
Cell-Based Treatment in Acute Myeloid Leukemia Relapsed after Allogeneic Stem Cell Transplantation

Abstract
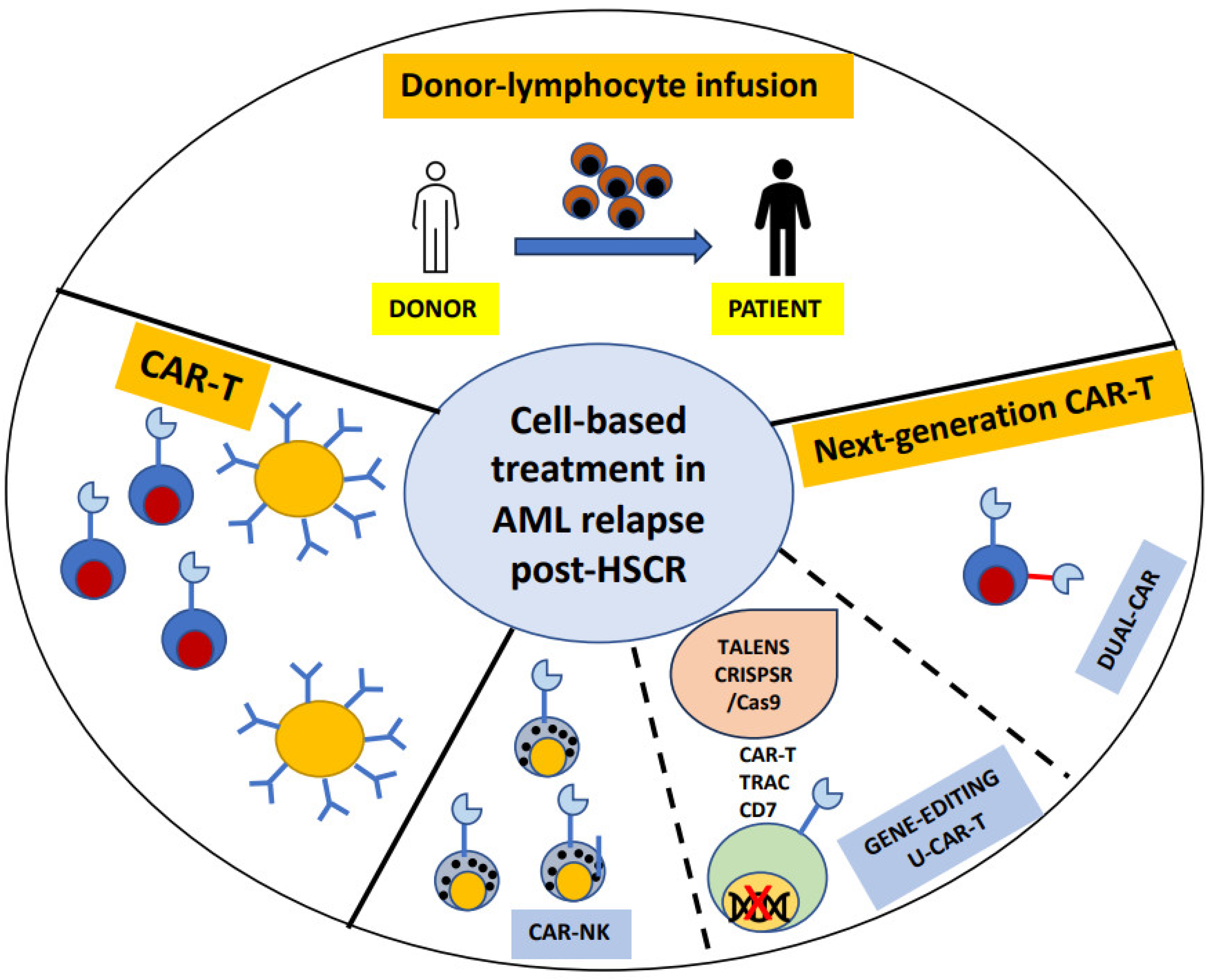
:1. Introduction
2. Donor Lymphocyte Infusions (DLI)
2.1. DLI, the Past
2.2. DLI, the Present
2.3. DLI, the Future
- The first infusion is recommended after cessation of immunosuppression for >30 days and is not recommended with active GvHD and infections;
- The median interval from ASCT to first DLI across all studies was 4–6 months, while an early application could be desirable in high-risk diseases;
- The infusions should have a dose increase of 0.5–1 log with intervals of 4–6 weeks;
- DLI frequencies should be guided by response (MRD, degree of chimerism) and the occurrence of GvHD has to be considered as limiting toxicity [11].
