


Beta2-Adrenergic Suppression of Neuroinflammation in Treatment of Parkinsonism, with Relevance for Neurodegenerative and Neoplastic Disorders

Abstract
:1. Introduction
1.1. Early Observations
1.2. α-Synuclein Gene (SNCA) and PD
1.3. PD Causation from Neuroinflammation
1.4. Epidemiological Associations between Beta2-Adrenergic Effects and PD
1.5. Life-Style Factors That May Influence the Incidence or Progression of PD
1.6. Genomic Modeling of the Potential Anti-Inflammatory Effects of Epinephrine
1.7. Anti-Inflammatory Studies in the Cytokine Release Syndrome (CRS) Assay
2. Materials and Methods
2.1. Analyses of Gene-Expression Signatures of β2-Adrenergic Agonists and Related Compounds in the Harvard/MIT Broad Institute Genomic Database, CLUE
2.2. CRS Assay Methodology
Study Design
- Mice were irradiated and injected with PBMCs from a single donor on Day 0.
- Animals were monitored for body weight and clinical observations once daily.
- Animals were euthanized before study end if they showed >20% body weight loss or a body condition score of ≥2.
- Treatment compounds were delivered in drinking water starting on Day 4 and continued for the duration of study.
- Mice were dosed on Day 6 with monoclonal antibodies OKT3 or anti-CD28 to induce cytokine release [41], or with PBS as control.
- Mice were bled via the retro-orbital (RO) method at 6 h post-dose on Day 6. Blood was processed and measured for human cytokines and chemokines using a MILLIPEX Human Cytokine/Chemokine/Growth Panel A Magnetic Bead Panel, Cat# HCYTOMAG-60K.
- Body weight and clinical CRS score were assessed daily starting on Day 6. Mice were euthanized by CO2 asphyxiation at 72 h post-dose on Day 9.

2.3. Statistics
3. Results
3.1. Analyses of Gene Expression Signatures of β2-Adrenergic Agonists and Related Compounds in the Harvard/MIT Broad Institute Genomic Database, CLUE
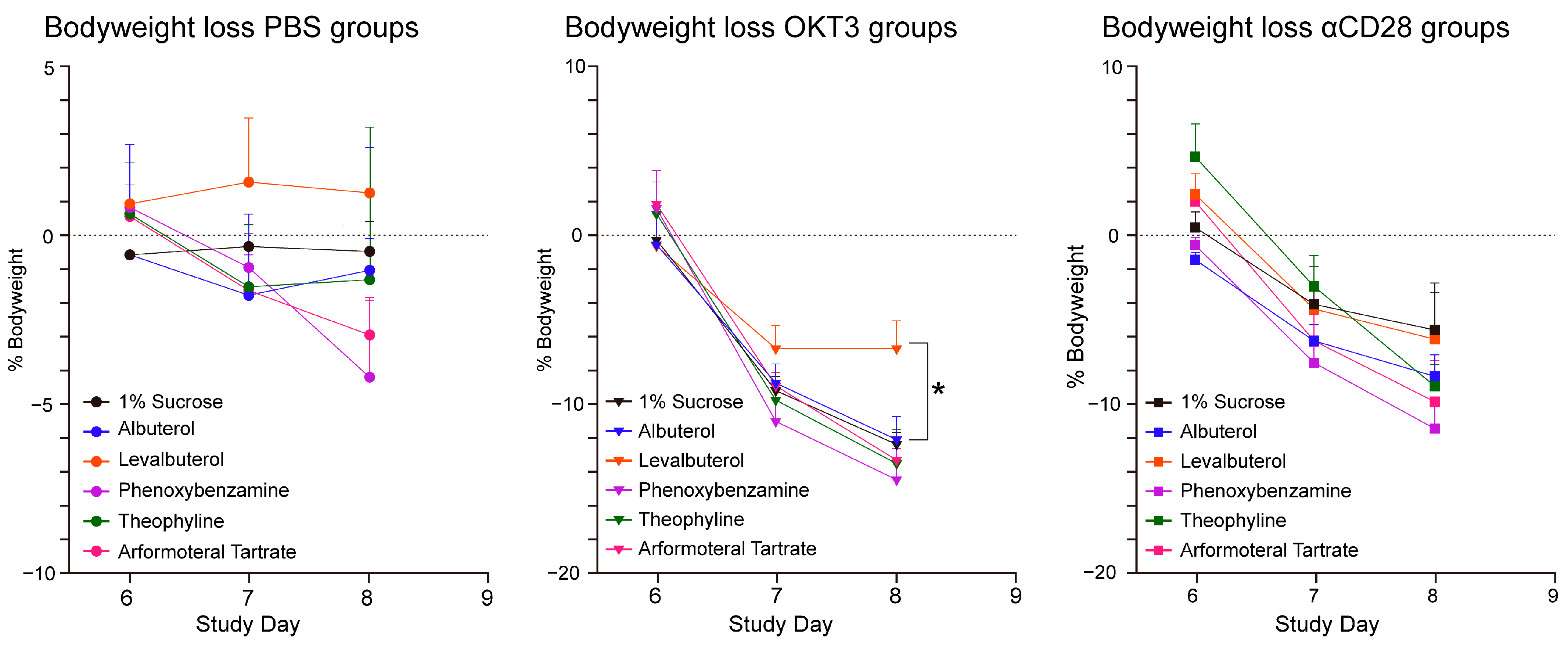
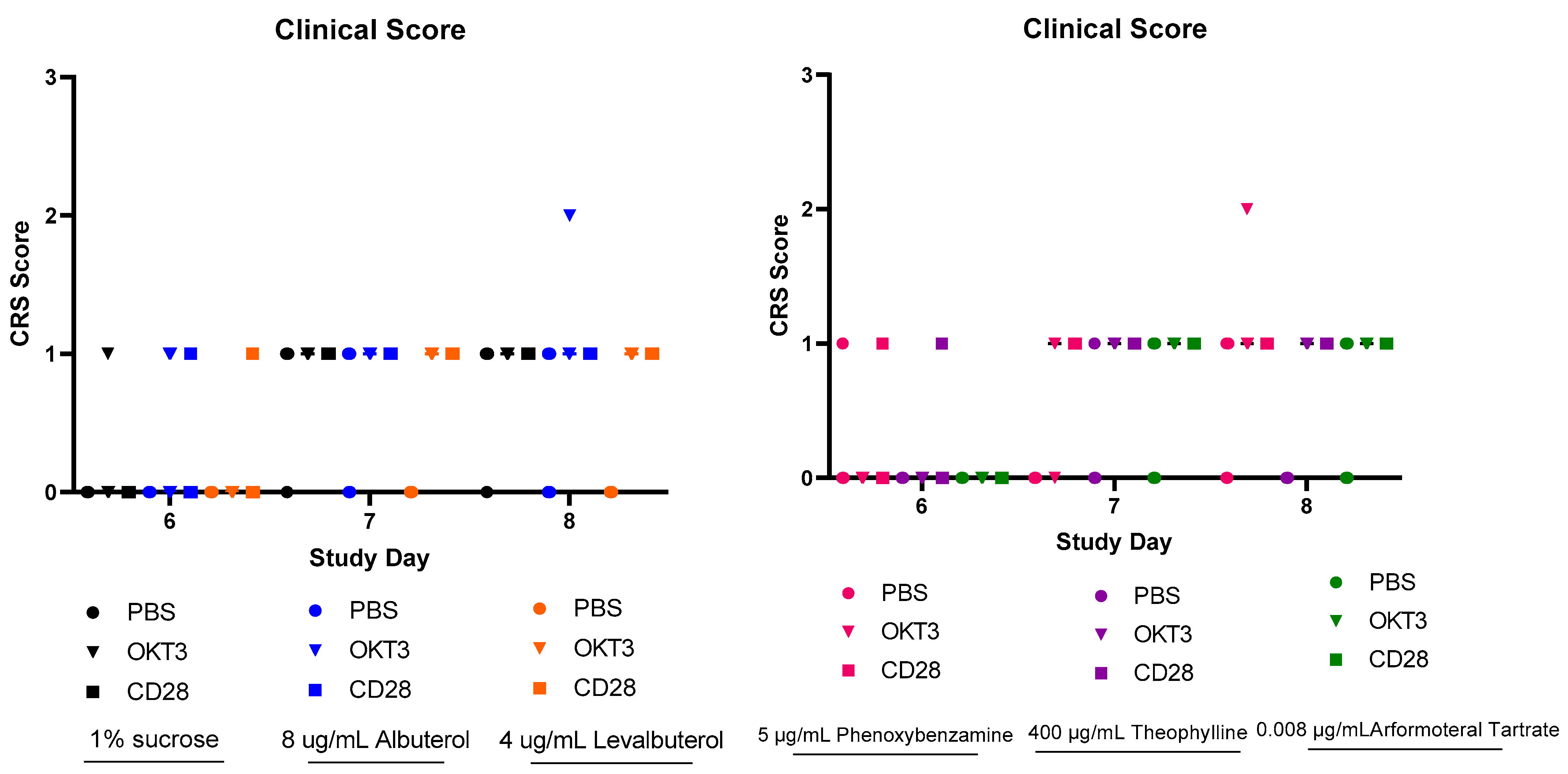
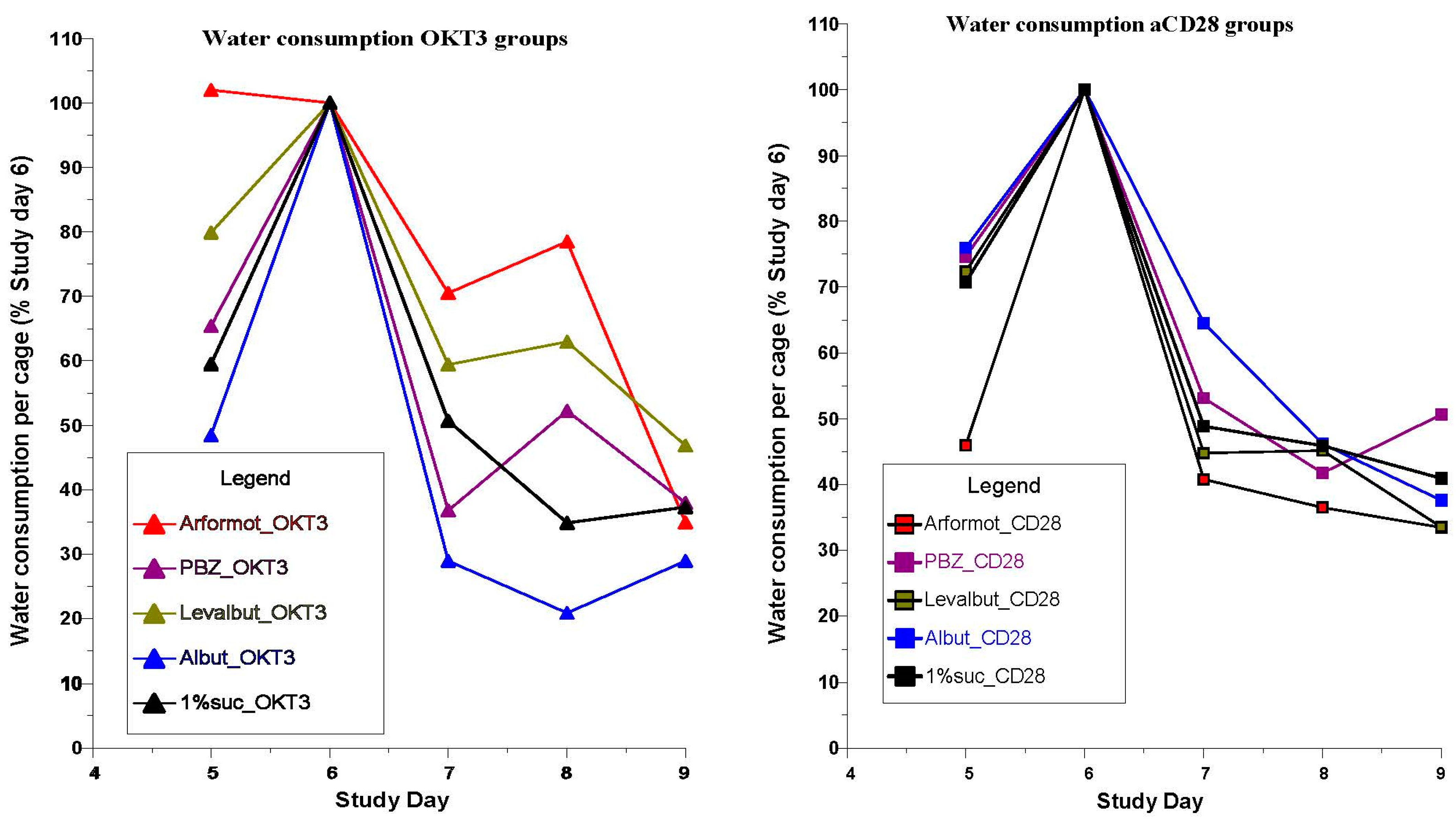
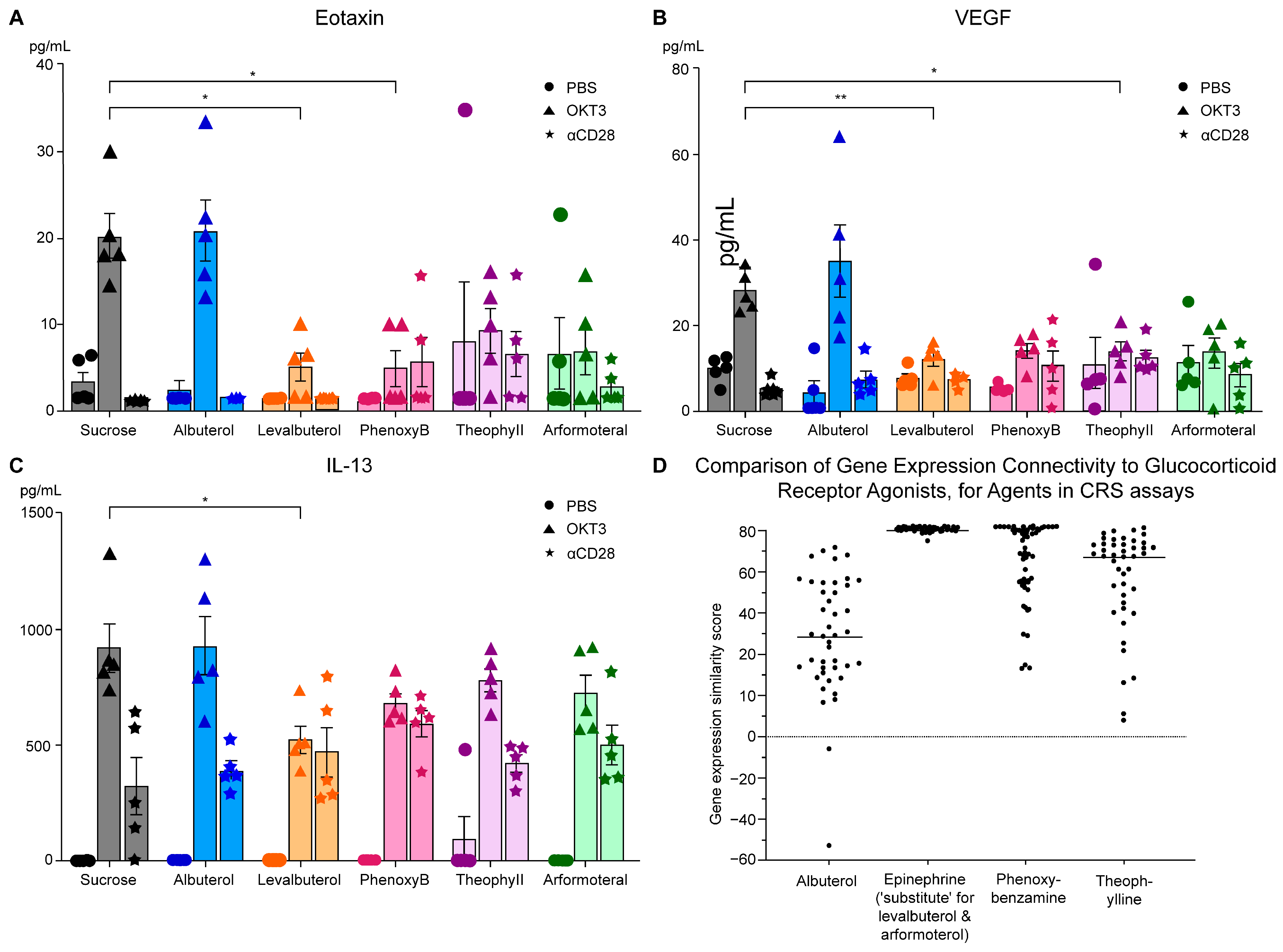
3.2. CRS Assays of Agents with β2-Adrenergic and Related Activities
- (a)
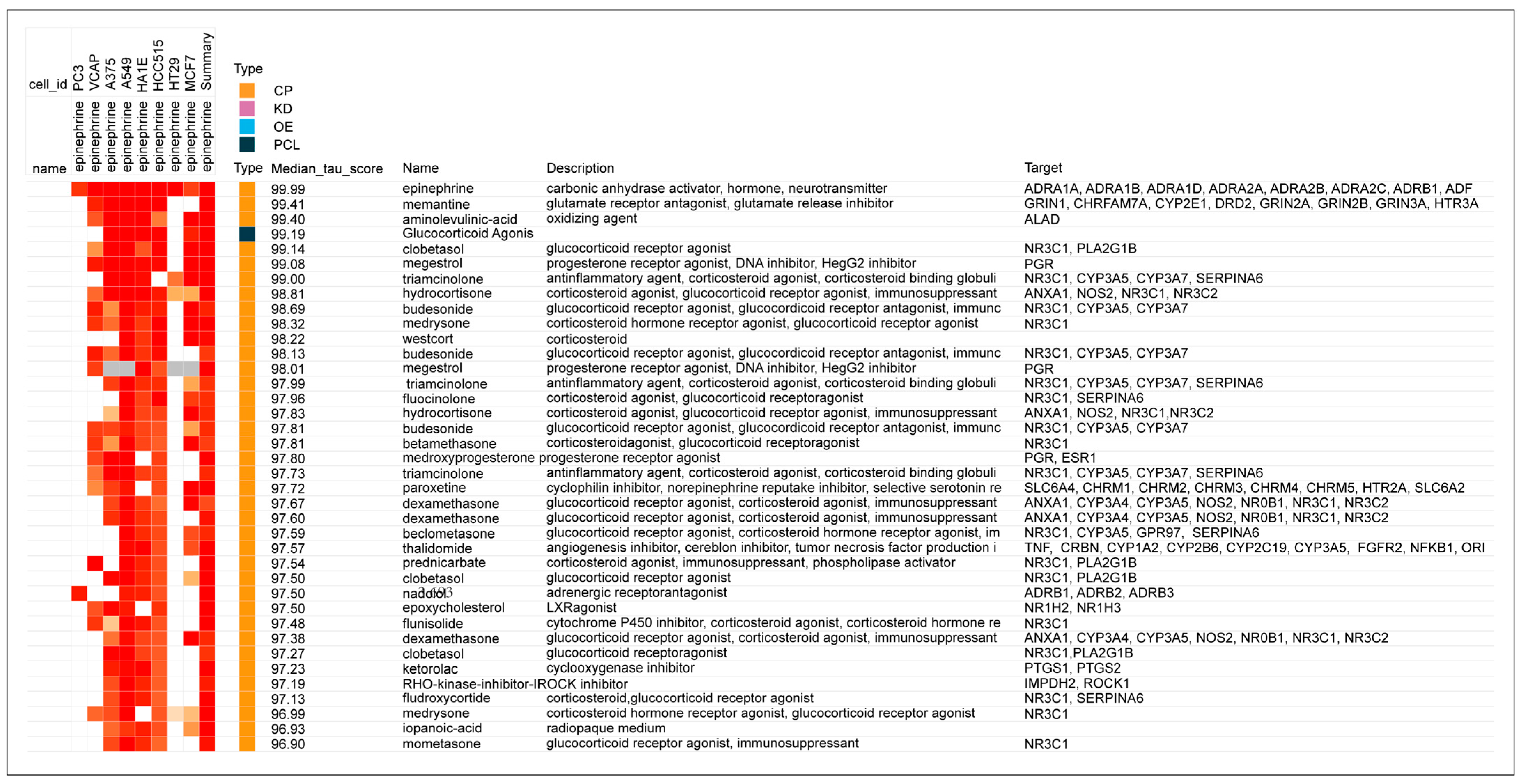
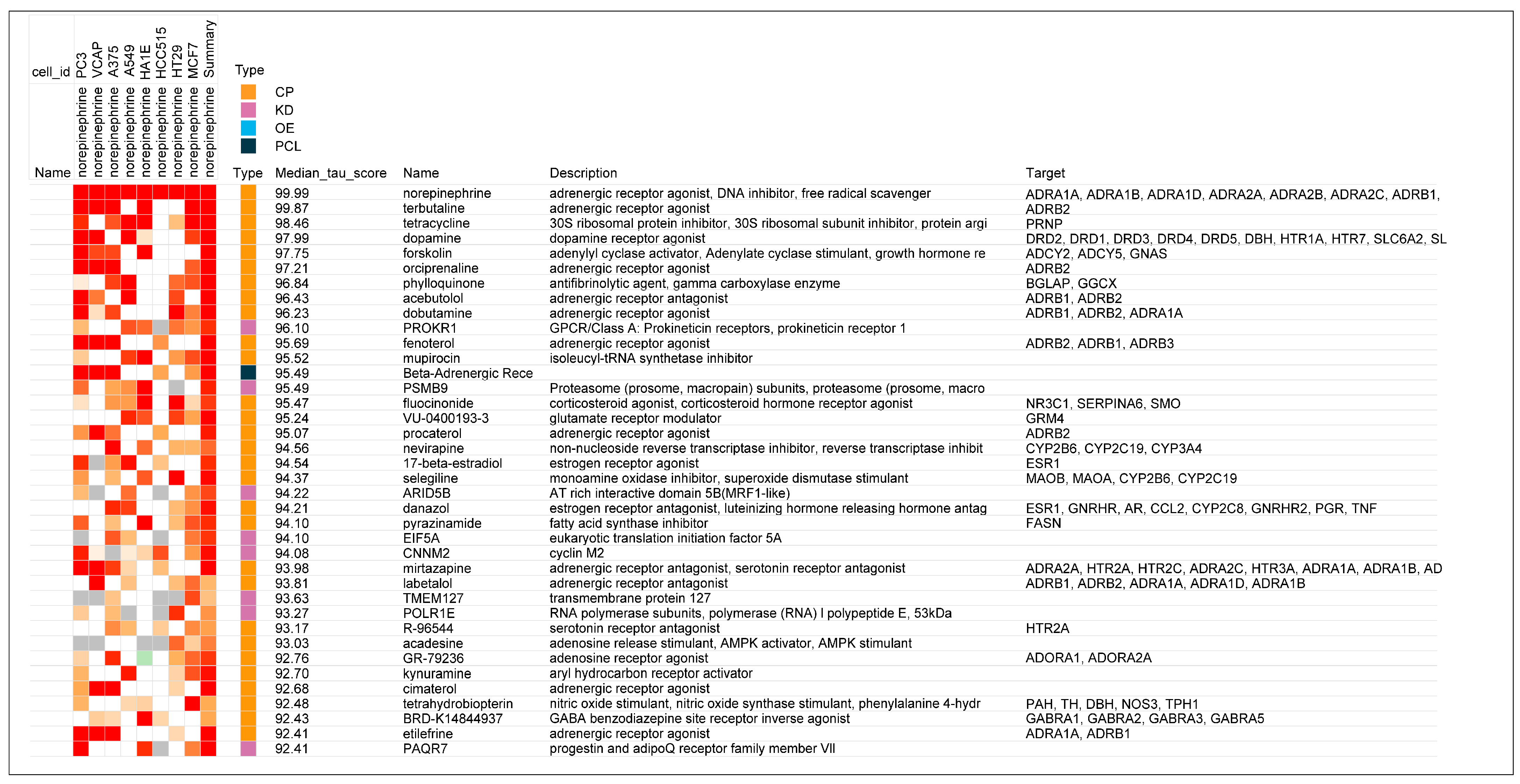
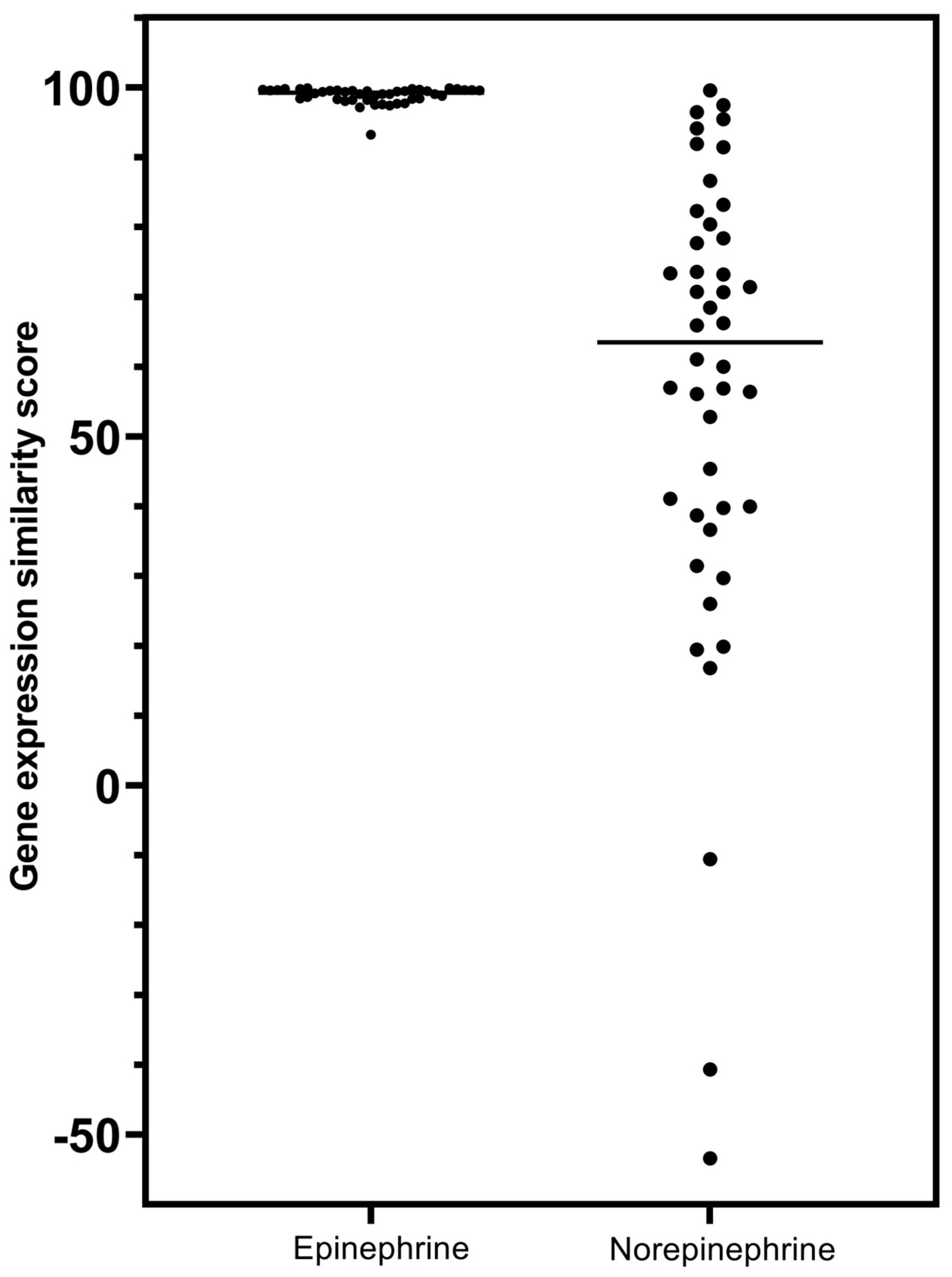
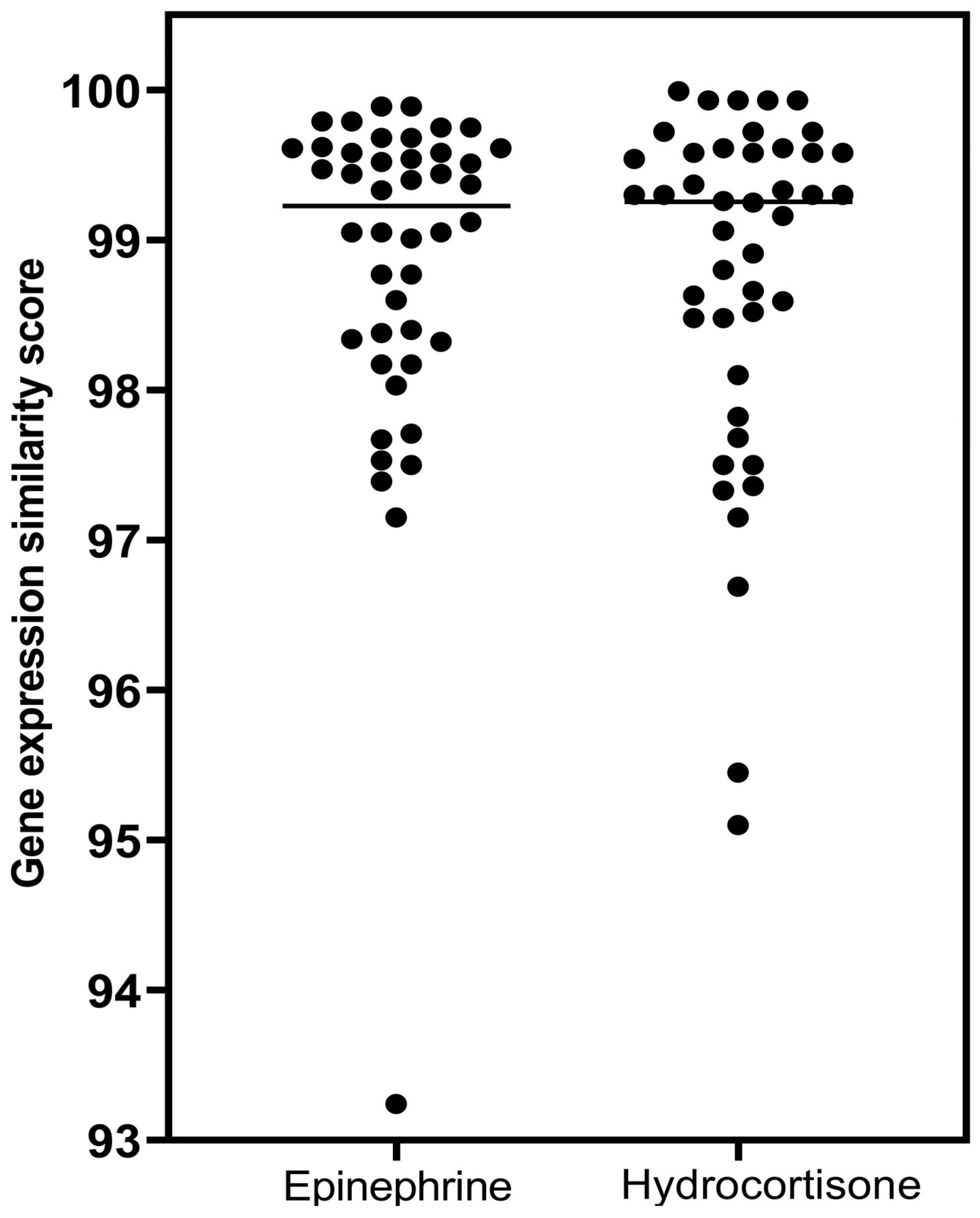
- The prototypical beta2-adrenergic agonist epinephrine, but not norepinephrine (minimal beta2-adrenergic activity), showed prominent similarity (connectivity) to the gene-expression of glucocorticoid receptor agonists when modeled in the Harvard/MIT genomic database, CLUE. This was seen consistently and progressively in the three data outputs from this cloud-based software, including a heat map of individual assays with eight malignant tumor cultures, cumulative scoring of gene-expression among the eight cultures for similarity to glucocorticoid receptor agonists, and the individual scores and ranks for epinephrine and norepinephrine gene expression for each member of the glucocorticoid class in the CLUE database;
- (b)
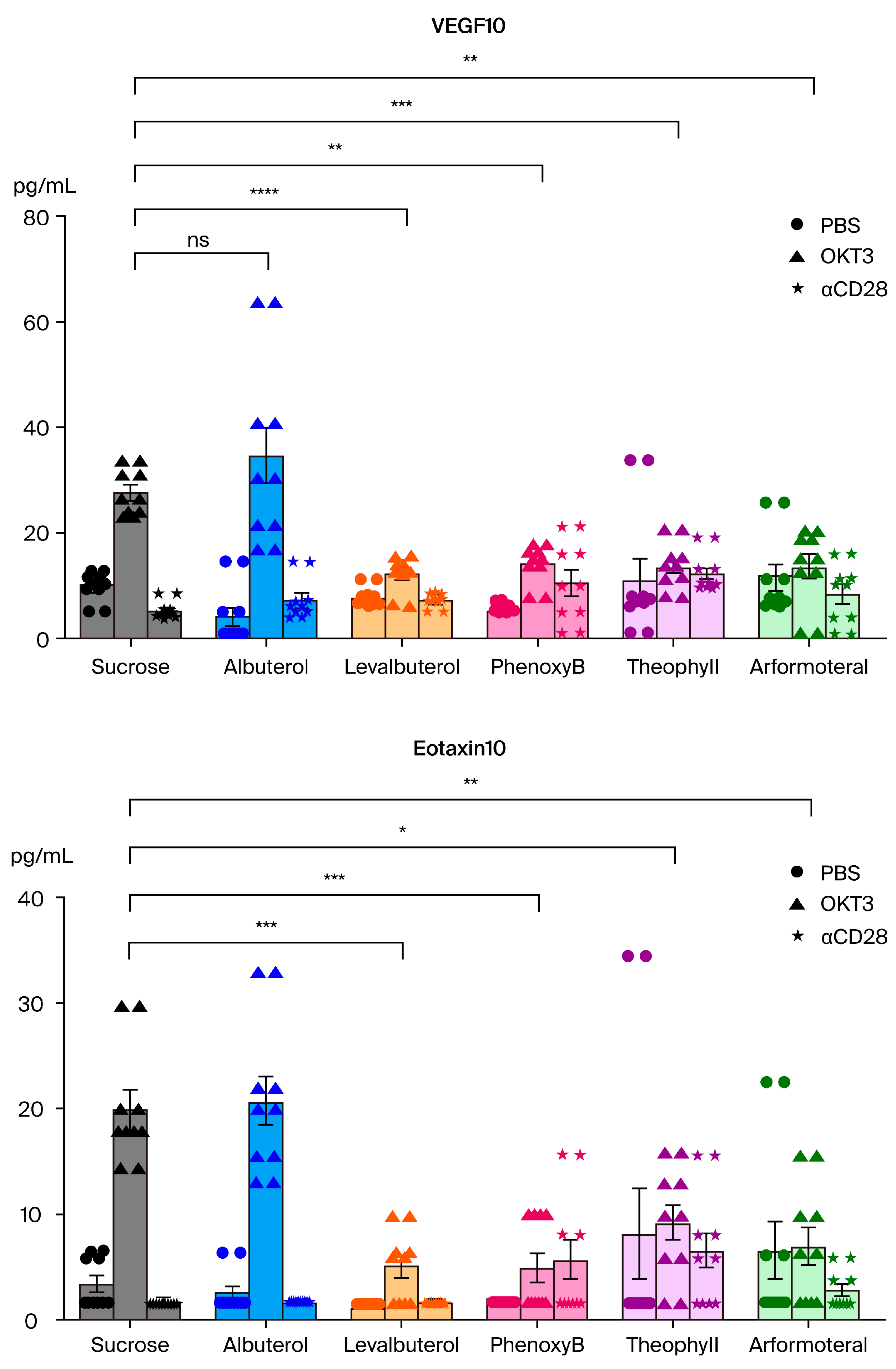
- The association of beta2-adrenergic activity with glucocorticoids suggested potential anti-inflammatory activity for these drugs and possible value in suppression of the neuroinflammation that is characteristic of the progression of PD and other neurodegenerative disorders. Several beta2-adrenergic agonists and adrenergic-related drugs were tested for anti-inflammatory activity in mice engrafted with human PBMCs. Pretreatment with these drugs prior to challenge with monoclonal antibodies known to result in activation and release of cytokines and chemokines inhibited elaboration of eotaxin-1/CCL11, VEGF165, and IL-13 to varying extents.
4. Discussion
4.1. Absence of Classical Glucocorticoid Toxicity
4.2. Continued Focus on Exercise as PD Therapy
4.3. Levalbuterol and Tyrosine Hydroxylase
4.4. Clinical Translation of the Findings
4.5. Limitations
4.6. Future Studies
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Magistrelli, L.; Comi, C. Beta2-Adrenoceptor Agonists in Parkinson’s Disease and Other Synucleinopathies. J. Neuroimmune Pharmacol. 2020, 15, 74–81. [Google Scholar] [CrossRef]
- Hishida, R.; Kurahashi, K.; Narita, S.; Baba, T.; Matsunaga, M. “Wearing-off” and β2-adrenoceptor agonist in Parkinson’s disease. Lancet 1992, 339, 870. [Google Scholar] [CrossRef]
- Alexander, G.M.; Schwartzman, R.J.; Nukes, T.A.; Grothusen, J.R.; Hooker, M.D. β2-adrenergic agonist as adjunct therapy to levodopa in Parkinson’s disease. Neurology 1994, 44, 1511–1513. [Google Scholar] [CrossRef]
- Uc, E.Y.; Lambert, C.P.; Harik, S.I.; Rodnitzky, R.L.; Evans, W.J. Albuterol improves response to levodopa and increases skeletal muscle mass in patients with fluctuating Parkinson disease. Clin. Neuropharmacol. 2003, 26, 207–212. [Google Scholar] [CrossRef]
