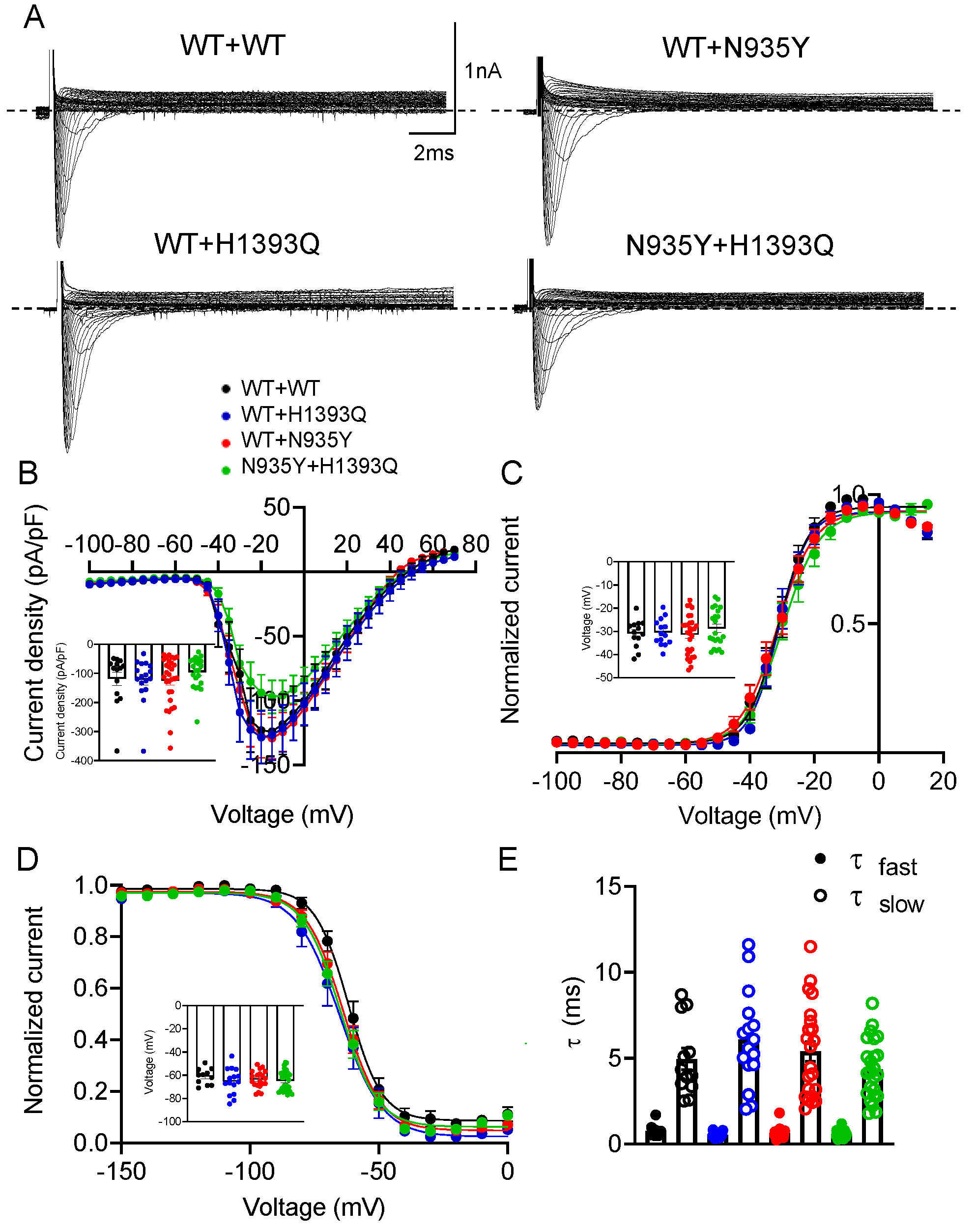
3.3. Functional Characterization of Mutant Nav1.1 Currents
4. Discussion
4.1. Genotype–Phenotype–Drug Response Correlation
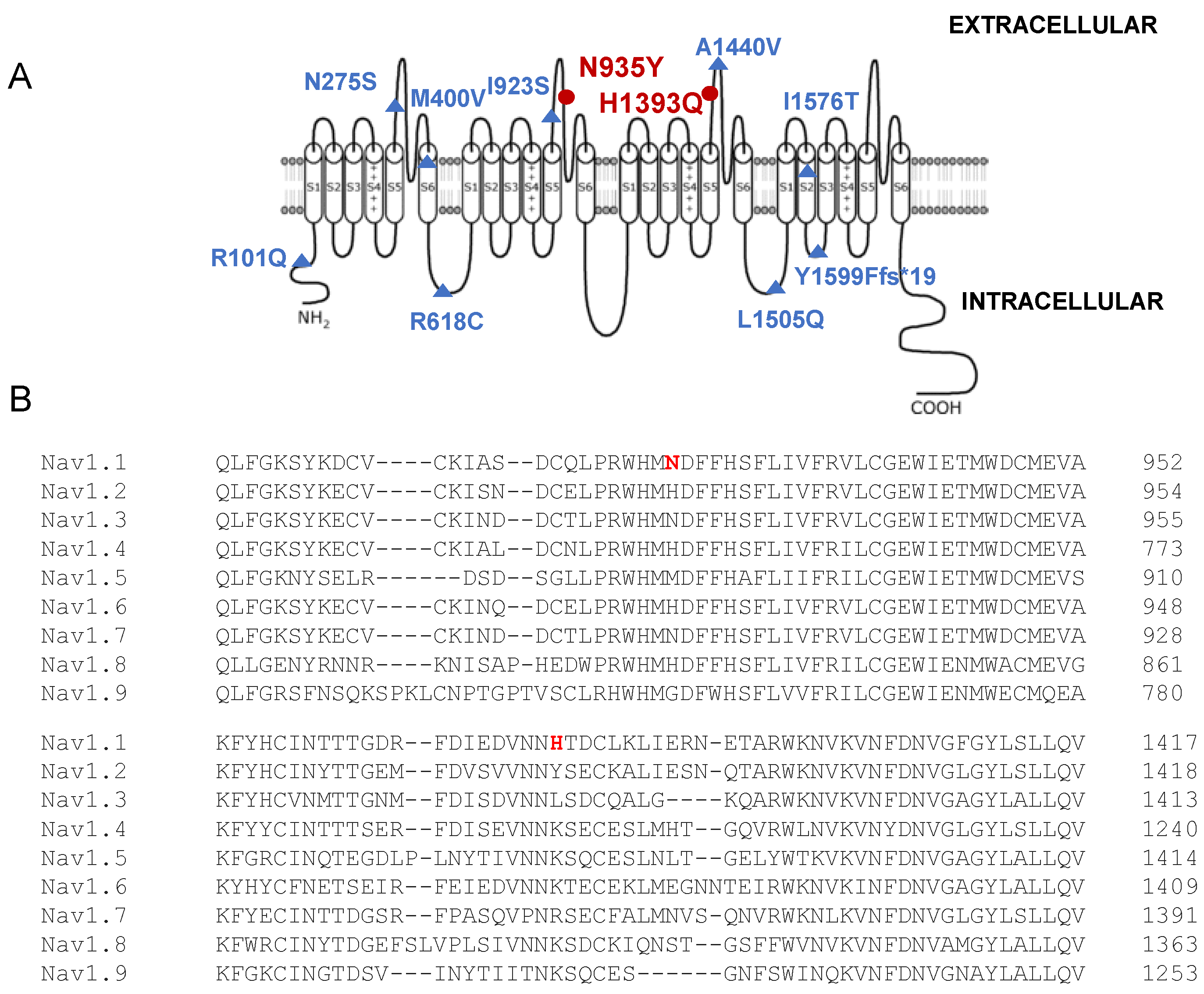
4.2. Structure–Function Correlation and Comparison with Similar SCN1A Variants
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brunklaus, A.; Lal, D. Sodium Channel Epilepsies and Neurodevelopmental Disorders: From Disease Mechanisms to Clinical Application. Dev. Med. Child Neurol. 2020, 62, 784–792. [Google Scholar] [CrossRef]
- Brunklaus, A.; Brünger, T.; Feng, T.; Fons, C.; Lehikoinen, A.; Panagiotakaki, E.; Vintan, M.-A.; Symonds, J.; Andrew, J.; Arzimanoglou, A.; et al. The Gain of Function SCN1A Disorder Spectrum: Novel Epilepsy Phenotypes and Therapeutic Implications. Brain 2022, 145, 3816–3831. [Google Scholar] [CrossRef]
- Bryson, A.; Petrou, S. SCN1A Channelopathies: Navigating from Genotype to Neural Circuit Dysfunction. Front. Neurol. 2023, 14, 1173460. [Google Scholar] [CrossRef]
- D’Adamo, M.C.; Liantonio, A.; Conte, E.; Pessia, M.; Imbrici, P. Ion Channels Involvement in Neurodevelopmental Disorders. Neuroscience 2020, 440, 337–359. [Google Scholar] [CrossRef]
- Sadleir, L.G.; Mountier, E.I.; Gill, D.; Davis, S.; Joshi, C.; DeVile, C.; Kurian, M.A.; Mandelstam, S.; Wirrell, E.; Nickels, K.C.; et al. Not All SCN1A Epileptic Encephalopathies Are Dravet Syndrome: Early Profound Thr226Met Phenotype. Neurology 2017, 89, 1035–1042. [Google Scholar] [CrossRef]
- Clatot, J.; Parthasarathy, S.; Cohen, S.; McKee, J.L.; Massey, S.; Somarowthu, A.; Goldberg, E.M.; Helbig, I. SCN1A Gain-of-Function Mutation Causing an Early Onset Epileptic Encephalopathy. Epilepsia 2023, 64, 1318–1330. [Google Scholar] [CrossRef]
- Brunklaus, A.; Feng, T.; Brünger, T.; Perez-Palma, E.; Heyne, H.; Matthews, E.; Semsarian, C.; Symonds, J.D.; Zuberi, S.M.; Lal, D.; et al. Gene Variant Effects across Sodium Channelopathies Predict Function and Guide Precision Therapy. Brain 2022, 145, 4275–4286. [Google Scholar] [CrossRef]