2.4. Beyond Unmanipulated DLI
- -
- Activation of cell subsets by cytokines or growth factor;
- -
- Selection and expansion of specific T cell subsets;
- -
- Infusion of natural killer (NK) cells.
2.4.1. Cytokine-Induced Killer (CIK) Cells
2.4.2. G-CSF-Stimulated DLI
2.4.3. Antigen-Specific DLI
2.4.4. DLI in Combination with Other Drugs
3. CAR-T
3.1. CD33-CAR-T
3.2. CD123-CAR-T
3.3. CLL1-CAR-T
3.4. FLT3-CAR-T
4. Novel Strategies for CAR-T in AML
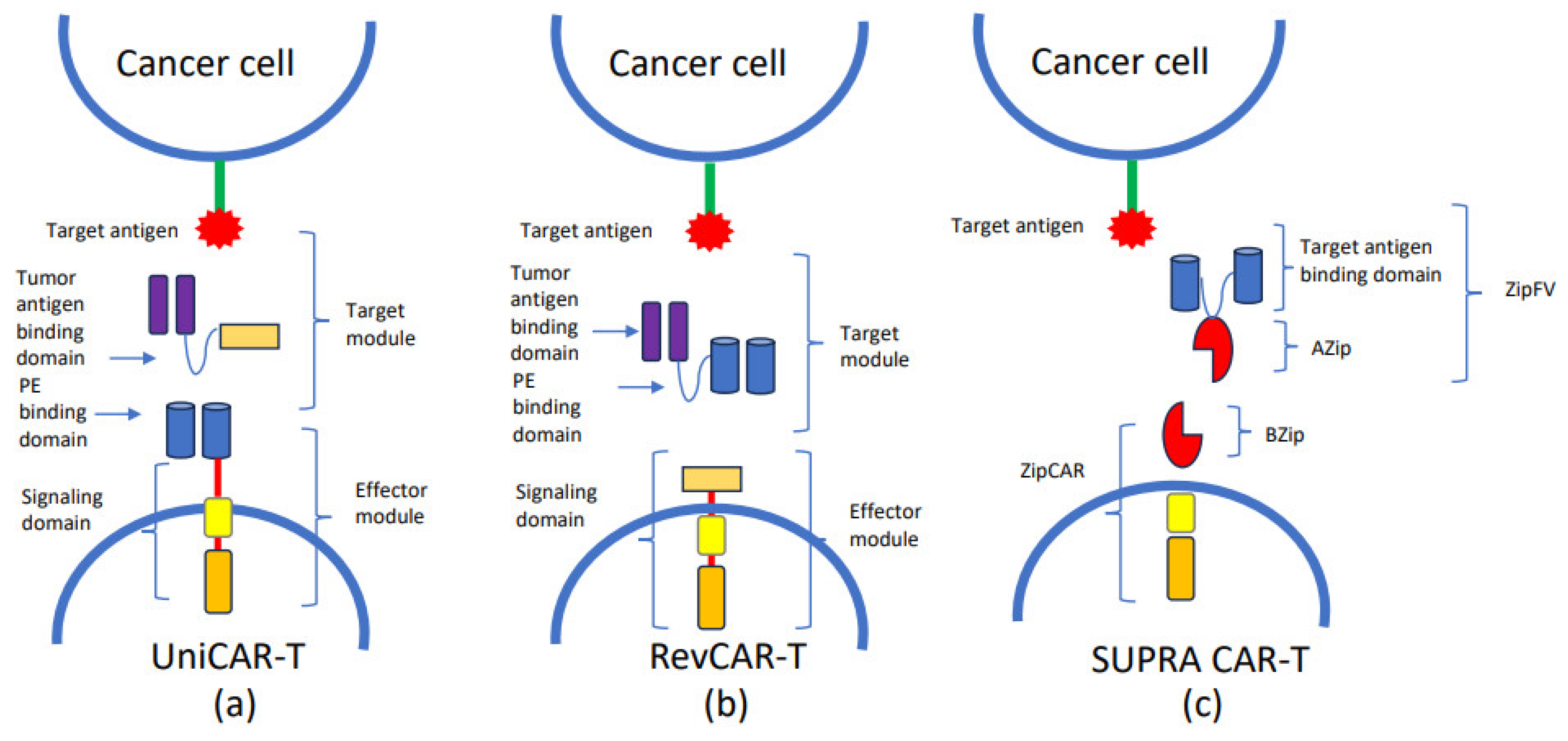
5. Strategies to Improve CAR-T Specificity
6. Dual CAR-T
7. Strategies to Reduce Toxicity: The Gene Editing Approach
8. UCAR-T
9. CAR-Natural Killers Cells
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 12, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- De Lima, M.; Porter, D.L.; Battiwalla, M.; Bishop, M.R.; Giralt, S.A.; Hardy, N.M. Proceedings from the National Cancer Institute’s Second International Workshop on the Biology, Prevention, and Treatment of Relapse after Hematopoietic Stem Cell Transplantation: Part III. Prevention and Treatment of Relapse after Allogeneic Transplantation. Biol. Blood Marrow Transplant. 2014, 20, 4–13. [Google Scholar] [PubMed]
- Christopeit, M.; Kuss, O.; Finke, J.; Bacher, U.; Beelen, D.W.; Bornhäuser, M. Second Allograft for Hematologic Relapse of Acute Leukemia After First Allogeneic Stem-Cell Transplantation from Related and Unrelated Donors: The Role of Donor Change. J. Clin. Oncol. 2013, 31, 3259–3271. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.; de Wreede, L.C.; van Biezen, A.; Finke, J.; Ehninger, G.; Ganser, A. Outcome after relapse of myelodysplastic syndrome and secondary acute myeloid leukemia following allogeneic stem cell transplantation: A retrospective registry analysis on 698 patients by the Chronic Malignancies Working Party of the European Society of Blood and Marrow Transplantation. Haematologica 2018, 103, 237–245. [Google Scholar] [PubMed]
- Schmid, C.; Labopin, M.; Nagler, A.; Bornhäuser, M.; Finke, J.; Fassas, A. Donor lymphocyte infusion in the treatment of first hematological relapse after allogeneic stem-cell transplantation in adults with acute myeloid leukemia: A retrospective risk factors analysis and comparison with other strategies by the EBMT Acute Leukemia Working Party. J. Clin. Oncol. 2007, 25, 4938–4945. [Google Scholar] [PubMed]