- Mittal, S.; Bjørnevik, K.; Im, D.S.; Flierl, A.; Dong, X.; Locascio, J.J.; Abo, K.M.; Long, E.; Jin, M.; Xu, B.; et al. β2-Adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson’s disease. Science 2017, 357, 891–898. [Google Scholar] [CrossRef]
- Devine, M.J.; Gwinn, K.; Singleton, A.; Hardy, J. Parkinson’s disease and alpha-synuclein expression. Mov. Disord. 2011, 26, 2160–2168. [Google Scholar] [CrossRef]
- Wang, S.; Liu, F.; Tan, K.S.; Ser, H.L.; Tan, L.T.; Lee, L.H.; Tan, W. Effect of (R)-salbutamol on the switch of phenotype and metabolic pattern in LPS-induced macrophage cells. J. Cell. Mol. Med. 2020, 24, 722–736. [Google Scholar] [CrossRef]
- Friere, M.; Pergolizzi, R.; Millon, C.; Dominguez, P.J. Cytokine, chemokine, and nitric oxide release in stimulated small airway epithelial cells treated with β2 agonist enantiomers of salbutamol. J. Allergy Clin. Immunol. 2000, 105, 292–293. [Google Scholar] [CrossRef]
- Severn, A.; Rapson, N.T.; Hunter, C.A.; Liew, F.Y. Regulation of tumor necrosis factor production by adrenaline and beta-adrenergic agonists. J. Immunol. 1992, 148, 3441–3445. [Google Scholar] [CrossRef]
- Elenkov, I.J.; Chrousos, G.P. Stress hormones, proinflammatory and antiinflammatory cytokines, and autoimmunity. Ann. N. Y. Acad. Sci. 2002, 966, 290–303. [Google Scholar] [CrossRef]
- Peterson, L.; Ismond, K.P.; Chapman, E.; Flood, P. Potential benefits of therapeutic use of β2-adrenergic receptor agonists in neuroprotection and Parkinsonmus disease. J. Immunol. Res. 2014, 2014, 103780. [Google Scholar] [CrossRef] [PubMed]
- Elenkov, I.J.; Wilder, R.L.; Chrousos, G.P.; Vizi, E.S. The sympathetic nerve--an integrative interface between two supersystems: The brain and the immune system. Pharmacol. Rev. 2000, 52, 595–638. [Google Scholar]
- Sharma, M.; Flood, P.M. β-arrestin2 regulates the anti-inflammatory effects of Salmeterol in lipopolysaccharide-stimulated BV2 cells. J. Neuroimmunol. 2018, 25, 10–19. [Google Scholar] [CrossRef]
- Mazzoni, L.; Naef, R.; Chapman, I.D.; Morley, J. Hyperresponsiveness of the airways to histamine following exposure of guinea pigs to racemic mixtures and distomers of β2-selective sympathomimetics. Pulm. Pharmacol. 1994, 7, 367–376. [Google Scholar] [CrossRef]
- Ibe, B.O.; Abdallah, M.F.; Raj, J.U. Mechanisms by which S-albuterol induces human bronchial smooth muscle cell proliferation. Int. Arch. Allergy Immunol. 2008, 146, 321–333. [Google Scholar] [CrossRef]
- Baramki, D.; Koester, J.; Anderson, A.J.; Borish, L. Modulation of T-cell function by (R)- and (S)-isomers of albuterol: Anti-inflammatory influences of (R)-isomers are negated in the presence of the (S)-isomer. J. Allergy Clin. Immunol. 2002, 109, 449–454. [Google Scholar] [CrossRef]
- Chorley, B.N.; Li, Y.; Fang, S.; Park, J.A.; Adler, K.B. (R)-albuterol elicits antiinflammatory effects in human airway epithelial cells via iNOS. Am. J. Respir. Cell. Mol. Biol. 2006, 34, 119–127. [Google Scholar] [CrossRef]
- Gronich, N.; Abernethy, D.R.; Auriel, E.; Lavi, I.; Rennert, G.; Saliba, W. β2-adrenoceptor agonists and antagonists and risk of Parkinson’s disease. Mov. Disord. 2018, 33, 1465–1471. [Google Scholar] [CrossRef]
- Chen, C.L.; Wang, S.Y.; Chen, T.C.; Chuang, C.S. Association between β2-Adrenoreceptor Medications and Risk of Parkinson’s Disease: A Meta-Analysis. Medicina 2021, 57, 1006. [Google Scholar] [CrossRef]
- Moore, C.G.; Schenkman, M.; Kohrt, W.M.; Delitto, A.; Hall, D.A.; Corcos, D. Study in Parkinson disease of exercise (SPARX): Translating high-intensity exercise from animals to humans. Contemp. Clin. Trials 2013, 36, 90–98. [Google Scholar] [CrossRef]
- Schenkman, M.; Moore, C.G.; Kohrt, W.M.; Hall, D.A.; Delitto, A.; Comella, C.L.; Josbeno, D.A.; Christiansen, C.L.; Berman, B.D.; Kluger, B.M.; et al. Effect of High-Intensity Treadmill Exercise on Motor Symptoms in Patients with De Novo Parkinson Disease: A Phase 2 Randomized Clinical Trial. JAMA Neurol. 2018, 75, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Kjaer, M. Epinephrine and some other hormonal responses to exercise in man: With special reference to physical training. Int. J. Sports Med. 1989, 10, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Baron, J.A. Cigarette smoking and Parkinson’s disease. Neurology 1986, 36, 1490–1496. [Google Scholar] [CrossRef] [PubMed]
- Mappin-Kasirer, B.; Pan, H.; Lewington, S.; Kizza, J.; Gray, R.; Clarke, R.; Peto, R. Tobacco smoking and the risk of Parkinson disease: A 65-year follow-up of 30,000 male British doctors. Neurology 2020, 94, e2132–e2138. [Google Scholar] [CrossRef] [PubMed]