- Matricardi, S.; Cestèle, S.; Trivisano, M.; Kassabian, B.; Leroudier, N.; Vittorini, R.; Nosadini, M.; Cesaroni, E.; Siliquini, S.; Marinaccio, C.; et al. Gain of Function SCN1A Disease-Causing Variants: Expanding the Phenotypic Spectrum and Functional Studies Guiding the Choice of Effective Antiseizure Medication. Epilepsia 2023, 64, 1331–1347. [Google Scholar] [CrossRef]
- Brunklaus, A.; Ellis, R.; Stewart, H.; Aylett, S.; Reavey, E.; Jefferson, R.; Jain, R.; Chakraborty, S.; Jayawant, S.; Zuberi, S.M. Homozygous Mutations in the SCN1A Gene Associated with Genetic Epilepsy with Febrile Seizures plus and Dravet Syndrome in 2 Families. Eur. J. Paediatr. Neurol. EJPN Off. J. Eur. Paediatr. Neurol. Soc. 2015, 19, 484–488. [Google Scholar] [CrossRef]
- Tuncer, F.N.; Gormez, Z.; Calik, M.; Altiokka Uzun, G.; Sagiroglu, M.S.; Yuceturk, B.; Yuksel, B.; Baykan, B.; Bebek, N.; Iscan, A.; et al. A Clinical Variant in SCN1A Inherited from a Mosaic Father Cosegregates with a Novel Variant to Cause Dravet Syndrome in a Consanguineous Family. Epilepsy Res. 2015, 113, 5–10. [Google Scholar] [CrossRef]
- Aslan, M.; Ozgor, B.; Kirik, S.; Gungor, S. A Novel SCN1A Mutation: A Case Report. J. Pediatr. Neurosci. 2020, 15, 120–123. [Google Scholar] [CrossRef]
- Moretti, R.; Arnaud, L.; Bouteiller, D.; Trouillard, O.; Moreau, P.; Buratti, J.; Rastetter, A.; Keren, B.; Des Portes, V.; Toulouse, J.; et al. SCN1A-Related Epilepsy with Recessive Inheritance: Two Further Families. Eur. J. Paediatr. Neurol. 2021, 33, 121–124. [Google Scholar] [CrossRef]
- Marco Hernández, A.V.; Tomás Vila, M.; Caro Llopis, A.; Monfort, S.; Martinez, F. Case Report: Novel Homozygous Likely Pathogenic SCN1A Variant With Autosomal Recessive Inheritance and Review of the Literature. Front. Neurol. 2021, 12, 784892. [Google Scholar] [CrossRef]
- Van, L.T.K.; Hien, H.T.D.; Kieu, H.T.T.; Hieu, N.L.T.; Vinh, L.S.; Hoa, G.; Hang, D.T.T. De Novo Homozygous Variant of the SCN1A Gene in a Patient with Severe Dravet Syndrome Complicated by Acute Encephalopathy. Neurogenetics 2021, 22, 133–136. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. DbSNP: The NCBI Database of Genetic Variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of Protein-Coding Genetic Variation in 60,706 Humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Glusman, G.; Caballero, J.; Mauldin, D.E.; Hood, L.; Roach, J.C. Kaviar: An Accessible System for Testing SNV Novelty. Bioinformatics 2011, 27, 3216–3217. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public Archive of Relationships among Sequence Variation and Human Phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. DbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Bertelli, S.; Barbieri, R.; Pusch, M.; Gavazzo, P. Gain of Function of Sporadic/Familial Hemiplegic Migraine-Causing SCN1A Mutations: Use of an Optimized CDNA. Cephalalgia 2019, 39, 477–488. [Google Scholar] [CrossRef]
- Imbrici, P.; Maggi, L.; Mangiatordi, G.F.; Dinardo, M.M.; Altamura, C.; Brugnoni, R.; Alberga, D.; Pinter, G.L.; Ricci, G.; Siciliano, G.; et al. ClC-1 Mutations in Myotonia Congenita Patients: Insights into Molecular Gating Mechanisms and Genotype-Phenotype Correlation. J. Physiol. 2015, 593, 4181–4199. [Google Scholar] [CrossRef]
- Balla, C.; Conte, E.; Selvatici, R.; Marsano, R.M.; Gerbino, A.; Farnè, M.; Blunck, R.; Vitali, F.; Armaroli, A.; Brieda, A.; et al. Functional Characterization of Two Novel Mutations in SCN5A Associated with Brugada Syndrome Identified in Italian Patients. Int. J. Mol. Sci. 2021, 22, 6513. [Google Scholar] [CrossRef]
- Farinato, A.; Altamura, C.; Imbrici, P.; Maggi, L.; Bernasconi, P.; Mantegazza, R.; Pasquali, L.; Siciliano, G.; Lo Monaco, M.; Vial, C.; et al. Pharmacogenetics of Myotonic HNav1.4 Sodium Channel Variants Situated near the Fast Inactivation Gate. Pharmacol. Res. 2019, 141, 224–235. [Google Scholar] [CrossRef]
- Martins Custodio, H.; Clayton, L.M.; Bellampalli, R.; Pagni, S.; Silvennoinen, K.; Caswell, R.; Brunklaus, A.; Guerrini, R.; Koeleman, B.P.C.; Lemke, J.R.; et al. Widespread Genomic Influences on Phenotype in Dravet Syndrome, a “monogenic” Condition. Brain 2023, 146, 3885–3897. [Google Scholar] [CrossRef]
- Rhodes, T.H.; Lossin, C.; Vanoye, C.G.; Wang, D.W.; George, A.L.J. Noninactivating Voltage-Gated Sodium Channels in Severe Myoclonic Epilepsy of Infancy. Proc. Natl. Acad. Sci. USA 2004, 101, 11147–11152. [Google Scholar] [CrossRef]
- Berecki, G.; Bryson, A.; Polster, T.; Petrou, S. Biophysical Characterization and Modelling of SCN1A Gain-of-Function Predicts Interneuron Hyperexcitability and a Predisposition to Network Instability through Homeostatic Plasticity. Neurobiol. Dis. 2023, 179, 106059. [Google Scholar] [CrossRef]