- Schmid, C.; Labopin, M.; Nagler, A.; Niederwieser, D.; Castagna, L.; Tabrizi, R.; Stadler, M.; Kuball, J.; Cornelissen, J.; Vorlicek, J.; et al. Treatment, risk factors, and outcome of adults with relapsed AML after reduced intensity conditioning for allogeneic stem cell transplantation. Blood 2012, 119, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.J.; Mittermüller, J.; Clemm, C.H.; Holler, E.; Ledderose, G.; Brehm, G.; Heim, M.; Wilmanns, W. Donorleukocyte transfusions for treatment of recurrent chronic myelogenous leukemia in marrow transplant patients. Blood 1990, 76, 2462–2465. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.H., Jr.; Shpilberg, O.; Drobyski, W.R.; Porter, D.L.; Giralt, S.; Champlin, R.; Goodman, S.A.; Wolff, S.N.; Hu, W.; Verfaillie, C.; et al. Donor leukocyte infusions in 140 patients with relapsed malignancy after allogeneic bone marrow transplantation. J. Clin. Oncol. 1997, 2, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.D.; Waller, E.K. Finding the sweet spot for donor lymphocyte infusions. Biol. Blood Marrow Transplant. 2013, 4, 507–508. [Google Scholar] [CrossRef]
- Schmid, C.; Kuball, J.; Bug, G. Defining the Role of Donor Lymphocyte Infusion in High-Risk Hematologic Malignancies. J. Clin. Oncol. 2021, 5, 397–418. [Google Scholar] [CrossRef]
- Frederik, F.J.H.; Schmid, C.; Kolb, H.J.; Locatelli, F.; Kuball, J. Delayed transfer of immune cells or the art of donor lymphocyte infusion. The EBMT Handbook: Hematopoietic Stem Cell Transplantation and Cellular Therapies; The EBMT: Maastricht, The Netherlands, 2019; pp. 443–448. [Google Scholar]
- Levine, J.E.; Braun, T.; Penza, S.L.; Beatty, P.; Cornetta, K.; Martino, R.; Drobyski, W.R.; Barrett, A.J.; Porter, D.L.; Giralt, S.; et al. Prospective trial of chemotherapy and donor leukocyte infusions for relapse of advanced myeloid malignancies after allogeneic stem-cell transplantation. J. Clin. Oncol. 2002, 2, 405–412. [Google Scholar] [CrossRef]
- Schmid, C.; Labopin, M.; Schaap, N.; Veelken, H.; Brecht, A.; Stadler, M.; Finke, J.; Baron, F.; Collin, M.; Bug, G.; et al. Long-term results and GvHD after prophylactic and preemptive donor lymphocyte infusion after allogeneic stem cell transplantation for acute leukemia. Bone Marrow Transplant. 2022, 2, 215–223. [Google Scholar] [CrossRef]
- Dominietto, A.; Pozzi, S.; Miglino, M.; Albarracin, F.; Piaggio, G.; Bertolotti, F.; Grasso, R.; Zupo, S.; Raiola, A.M.; Gobbi, M.; et al. Donor lymphocyte infusions for the treatment of minimal residual disease in acute leukemia. Blood 2007, 109, 5063–5064. [Google Scholar] [CrossRef]
- Di Grazia, C.; Pozzi, S.; Geroldi, S.; Grasso, R.; Miglino, M.; Colombo, N.; Tedone, E.; Luchetti, S.; Lamparelli, T.; Gualandi, F.; et al. Wilms Tumor 1 Expression and Pre-emptive Immunotherapy in Patients with Acute Myeloid Leukemia Undergoing an Allogeneic Hemopoietic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2016, 22, 1242–1246. [Google Scholar] [CrossRef]