- Fratiglioni, L.; Wang, H.X. Smoking and Parkinson’s and Alzheimer’s disease: Review of the epidemiological studies. Behav. Brain Res. 2000, 113, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Allam, M.F.; Campbell, M.J.; Hofman, A.; Del Castillo, A.S.; Fernandez-Crehuet Navajas, R. Smoking and Parkinson’s disease: Systematic review of prospective studies. Mov. Disord. 2004, 19, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Hernan, M.A.; Takkouche, B.; Caamano-Isorna, F.; Gestal-Otero, J.J. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann. Neurol. 2002, 52, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Cryer, P.E.; Haymond, M.W.; Santiago, J.V.; Shah, S.D. Norepinephrine and epinephrine release and adrenergic mediation of smoking-associated hemodynamic and metabolic events. N. Engl. J. Med. 1976, 295, 573–577. [Google Scholar] [CrossRef]
- Grassi, G.; Seravalle, G.; Calhoun, D.A.; Bolla, G.; Mancia, G. Cigarette smoking and the adrenergic nervous system. Clin. Exp. Hypertens. A 1992, 14, 251–260. [Google Scholar] [CrossRef]
- Grassi, G.; Seravalle, G.; Calhoun, D.A.; Bolla, G.B.; Giannattasio, C.; Marabini, M.; Del Bo, A.; Mancia, G. Mechanisms responsible for sympathetic activation by cigarette smoking in humans. Circulation 1994, 90, 248–253. [Google Scholar] [CrossRef]
- Ragonese, P.; Salemi, G.; Morgante, L.; Aridon, P.; Epifanio, A.; Buffa, D.; Scoppa, F.; Savettieri, G. A case-control study on cigarette, alcohol, and coffee consumption preceding Parkinson’s disease. Neuroepidemiology 2003, 22, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.K.; Tan, C.; Fook-Chong, S.M.; Lum, S.Y.; Chai, A.; Chung, H.; Shen, H.; Zhao, Y.; Teoh, M.L.; Yih, Y.; et al. Dose-dependent protective effect of coffee, tea, and smoking in Parkinson’s disease: A study in ethnic Chinese. J. Neurol. Sci. 2003, 216, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Zhang, S.M.; Hernán, M.A.; Kawachi, I.; Colditz, G.A.; Speizer, F.E.; Willett, W.C. Prospective study of caffeine consumption and risk of Parkinson’s disease in men and women. Ann. Neurol. 2001, 50, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Palacios, N.; Gao, X.; McCullough, M.L.; Schwarzschild, M.A.; Shah, R.; Gapstur, S.; Ascherio, A. Caffeine and risk of Parkinson’s disease in a large cohort of men and women. Mov. Disord. 2012, 27, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Li, S. Dose-response meta-analysis on coffee, tea and caffeine consumption with risk of Parkinson’s disease. Geriatr. Gerontol. Int. 2014, 14, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Chen, J.F. Caffeine and Parkinson’s Disease: Multiple Benefits and Emerging Mechanisms. Front. Neurosci. 2020, 14, 602697. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.; Frölich, J.C.; Carr, R.K.; Watson, J.T.; Hollifield, J.W.; Shand, D.G.; Oates, J.A. Effects of caffeine on plasma renin activity, catecholamines and blood pressure. N. Engl. J. Med. 1978, 298, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Benowitz, N.L.; Jacob, P., 3rd; Mayan, H.; Denaro, C. Sympathomimetic effects of paraxanthine and caffeine in humans. Clin. Pharmacol. Ther. 1995, 58, 684–691. [Google Scholar] [CrossRef]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef]
- Shultz, L.D.; Keck, J.; Burzenski, L.; Jangalwe, S.; Vaidya, S.; Greiner, D.L.; Brehm, M.A. Humanized mouse models of immunological diseases and precision medicine. Mamm. Genome 2019, 30, 123–142. [Google Scholar] [CrossRef]
- Ye, C.; Yang, H.; Cheng, M.; Shultz, L.D.; Greiner, D.L.; Brehm, M.A.; Keck, J.G. A rapid, sensitive, and reproducible in vivo PBMC humanized murine model for determining therapeutic-related cytokine release syndrome. FASEB J. 2020, 34, 12963–12975. [Google Scholar] [CrossRef] [PubMed]
- Perez, D.M. α1-Adrenergic Receptors in Neurotransmission, Synaptic Plasticity, and Cognition. Front. Pharmacol. 2020, 11, 581098. [Google Scholar] [CrossRef] [PubMed]
- Inchiosa, M.A., Jr. Anti-tumor activity of phenoxybenzamine and its inhibition of histone deacetylases. PLoS ONE 2018, 13, e0198514. [Google Scholar] [CrossRef] [PubMed]
- Inchiosa, M.A., Jr. Further investigation of the potential anti-neoplastic, anti-inflammatory and immunomodulatory actions of phenoxybenzamine using the Broad Institute CLUE platform. In The Preprint Server for Biology; Cold Spring Harbor Laboratory: Laurel Hollow, NY, USA, 2023. [Google Scholar]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Lanau, F.; Zenner, M.T.; Civelli, O.; Hartman, D.S. Epinephrine and norepinephrine act as potent agonists at the recombinant human dopamine D4 receptor. J. Neurochem. 1997, 68, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zeng, L.; Zhao, S. Ligands of Adrenergic Receptors: A Structural Point of View. Biomolecules 2021, 11, 936. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Tan, E.K. Targeting LRRK2 in Parkinson’s disease. Cell Rep. Med. 2022, 3, 100778. [Google Scholar] [CrossRef] [PubMed]