- Koch, N.A.; Sonnenberg, L.; Hedrich, U.B.S.; Lauxmann, S.; Benda, J. Loss or Gain of Function? Effects of Ion Channel Mutations on Neuronal Firing Depend on the Neuron Type. Front. Neurol. 2023, 14, 1194811. [Google Scholar] [CrossRef]
- Ogiwara, I.; Miyamoto, H.; Morita, N.; Atapour, N.; Mazaki, E.; Inoue, I.; Takeuchi, T.; Itohara, S.; Yanagawa, Y.; Obata, K.; et al. Nav1.1 Localizes to Axons of Parvalbumin-Positive Inhibitory Interneurons: A Circuit Basis for Epileptic Seizures in Mice Carrying an Scn1a Gene Mutation. J. Neurosci. 2007, 27, 5903–5914. [Google Scholar] [CrossRef]
- Han, S.; Tai, C.; Westenbroek, R.E.; Yu, F.H.; Cheah, C.S.; Potter, G.B.; Rubenstein, J.L.; Scheuer, T.; de la Iglesia, H.O.; Catterall, W.A. Autistic-like Behaviour in Scn1a+/− Mice and Rescue by Enhanced GABA-Mediated Neurotransmission. Nature 2012, 489, 385–390. [Google Scholar] [CrossRef]
- Bender, A.C.; Morse, R.P.; Scott, R.C.; Holmes, G.L.; Lenck-Santini, P.-P. SCN1A Mutations in Dravet Syndrome: Impact of Interneuron Dysfunction on Neural Networks and Cognitive Outcome. Epilepsy Behav. 2012, 23, 177–186. [Google Scholar] [CrossRef]
- van Hugte, E.J.H.; Lewerissa, E.I.; Wu, K.M.; Scheefhals, N.; Parodi, G.; van Voorst, T.W.; Puvogel, S.; Kogo, N.; Keller, J.M.; Frega, M.; et al. SCN1A-Deficient Excitatory Neuronal Networks Display Mutation-Specific Phenotypes. Brain 2023, 146, 5153–5167. [Google Scholar] [CrossRef]
- Balestrini, S.; Mei, D.; Sisodiya, S.M.; Guerrini, R. Steps to Improve Precision Medicine in Epilepsy. Mol. Diagn. Ther. 2023, 27, 661–672. [Google Scholar] [CrossRef]
- Guerrini, R.; Conti, V.; Mantegazza, M.; Balestrini, S.; Galanopoulou, A.S.; Benfenati, F. Developmental and Epileptic Encephalopathies: From Genetic Heterogeneity to Phenotypic Continuum. Physiol. Rev. 2023, 103, 433–513. [Google Scholar] [CrossRef]
- Volnova, A.; Tsytsarev, V.; Ganina, O.; Vélez-Crespo, G.E.; Alves, J.M.; Ignashchenkova, A.; Inyushin, M. The Anti-Epileptic Effects of Carbenoxolone In Vitro and In Vivo. Int. J. Mol. Sci. 2022, 23, 663. [Google Scholar] [CrossRef]