- Yan, C.-H.; Liu, D.-H.; Liu, K.-Y.; Xu, L.-P.; Liu, Y.-R.; Chen, H.; Han, W.; Wang, Y.; Qin, Y.-Z.; Huang, X.-J. Risk stratification- directed donor lymphocyte infusion could reduce relapse of standard-risk acute leukemia patients after allogeneic hematopoietic stem cell transplantation. Blood 2012, 119, 3256–3262. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.; Labopin, M.; Schaap, N.; Veelken, H.; Schleuning, M.; Stadler, M.; Finke, J.; Hurst, E.; Baron, F.; Ringden, O.; et al. EBMT Acute Leukaemia Working Party. Prophylactic donor lymphocyte infusion after allogeneic stem cell transplantation in acute leukaemia—A matched pair analysis by the Acute Leukaemia Working Party of EBMT. Br. J. Haematol. 2019, 5, 782–787. [Google Scholar] [CrossRef]
- Legrand, F.; Le Floch, A.C.; Granata, A.; Fürst, S.; Faucher, C.; Lemarie, C.; Harbi, S.; Bramanti, S.; Calmels, B.; El-Cheikh, J.; et al. Prophylactic donor lymphocyte infusion after allogeneic stem cell transplantation for high-risk AML. Bone Marrow Transplant. 2017, 52, 620–621. [Google Scholar] [CrossRef] [PubMed]
- Jedlickova, Z.; Schmid, C.; Koenecke, C.; Hertenstein, B.; Baurmann, H.; Schwerdtfeger, R.; Tischer, J.; Kolb, H.J.; Schleuning, M. Long-term results of adjuvant donor lymphocyte transfusion in AML after allogeneic stem cell transplantation. Bone Marrow Transplant. 2016, 51, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Cauchois, R.; Castagna, L.; Pagliardini, T.; Harbi, S.; Calmels, B.; Bramanti, S.; Granata, A.; Lemarie, C.; Maisano, V.; Legrand, F.; et al. Prophylactic donor lymphocyte infusions after haploidentical haematopoietic stem cell transplantation for high risk haematological malignancies: A retrospective bicentric analysis of serial infusions of increasing doses of CD3+ cells. Br. J. Haematol. 2019, 3, 570–573. [Google Scholar] [CrossRef]
- Santoro, N.; Mooyaart, J.E.; Devillier, R.; Koc, Y.; Vydra, J.; Castagna, L.; Gülbas, Z.; Martin, J.D.; Araujo, M.C.; Kulagin, A.; et al. Donor lymphocyte infusions after haploidentical stem cell transplantation with PTCY: A study on behalf of the EBMT cellular therapy & immunobiology working party. Bone Marrow Transplant. 2023, 58, 54–60. [Google Scholar]
- Schmidt-Wolf, I.G.; Negrin, R.S.; Kiem, H.P.; Blume, K.G.; Weissman, I.L. Use of a SCID mouse/human lymphoma model to evaluate cytokine-induced killer cells with potent antitumor cell activity. J. Exp. Med. 1991, 174, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Pievani, A.; Borleri, G.; Pende, D.; Moretta, L.; Rambaldi, A.; Golay, J.; Introna, M. Dual-functional capability of CD3+CD56+ CIK cells, a T-cell subset that acquires NK function and retains TCR-mediated specific cytotoxicity. Blood 2011, 118, 3301–3310. [Google Scholar] [CrossRef] [PubMed]