- Cresto, N.; Gardier, C.; Gubinelli, F.; Gaillard, M.C.; Liot, G.; West, A.B.; Brouillet, E. The unlikely partnership between LRRK2 and alpha-synuclein in Parkinson’s disease. Eur. J. Neurosci. 2019, 49, 339–363. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Dzamko, N. Recent Developments in LRRK2-Targeted Therapy for Parkinson’s Disease. Drugs 2019, 79, 1037–1051. [Google Scholar] [CrossRef]
- Jennings, D.; Huntwork-Rodriguez, S.; Henry, A.G.; Sasaki, J.C.; Meisner, R.; Diaz, D.; Solanoy, H.; Wang, X.; Negrou, E.; Bondar, V.V.; et al. Preclinical and clinical evaluation of the LRRK2 inhibitor DNL201 for Parkinson’s disease. Sci. Transl. Med. 2022, 14, eabj2658. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Purvis, G.S.D.; Solito, E.; Thiemermann, C. Annexin-A1: Therapeutic Potential in Microvascular Disease. Front. Immunol. 2019, 10, 938. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.M.; Wolters, P.L.; Dombi, E.; Baldwin, A.; Whitcomb, P.; Fisher, M.J.; Weiss, B.; Kim, A.; Bornhorst, M.; Shah, A.C.; et al. Selumetinib in Children with Inoperable Plexiform Neurofibromas. N. Engl. J. Med. 2020, 382, 1430–1442. [Google Scholar] [CrossRef] [PubMed]
- Ciombor, K.K.; Bekaii-Saab, T. Selumetinib for the treatment of cancer. Expert. Opin. Investig. Drugs 2015, 24, 111–123. [Google Scholar] [CrossRef]
- Davies, A.O.; Lefkowitz, R.J. Regulation of β-adrenergic receptors by steroid hormones. Annu. Rev. Physiol. 1984, 46, 119–130. [Google Scholar] [CrossRef]
- Aksoy, M.O.; Mardini, I.A.; Yang, Y.; Bin, W.; Zhou, S.; Kelsen, S.G. Glucocorticoid effects on the β-adrenergic receptor-adenylyl cyclase system of human airway epithelium. J. Allergy Clin. Immunol. 2002, 109, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Charo, I.F.; Ransohoff, R.M. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 2006, 354, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Doran, E.; Cai, F.; Holweg, C.T.J.; Wong, K.; Brumm, J.; Arron, J.R. Interleukin-13 in Asthma and Other Eosinophilic Disorders. Front. Med. 2017, 4, 139. [Google Scholar] [CrossRef]
- Teixeira, A.L.; Gama, C.S.; Rocha, N.P.; Teixeira, M.M. Revisiting the Role of Eotaxin-1/CCL11 in Psychiatric Disorders. Front. Psychiatry 2018, 9, 241. [Google Scholar] [CrossRef]
- Chandra, G.; Rangasamy, S.B.; Roy, A.; Kordower, J.H.; Pahan, K. Neutralization of RANTES and Eotaxin Prevents the Loss of Dopaminergic Neurons in a Mouse Model of Parkinson Disease. J. Biol. Chem. 2016, 291, 15267–15281. [Google Scholar] [CrossRef]
- Bhatt, S.P.; Rabe, K.F.; Hanania, N.A.; Vogelmeier, C.F.; Cole, J.; Bafadhel, M.; Christenson, S.A.; Papi, A.; Singh, D.; Laws, E.; et al. Dupilumab for COPD with Type 2 Inflammation Indicated by Eosinophil Counts. N. Engl. J. Med. 2023, 389, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Goel, H.L.; Burkart, C.; Burman, L.; Chong, Y.E.; Barber, A.G.; Geng, Y.; Zhai, L.; Wang, M.; Kumar, A.; et al. Inhibition of VEGF binding to neuropilin-2 enhances chemosensitivity and inhibits metastasis in triple-negative breast cancer. Sci. Transl. Med. 2023, 15, eadf1128. [Google Scholar] [CrossRef] [PubMed]
- Mabeta, P.; Steenkamp, V. The VEGF/VEGFR Axis Revisited: Implications for Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 15585. [Google Scholar] [CrossRef] [PubMed]
- Schacke, H.; Docke, W.D.; Asadullah, K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol. Ther. 2002, 96, 23–43. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, C.; Wellstein, A.; Riegel, A.T. Hormones and Related Agents in the Therapy of Cancer. In Goodman & Gilman’s The Pharmacological Basis of Therapeutics; Brunton, L.L., Ed.; McGraw Hill: New York, NY, USA, 2018; pp. 1237–1247. [Google Scholar]
- Lin, K.T.; Wang, L.H. New dimension of glucocorticoids in cancer treatment. Steroids 2016, 111, 84–88. [Google Scholar] [CrossRef]
- Greenstein, S.; Ghias, K.; Krett, N.L.; Rosen, S.T. Mechanisms of glucocorticoid-mediated apoptosis in hematological malignancies. Clin. Cancer Res. 2002, 8, 1681–1694. [Google Scholar]
- Schacke, H.; Rehwinkel, H. Dissociated glucocorticoid receptor ligands. Curr. Opin. Investig. Drugs 2004, 5, 524–528. [Google Scholar]
- Schacke, H.; Berger, M.; Rehwinkel, H.; Asadullah, K. Selective glucocorticoid receptor agonists (SEGRAs): Novel ligands with an improved therapeutic index. Mol. Cell Endocrinol. 2007, 275, 109–117. [Google Scholar] [CrossRef]
- Keenan, C.R.; Lew, M.J.; Stewart, A.G. Biased signaling from the glucocorticoid receptor: Renewed opportunity for tailoring glucocorticoid activity. Biochem. Pharmacol. 2016, 112, 6–12. [Google Scholar] [CrossRef]
- Kleiman, A.; Tuckermann, J.P. Glucocorticoid receptor action in beneficial and side effects of steroid therapy: Lessons from conditional knockout mice. Mol. Cell Endocrinol. 2007, 275, 98–108. [Google Scholar] [CrossRef] [PubMed]