- Uchino, K.; Tanaka, Y.; Ikezawa, W.; Deshimaru, M.; Kubota, K.; Watanabe, T.; Katsurabayashi, S.; Iwasaki, K.; Hirose, S. Astrocyte Ca2+ Signaling Is Facilitated in Scn1a+/− Mouse Model of Dravet Syndrome. Biochem. Biophys. Res. Commun. 2023, 643, 169–174. [Google Scholar] [CrossRef]
- Goisis, R.C.; Chiavegato, A.; Gomez-Gonzalo, M.; Marcon, I.; Requie, L.M.; Scholze, P.; Carmignoto, G.; Losi, G. GABA Tonic Currents and Glial Cells Are Altered during Epileptogenesis in a Mouse Model of Dravet Syndrome. Front. Cell. Neurosci. 2022, 16, 919493. [Google Scholar] [CrossRef]
- Contreras-García, I.J.; Cárdenas-Rodríguez, N.; Romo-Mancillas, A.; Bandala, C.; Zamudio, S.R.; Gómez-Manzo, S.; Hernández-Ochoa, B.; Mendoza-Torreblanca, J.G.; Pichardo-Macías, L.A. Levetiracetam Mechanisms of Action: From Molecules to Systems. Pharmaceuticals 2022, 15, 475. [Google Scholar] [CrossRef]
- Fukuyama, K.; Okada, M. Brivaracetam and Levetiracetam Suppress Astroglial L-Glutamate Release through Hemichannel via Inhibition of Synaptic Vesicle Protein. Int. J. Mol. Sci. 2022, 23, 4473. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Li, Z.; Jin, X.; Zhao, Y.; Huang, G.; Huang, X.; Shen, Z.; Cao, Y.; Dong, M.; Lei, J.; et al. Comparative Structural Analysis of Human Nav1.1 and Nav1.5 Reveals Mutational Hotspots for Sodium Channelopathies. Proc. Natl. Acad. Sci. USA 2021, 118, e2100066118. [Google Scholar] [CrossRef] [PubMed]
- Stefanaki, E.; Aggelakou, V.; Orfanou, M.; Kokori, E.; Boutoufianakis, S. Epilepsy with a de Novo Missense Mutation in the Sodium Channel A1 Subunit: A Case Report. Acta Paediatr. 2006, 95, 1703–1706. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhang, Y.; Liang, J.; Liu, X.; Ma, X.; Wu, H.; Xu, K.; Qin, J.; Qi, Y.; Wu, X. SCN1A, SCN1B, and GABRG2 Gene Mutation Analysis in Chinese Families with Generalized Epilepsy with Febrile Seizures Plus. J. Hum. Genet. 2008, 53, 769–774. [Google Scholar] [CrossRef]
- Mantegazza, M.; Broccoli, V. SCN1A/Na(V) 1.1 Channelopathies: Mechanisms in Expression Systems, Animal Models, and Human IPSC Models. Epilepsia 2019, 60 (Suppl. S3), S25–S38. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleotide Change | Amino Acid Change | Protein Location | Biallelic Inheritance | N. of Patients and Phenotype | Pathogenicity Score or Functional Study | Reference |