- De Bosscher, K.; Haegeman, G.; Elewaut, D. Targeting inflammation using selective glucocorticoid receptor modulators. Curr. Opin. Pharmacol. 2010, 10, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Nicolaides, N.C.; Chrousos, G.; Kino, T. Glucocorticoid Receptor. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Lesovaya, E.A.; Chudakova, D.; Baida, G.; Zhidkova, E.M.; Kirsanov, K.I.; Yakubovskaya, M.G.; Budunova, I.V. The long winding road to the safer glucocorticoid receptor (GR) targeting therapies. Oncotarget 2022, 13, 408–424. [Google Scholar] [CrossRef] [PubMed]
- Haming, D.; Simoes-Costa, M.; Uy, B.; Valencia, J.; Sauka-Spengler, T.; Bronner-Fraser, M. Expression of sympathetic nervous system genes in Lamprey suggests their recruitment for specification of a new vertebrate feature. PLoS ONE 2011, 6, e26543. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.O.; Lefkowitz, R.J. Agonist-promoted high affinity state of the β-adrenergic receptor in human neutrophils: Modulation by corticosteroids. J. Clin. Endocrinol. Metab. 1981, 53, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J. Clinical physiology of adrenergic receptor regulation. Am. J. Physiol. 1982, 243, E43–E47. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, P.J.; Middleton, E.; Jr Coffey, R.G. Stimulation of leukocyte adenyl cyclase by hydrocortisone and isoproterenol in asthmatic and no asthmatic subjects. J. Allergy Clin. Immunol. 1972, 50, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Marone, G.; Lichtenstein, L.M.; Plaut, M. Hydrocortisone and human lymphocytes: Increases in cyclic adenosine 3′:5′-monophosphate and potentiation of adenylate cyclase-activating agents. J. Pharmacol. Exp. Ther. 1980, 215, 469–478. [Google Scholar] [PubMed]
- Parker, C.W.; Huber, M.G.; Baumann, M.L. Alterations in cyclic AMP metabolism in human bronchial asthma. 3. Leukocyte and lymphocyte responses to steroids. J. Clin. Investig. 1973, 52, 1342–1348. [Google Scholar] [CrossRef]
- Adams, M.; Meijer, O.C.; Wang, J.; Bhargava, A.; Pearce, D. Homodimerization of the glucocorticoid receptor is not essential for response element binding: Activation of the phenylethanolamine N-methyltransferase gene by dimerization-defective mutants. Mol. Endocrinol. 2003, 17, 2583–2592. [Google Scholar] [CrossRef]
- Tai, T.C.; Claycomb, R.; Her, S.; Bloom, A.K.; Wong, D.L. Glucocorticoid responsiveness of the rat phenylethanolamine N-methyltransferase gene. Mol. Pharmacol. 2002, 61, 1385–1392. [Google Scholar] [CrossRef]
- Dolhun, R.; Macpherson, C.; Long, K.; Quinn, L. Make Your Move; Exercise_Guide_Singles_02.01.24; The Michael J. Fox Foundation for Parkinson’s Research: New York, NY, USA, 2024. [Google Scholar]
- Chauquet, S.; Willis, E.F.; Grice, L.; Harley, S.B.; Powell, J.E.; Wray, N.R.; Nguyen, Q.; Ruitenberg, M.J.; Shah, S.; Vukovic, J. Exercise rejuvenates microglia and reverses T cell accumulation in the aged female mouse brain. Aging Cell 2024, 23, e14172. [Google Scholar] [CrossRef] [PubMed]
- Augusto-Oliveira, M.; Arrifano, G.P.; Leal-Nazaré, C.G.; Santos-Sacramento, L.; Lopes-Araújo, A.; Royes, L.F.F.; Crespo-Lopez, M.E. Exercise Reshapes the Brain: Molecular, Cellular, and Structural Changes Associated with Cognitive Improvements. Mol. Neurobiol. 2023, 60, 6950–6974. [Google Scholar] [CrossRef]
- Flydal, M.I.; Krakenes, T.-A.; Tai, M.D.S.; Tran, M.P.A.; Teigen, K.; Martinez, A. Levalbuterol lowers the feedback inhibition by dopamine and delays misfolding and aggregation in tyrosine hydroxylase. Biochimie 2021, 183, 126–132. [Google Scholar] [CrossRef]
- Milgrom, H.; Skoner, D.P.; Bensch, G.; Kim, K.T.; Claus, R.; Baumgartner, R.A. Low-dose levalbuterol in children with asthma: Safety and efficacy in comparison with placebo and racemic albuterol. J. Allergy Clin. Immunol. 2001, 108, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Sriprasart, T.; Waterer, G.; Garcia, G.; Rubin, A.; Andrade, M.A.; Roguska, A.; Phansalkar, A.; Fulmali, S.; Martin, A.; Mittal, L.; et al. Safety of SABA monotherapy in asthma management: A systematic review and meta-analysis. Adv. Ther. 2023, 40, 133–158. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Description |
|---|---|
| A375 | Human malignant melanoma |
| A549 | Human non-small cell carcinoma |
| HA1E | Human kidney epithelial immortalized |
| HCC515 | Human non-small cell lung adenocarcinoma |
| HEPG2 | Human hepatocellular carcinoma cell line |
| MCF7 | Human breast adenocarcinoma |
| PC3 | Human prostate adenocarcinoma |
| VCAP | Human metastatic prostate cancer |
| HT29 | Human colorectal adenocarcinoma |
| Group | N | Drinking Water * (D4) | Compound (D6) | Dose (mg/kg) | Dosing Route | Dosing Frequency | Bleed ** | |
|---|---|---|---|---|---|---|---|---|
| RO | Terminal | |||||||
| 1 | 5 | No pre-treatment | PBS | N/A | IV | Single dose | D6 | D9 |
| 2 | 5 | OKT3 | 0.25 | IV | Single dose | D6 | D9 | |
| 3 | 5 | Anti-CD28 | 1 | IV | Single dose | D6 | D9 | |
| 4 | 5 | Albuterol 8 ug/mL | PBS | N/A | IV | Single dose | D6 | D9 |
| 5 | 5 | OKT3 | 0.25 | IV | Single dose | D6 | D9 | |
| 6 | 5 | Anti-CD28 | 1 | IV | Single dose | D6 | D9 | |
| 7 | 5 | Levalbuterol 4 ug/mL | PBS | N/A | IV | Single dose | D6 | D9 |
| 8 | 5 | OKT3 | 0.25 | IV | Single dose | D6 | D9 | |
| 9 | 5 | Anti-CD28 | 1 | IV | Single dose | D6 | D9 | |
| 10 | 5 | Phenoxy-benzamine 5 ug/mL | PBS | N/A | IV | Single dose | D6 | D9 |
| 11 | 5 | OKT3 | 0.25 | IV | Single dose | D6 | D9 | |
| 12 | 5 | Anti-CD28 | 1 | IV | Single dose | D6 | D9 | |
| 13 | 5 | Theophylline 400 ug/mL | PBS | N/A | IV | Single dose | D6 | D9 |
| 14 | 5 | OKT3 | 0.25 | IV | Single dose | D6 | D9 | |
| 15 | 5 | Anti-CD28 | 1 | IV | Single dose | D6 | D9 | |
| 16 | 5 | Arformoterol tartrate 0.008 ug/mL | PBS | N/A | IV | Single dose | D6 | D9 |
| 17 | 5 | OKT3 | 0.25 | IV | Single dose | D6 | D9 | |
| 18 | 5 | Anti-CD28 | 1 | IV | Single dose | D6 | D9 | |
| Total | 90 | |||||||
| Perturbagen Class (PCL) | Group Enrichment Score |
|---|---|
| Progesterone receptor agonist × 12 | 100.00 |
| Glucocorticoid receptor agonist × 44 | 99.67 |
| Aromatase inhibitor × 3 | 97.00 |
| Beta-adrenergic receptor agonist × 11 | 96.12 |
| FLT3 inhibitor × 6 | 95.46 |
| Progesterone receptor antagonist × 5 | 95.13 |
| Leucine rich repeat kinase inhibitor × 3 | 94.15 |
| Bromodomain inhibitor × 6 | 93.59 |
| DNA synthesis inhibitor × 3 | 92.13 |
| VEGFR inhibitor × 13 | 91.84 |
| RAF inhibitor × 9 | 91.19 |
| Perturbagen Class (PCL) | Group Enrichment Score |
|---|---|
| Beta-adrenergic receptor agonist × 11 | 99.95 |
| Adenosine receptor agonist × 6 | 99.58 |
| Reverse transcriptase inhibitor × 4 | 99.27 |
| Bacterial 30S ribosomal subunit inhibitor × 5 | 99.26 |
| GABA receptor antagonist × 5 | 95.90 |
| Sigma receptor antagonist × 3 | 95.58 |
| Bile acid × 4 | 93.38 |
| PPAR receptor agonist × 16 | 92.04 |
| Benzodiazepine receptor agonist × 4 | 90.55 |
| FXR antagonist × 3 | 90.27 |
| Estrogen receptor agonist × 17 | 90.13 |
| Rank | Score | Name | Description |
|---|---|---|---|
| 4 | 99.89 | Hydrocortisone | Glucocorticoid receptor agonist |
| 5 | 99.89 | Betamethasone | Glucocorticoid receptor agonist |
| 7 | 99.79 | Budesonide | Glucocorticoid receptor agonist |
| 8 | 99.79 | Flunisolide | Cytochrome P450 inhibitor |
| 9 | 99.75 | Mometasone | Glucocorticoid receptor agonist |
| 11 | 99.75 | Fluocinonide | Glucocorticoid receptor agonist |
| 13 | 99.68 | Triamcinolone | Glucocorticoid receptor agonist |
| 14 | 99.68 | Fludroxycortide | Glucocorticoid receptor agonist |
| 19 | 99.62 | Clobetasol | Glucocorticoid receptor agonist |
| 21 | 99.61 | Triamcinolone | Glucocorticoid receptor agonist |
| 22 | 99.61 | Flumetasone | Glucocorticoid receptor agonist |
| 23 | 99.58 | Fluticasone | Glucocorticoid receptor agonist |
| 24 | 99.58 | Fluorometholone | Glucocorticoid receptor agonist |
| 26 | 99.54 | Dexamethasone | Glucocorticoid receptor agonist |
| 27 | 99.52 | Prednicarbate | Phospholipase activator |
| 28 | 99.51 | Isoflupredone | Glucocorticoid receptor agonist |
| 29 | 99.47 | Hydrocortisone | Glucocorticoid receptor agonist |
| 30 | 99.44 | Diflorasone | Corticosteroid agonist |
| 31 | 99.44 | Dexamethasone | Glucocorticoid receptor agonist |
| 32 | 99.4 | Medrysone | Glucocorticoid receptor agonist |
| 33 | 99.37 | Prednisolone | Glucocorticoid receptor agonist |
| 34 | 99.33 | Halcinonide | Glucocorticoid receptor agonist |
| 37 | 99.12 | Betamethasone | Glucocorticoid receptor agonist |
| 39 | 99.05 | Beclomethasone | Glucocorticoid receptor agonist |
| 40 | 99.05 | Fluocinolone | Glucocorticoid receptor agonist |
| 42 | 99.05 | Hydrocortisone | Glucocorticoid receptor agonist |
| 43 | 99.01 | Hydrocortisone | Glucocorticoid receptor agonist |
| 45 | 98.77 | Clocortolone | Glucocorticoid receptor agonist |
| 46 | 98.77 | Beclomethasone | Glucocorticoid receptor agonist |
| 48 | 98.6 | Westcort | Glucocorticoid receptor agonist |
| 51 | 98.4 | Prednisolone | Glucocorticoid receptor agonist |
| 52 | 98.38 | Fluticasone | Glucocorticoid receptor agonist |
| 55 | 98.34 | Hydrocortisone | Glucocorticoid receptor agonist |
| 56 | 98.32 | Rimexolone | Glucocorticoid receptor agonist |
| 58 | 98.17 | Fludrocortisone | Glucocorticoid receptor agonist |
| 59 | 98.17 | Amcinonide | Glucocorticoid receptor agonist |
| 61 | 98.03 | Methylprednisolone | Glucocorticoid receptor agonist |
| 67 | 97.71 | Desoximetasone | Glucocorticoid receptor agonist |
| 75 | 97.67 | Halometasone | Glucocorticoid receptor agonist |
| 88 | 97.53 | Loteprednol | Glucocorticoid receptor agonist |
| 96 | 97.5 | Depomedrol | Glucocorticoid receptor agonist |
| 117 | 97.39 | Prednisolone | Glucocorticoid receptor agonist |