|---|---|---|---|---|---|---|
| c.302G>A c.4727T>C | R101Q I1576T | Intracellular N-term DIV S2 | Compound heterozygous | 2, Dravet syndrome | Pathogenic Likely pathogenic | [10] |
| c.824A>G | N275S | DI S5-S6 extracellular loop | Homozygous | 1, GEFS+ | Likely pathogenic | [12] |
| c.1198A>G | M400V | DI S6 | Homozygous | 1, Dravet syndrome; and 1, febrile seizures | Pathogenic | [9] |
| c.1852C>T | R618C | DI-DII cytoplasmic linker | Homozygous | 2, febrile seizures | Uncertain significance/Likely pathogenic | [9] |
| c.2768T>G | I923S | DII S5-S6 extracellular loop | Homozygous | 1, GEFS+ | Uncertain significance | [12] |
| c.2770A>T c.4146T>A | N935Y H1393Q | DII S5-S6 extracellular loop DIII S5-S6 extracellular loop | Compound heterozygous | 2, developmental and epileptic encephalopathy | Likely pathogenic Likely pathogenic | This study |
| c.4319C>T | A1440V | DIII S5-S6 extracellular loop | Homozygous de novo | 1, Dravet syndrome and acute encephalopathy | Likely pathogenic | [14] |
| c.4513A>C | K1505Q | DIII-DIV cytoplasmic linker | Homozygous | 2, GEFS+ | Likely pathogenic | [13] |
| c.4796delA | Y1599Ffs*19 | DIV S2-S3 intracellular loop | Homozygous | 1, Dravet syndrome | Pathogenic | [11] |
| Channel Types | Current Density (−10 mV) | Voltage-Dependent Activation | Steady-State Inactivation | Kinetics of Fast Inactivation (−10 mV) | Recovery from Inactivation | |||
|---|---|---|---|---|---|---|---|---|
| (pA/pF) | V1/2 (mV) | k (mV) | V1/2 (mV) | k (mV) | tfast (ms) | tslow (ms) | t (ms) | |
| Nav1.1 WT 7 μg | −56.0 ± 9.9 (12) | −26.2 ± 3.0 (10) | 3.9 ± 0.7 (10) | −59.5 ± 2.2 (11) | 6.6 ± 0.8 (11) | 0.72 ± 0.12 (14) | 4.25 ± 0.67 (14) | 2.01 ± 0.4 (6) |
| H1393Q 7 μg | −67.5 ± 16.9 (11) | −24.9 ± 2.2 (9) | 4.7 ± 0.7 (9) | −60.8 ± 2.3 (9) | 6.9 ± 0.9 (9) | 0.70 ± 0.12 (14) | 4.69 ± 0.57 (14) | 1.77 ± 0.15 (4) |
| N935Y 7 μg | −64.7 ± 10.8 (10) | −25.5 ± 2.6 (9) | 3.1 ± 0.5 (9) | −61.2 ± 2.4 (14) | 5.3 ± 0.3 (14) | 0.66 ± 0.10 (14) | 5.02 ± 0.62 (14) | 1.44 ± 0.25 (4) |
| WT + WT 7 μg + 7 μg | −118.4 ± 23.3 (14) | −30.9 ± 1.7 (13) | 3.3 ± 0.4 (13) | −61.1 ± 2.1 (11) | 5.0 ± 0.2 (11) | 0.78 ± 0.10 (12) | 4.96 ± 0.63 (12) | 1.55 ± 0.13 (10) |