| 137 | 97.15 | Fluocinonide | Glucocorticoid receptor agonist |
| 632 | 93.24 | Alclometasone | Glucocorticoid receptor agonist |
| Rank | Score | Name | Description |
|---|---|---|---|
| 23 | 99.55 | Fluocinonide | Glucocorticoid receptor agonist |
| 244 | 97.47 | Depomedrol | Glucocorticoid receptor agonist |
| 376 | 96.44 | Beclometasone | Glucocorticoid receptor agonist |
| 471 | 95.46 | Fluocinolone | Glucocorticoid receptor agonist |
| 615 | 94.13 | Prednisolone | Glucocorticoid receptor agonist |
| 890 | 91.89 | Triamcinolone | Glucocorticoid receptor agonist |
| 943 | 91.4 | Amcinonide | Glucocorticoid receptor agonist |
| 1479 | 86.61 | Rimexolone | Glucocorticoid receptor agonist |
| 1810 | 83.17 | Hydrocortisone | Glucocorticoid receptor agonist |
| 1959 | 82.28 | Halometasone | Glucocorticoid receptor agonist |
| 2159 | 80.37 | Beclometasone | Glucocorticoid receptor agonist |
| 2349 | 78.38 | Triamcinolone | Glucocorticoid receptor agonist |
| 2445 | 77.7 | Prednisolone | Glucocorticoid receptor agonist |
| 2855 | 73.57 | Betamethasone | Glucocorticoid receptor agonist |
| 2864 | 73.36 | Hydrocortisone | Glucocorticoid receptor agonist |
| 2877 | 73.18 | Alclometasone | Glucocorticoid receptor agonist |
| 3084 | 71.41 | Westcort | Glucocorticoid receptor agonist |
| 3155 | 70.7 | Fluticasone | Glucocorticoid receptor agonist |
| 3160 | 70.64 | Loteprednol | Glucocorticoid receptor agonist |
| 3348 | 68.45 | Dexamethasone | Glucocorticoid receptor agonist |
| 3561 | 66.22 | Fluocinonide | Glucocorticoid receptor agonist |
| 3591 | 65.9 | Desoximetasone | Glucocorticoid receptor agonist |
| 4051 | 61.03 | Betamethasone | Glucocorticoid receptor agonist |
| 4147 | 60 | Prednicarbate | Phospholipase activator |
| 4393 | 56.96 | Clocortolone | Glucocorticoid receptor agonist |
| 4404 | 56.86 | Hydrocortisone | Glucocorticoid receptor agonist |
| 4465 | 56.37 | Fluorometholone | Glucocorticoid receptor agonist |
| 4489 | 56.05 | Flunisolide | Cytochrome P450 inhibitor |
| 4701 | 52.82 | Mometasone | Glucocorticoid receptor agonist |
| 5218 | 45.35 | Dexamethasone | Glucocorticoid receptor agonist |
| 5512 | 41.08 | Prednisolone | Glucocorticoid receptor agonist |
| 5594 | 39.97 | Fludroxycortide | Glucocorticoid receptor agonist |
| 5618 | 39.72 | Methylprednisolone | Glucocorticoid receptor agonist |
| 5696 | 38.68 | Fluticasone | Glucocorticoid receptor agonist |
| 5837 | 36.6 | Hydrocortisone | Glucocorticoid receptor agonist |
| 6223 | 31.46 | Medrysone | Glucocorticoid receptor agonist |
| 6329 | 29.69 | Budesonide | Glucocorticoid receptor agonist |
| 6560 | 25.98 | Isoflupredone | Glucocorticoid receptor agonist |
| 6962 | 19.87 | Clobetasol | Glucocorticoid receptor agonist |
| 7001 | 19.45 | Fludrocortisone | Glucocorticoid receptor agonist |
| 7129 | 16.81 | Halcinonide | Glucocorticoid receptor agonist |
| 8126 | −10.56 | Flumetasone | Glucocorticoid receptor agonist |
| 8351 | −40.7 | Hydrocortisone | Glucocorticoid receptor agonist |
| 8433 | −53.42 | Diflorasone | Corticosteroid agonist |
| Receptor | Agonists | Antagonists | Tissue | Responses |
|---|---|---|---|---|
| Alpha1 | Epi ≥ NE >> Iso Drug example, phenylephrine | Prazosin | Vascular smooth muscle | Contraction |
| Genitourinary smooth muscle | Contraction | |||
| Liver | Glycogenolysis; gluconeogenesis | |||
| Intestinal smooth muscle | Hyperpolarization and relaxation | |||
| Heart | Increased contractile force, arrhythmias | |||
| Alpha2 | Epi ≥ NE >> Iso Drug example, Clonidine | Yohimbine | Pancreatic islet (b cells) | Decreased insulin secretion |
| Platelets | Aggregation | |||
| Nerve terminals | Decreased release of NE | |||
| Vascular Smooth Muscle | Contraction | |||
| Beta1 | Iso >> Epi = NE Drug example, Dobutamine | Metoprolol | Heart Juxtaglomerular cell | Increased force and rate of contraction and AV nodal conduction velocity Increased renin secretion |
| Beta2 | Iso >> Epi >>> NE Drug example, Terbutaline | ICI 118551 (Inverse agonist) | Smooth muscle (vascular, bronchial, gastrointestinal, and genitourinary) | Relaxation |
| Skeletal muscle | Glycogenolysis; uptake of K+ | |||
| Liver | Glycogenolysis; gluconeogenesis | |||
| Beta3 | Iso = NE > Epi | ICI 118551 | Adipose tissue | Lipolysis |
| Perturbagen Class (PCL) | Group Enrichment Score |
|---|---|
| Glucocorticoid receptor agonist × 44 | 99.89 |
| Cyclooxygenase inhibitor × 6 | 97.34 |
| Beta-adrenergic receptor agonist × 11 | 95.88 |
| Adenosine receptor agonist × 6 | 95.49 |
| Na-K-Cl transporter inhibitor × 3 | 94.89 |
| Rho associated kinase inhibitor × 4 | 93.79 |
| MEK inhibitor × 8 | 92.61 |
| Rank | Score | Name | Description |
|---|---|---|---|
| 1 | 99.99 | Hydrocortisone | Glucocorticoid receptor agonist |
| 2 | 99.93 | Triamcinolone | Glucocorticoid receptor agonist |
| 3 | 99.93 | Budesonide | Glucocorticoid receptor agonist |
| 4 | 99.93 | Fluocinonide | Glucocorticoid receptor agonist |
| 5 | 99.93 | Fluorometholone | Glucocorticoid receptor agonist |
| 7 | 99.72 | Mometasone | Glucocorticoid receptor agonist |
| 8 | 99.72 | Fluticasone | Glucocorticoid receptor agonist |
| 9 | 99.72 | Fludrocortisone | Glucocorticoid receptor agonist |
| 11 | 99.61 | Betamethasone | Glucocorticoid receptor agonist |
| 12 | 99.61 | Betamethasone | Glucocorticoid receptor agonist |
| 13 | 99.58 | Clocortolone | Glucocorticoid receptor agonist |
| 14 | 99.58 | Flunisolide | Cytochrome P450 inhibitor |
| 15 | 99.58 | Dexamethasone | Glucocorticoid receptor agonist |
| 16 | 99.58 | Clobetasol | Glucocorticoid receptor agonist |
| 17 | 99.54 | Isoflupredone | Glucocorticoid receptor agonist |
| 20 | 99.37 | Fludroxycortide | Glucocorticoid receptor agonist |
| 21 | 99.33 | Prednisolone | Glucocorticoid receptor agonist |
| 22 | 99.3 | Beclomethasone | Glucocorticoid receptor agonist |
| 23 | 99.3 | Fluticasone | Glucocorticoid receptor agonist |
| 24 | 99.3 | Alclometasone | Glucocorticoid receptor agonist |
| 26 | 99.3 | Hydrocortisone | Glucocorticoid receptor agonist |
| 27 | 99.26 | Fluocinolone | Glucocorticoid receptor agonist |
| 28 | 99.25 | Flumetasone | Glucocorticoid receptor agonist |
| 30 | 99.16 | Rimexolone | Glucocorticoid receptor agonist |
| 32 | 99.06 | Loteprednol | Glucocorticoid receptor agonist |
| 34 | 98.91 | Hydrocortisone | Glucocorticoid receptor agonist |
| 35 | 98.8 | Halcinonide | Glucocorticoid receptor agonist |
| 36 | 98.66 | Triamcinolone | Glucocorticoid receptor agonist |
| 37 | 98.63 | Dexamethasone | Glucocorticoid receptor agonist |
| 38 | 98.59 | Beclomethasone | Glucocorticoid receptor agonist |
| 39 | 98.52 | Prednicarbate | Phospholipase activator |
| 44 | 98.48 | Prednisolone | Glucocorticoid receptor agonist |
| 45 | 98.48 | Amcinonide | Glucocorticoid receptor agonist |
| 46 | 98.1 | Halometasone | Glucocorticoid receptor agonist |
| 48 | 97.82 | Methylprednisolone | Glucocorticoid receptor agonist |
| 50 | 97.68 | Diflorasone | Corticosteroid agonist |
| 52 | 97.5 | Hydrocortisone | Glucocorticoid receptor agonist |
| 53 | 97.5 | Fluocinonide | Glucocorticoid receptor agonist |
| 55 | 97.36 | Desoximetasone | Glucocorticoid receptor agonist |
| 58 | 97.33 | Prednisolone | Glucocorticoid receptor agonist |
| 66 | 97.15 | Depomedrol | Glucocorticoid receptor agonist |
| 83 | 96.69 | Medrysone | Glucocorticoid receptor agonist |
| 141 | 95.45 | Westcort | Glucocorticoid receptor agonist |
| 187 | 95.1 | Hydrocortisone | Glucocorticoid receptor agonist |
| Perturbagen Class (PCL) | Group Enrichment Score |
|---|---|
| Progesterone receptor agonist × 12 | 99.50 |
| Glucocorticoid receptor agonist × 44 | 98.78 |
| Aromatase inhibitor × 3 | 97.98 |
| Glycogen synthase kinase inhibitor × 11 | 97.63 |
| HSP inhibitor × 7 | 96.76 |
| EGFR inhibitor × 14 | 96.33 |
| PPAR receptor agonist × 16 | 95.40 |
| DNA synthesis inhibitor × 3 | 95.02 |
| PKC inhibitor × 7 | 93.75 |
| JAK inhibitor × 5 | 92.59 |
| HIV protease inhibitor × 4 | 92.34 |
| Cannabinoid receptor agonist × 8 | 91.13 |
| Perturbagen Class (PCL) | Group Enrichment Score |
|---|---|
| Dihydrofolate reductase inhibitor × 4 | 99.78 |
| Reverse transcriptase inhibitor × 4 | 99.40 |
| Glucocorticoid receptor agonist × 44 | 98.37 |
| Vitamin D receptor agonist × 6 | 97.36 |
| Nucleoside reverse transcriptase inhibitor × 3 | 97.07 |
| IMPDH inhibitor × 3 | 95.65 |
| Bacterial 30S ribosomal subunit inhibitor × 5 | 94.42 |
| FXR antagonist × 3 | 93.95 |
| Calmodulin antagonist × 3 | 93.51 |
| Bacterial cell wall synthesis inhibitor × 9 | 90.77 |
| Perturbagen Class (PCL) | Group Enrichment Score |
|---|---|
| Bacterial DNA gyrase inhibitor × 5 | 95.54 |
| Aromatase inhibitor × 3 | 93.32 |
| Dihydrofolate reductase inhibitor × 4 | 91.76 |
| Cyclooxygenase inhibitor × 6 | 90.65 |
| Glucocorticoid receptor agonist × 44 | 90.24 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inchiosa, M.A., Jr. Beta2-Adrenergic Suppression of Neuroinflammation in Treatment of Parkinsonism, with Relevance for Neurodegenerative and Neoplastic Disorders. Biomedicines 2024, 12, 1720. https://doi.org/10.3390/biomedicines12081720
Inchiosa MA Jr. Beta2-Adrenergic Suppression of Neuroinflammation in Treatment of Parkinsonism, with Relevance for Neurodegenerative and Neoplastic Disorders. Biomedicines. 2024; 12(8):1720. https://doi.org/10.3390/biomedicines12081720
Chicago/Turabian StyleInchiosa, Mario A., Jr. 2024. "Beta2-Adrenergic Suppression of Neuroinflammation in Treatment of Parkinsonism, with Relevance for Neurodegenerative and Neoplastic Disorders" Biomedicines 12, no. 8: 1720. https://doi.org/10.3390/biomedicines12081720