| WT + H1393Q 7 μg + 7 μg | −121.6 ± 18.3 (17) | −30.4 ± 1.5 (14) | 3.2 ± 0.4 (14) | −65.3 ± 2.9 (15) | 5.02 ± 0.2 (15) | 0.54 ± 0.04 (16) | 6.08 ± 0.69 (16) | 1.33 ± 0.15 (9) |
| WT + N935Y 7 μg + 7 μg | −124.4 ± 15.1 (18) | −31.3 ± 1.6 (29) | 3.5 ± 0.2 (29) | −63.6 ± 1.7 (20) | 5.8 ± 0.3 (20) | 0.66 ± 0.06 (22) | 5.4 ± 0.58 (22) | 2.06 ± 0.49 (8) |
| N935Y + H1393Q 7 μg + 7 μg | −95.8 ± 11.7 (22) | −28.7 ± 1.7 (22) | 4.0 ± 0.4 (22) | −64.7 ± 1.7 (27) | −5.3 ± 0.2 (27) | 0.60 ± 0.04 (25) | 4.36 ± 0.34 (25) | 2.71 ± 0.56 (8) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dinoi, G.; Conte, E.; Palumbo, O.; Benvenuto, M.; Coppola, M.A.; Palumbo, P.; Lastella, P.; Boccanegra, B.; Di Muro, E.; Castori, M.; et al. The Biallelic Inheritance of Two Novel SCN1A Variants Results in Developmental and Epileptic Encephalopathy Responsive to Levetiracetam. Biomedicines 2024, 12, 1698. https://doi.org/10.3390/biomedicines12081698
Dinoi G, Conte E, Palumbo O, Benvenuto M, Coppola MA, Palumbo P, Lastella P, Boccanegra B, Di Muro E, Castori M, et al. The Biallelic Inheritance of Two Novel SCN1A Variants Results in Developmental and Epileptic Encephalopathy Responsive to Levetiracetam. Biomedicines. 2024; 12(8):1698. https://doi.org/10.3390/biomedicines12081698
Chicago/Turabian StyleDinoi, Giorgia, Elena Conte, Orazio Palumbo, Mario Benvenuto, Maria Antonietta Coppola, Pietro Palumbo, Patrizia Lastella, Brigida Boccanegra, Ester Di Muro, Marco Castori, and et al. 2024. "The Biallelic Inheritance of Two Novel SCN1A Variants Results in Developmental and Epileptic Encephalopathy Responsive to Levetiracetam" Biomedicines 12, no. 8: 1698. https://doi.org/10.3390/biomedicines12081698
APA StyleDinoi, G., Conte, E., Palumbo, O., Benvenuto, M., Coppola, M. A., Palumbo, P., Lastella, P., Boccanegra, B., Di Muro, E., Castori, M., Carella, M., Sciruicchio, V., de Tommaso, M., Liantonio, A., De Luca, A., La Neve, A., & Imbrici, P. (2024). The Biallelic Inheritance of Two Novel SCN1A Variants Results in Developmental and Epileptic Encephalopathy Responsive to Levetiracetam. Biomedicines, 12(8), 1698. https://doi.org/10.3390/biomedicines12081698