- Introna, M.; Lussana, F.; Algarotti, A.; Gotti, E.; Valgardsdottir, R.; Micò, C.; Grassi, A.; Pavoni, C.; Ferrari, M.L.; Delaini, F.; et al. Phase II study of sequential infusion of donor lymphocyte infusion and cytokine-induced killer cells for patients relapsed after allogeneic hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 2017, 23, 2070–2078. [Google Scholar] [CrossRef] [PubMed]
- Merker, M.; Salzmann-Manrique, E.; Katzki, V.; Huenecke, S.; Bremm, M.; Bakhtiar, S.; Willasch, A.; Jarisch, A.; Soerensen, J.; Schulz, A.; et al. Clearance of Hematologic Malignancies by Allogeneic Cytokine-Induced Killer Cell or Donor Lymphocyte Infusions. Biol. Blood Marrow Transplant. 2019, 7, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.J.; Wang, Y.; Liu, D.H.; Xu, L.P.; Chen, H.; Chen, Y.H.; Han, W.; Shi, H.X.; Liu, K.Y. Modified donor lymphocyte infusion (DLI) for the prophylaxis of leukemia relapse after hematopoietic stem cell transplantation in patients with advanced leukemia--feasibility and safety study. J. Clin. Immunol. 2008, 4, 390–397. [Google Scholar] [CrossRef]
- Schmid, C.; Schleuning, M.; Aschan, J.; Ringdén, O.; Hahn, J.; Holler, E.; Hegenbart, U.; Niederwieser, D.; Dugas, M.; Ledderose, G.; et al. Low-dose ARAC, donor cells, and GM-CSF for treatment of recurrent acute myeloid leukemia after allogeneic stem cell transplantation. Leukemia 2004, 18, 1430–1433. [Google Scholar] [CrossRef] [PubMed]
- Anguille, S.; Van Tendeloo, V.F.; Berneman, Z.N. Leukemia-associated antigens and their relevance to the immunotherapy of acute myeloid leukemia. Leukemia 2012, 26, 2186–2196. [Google Scholar] [CrossRef] [PubMed]
- Greiner, J.; Schneider, V.; Schmitt, M.; Götz, M.; Döhner, K.; Wiesneth, M.; Döhner, H.; Hofmann, S. Immune responses against the mutated region of cytoplasmatic NPM1 might contribute to the favorable clinical outcome of AML patients with NPM1 mutations (NPM1mut). Blood 2013, 6, 1087–1088. [Google Scholar] [CrossRef]
- Guillaume, T.; Thépot, S.; Peterlin, P.; Ceballos, P.; Le Bourgeois, A.; Garnier, A.; Orvain, C.; Giltat, A.; François, S.; Le Bris, Y.; et al. Prophylactic or Preemptive Low-Dose Azacitidine and Donor Lymphocyte Infusion to Prevent Disease Relapse following Allogeneic Transplantation in Patients with High-Risk Acute Myelogenous Leukemia or Myelodysplastic Syndrome. Transplant. Cell. Ther. 2021, 10, 839.e1–839.e6. [Google Scholar] [CrossRef]
- Booth, N.; Mirea, L.; Huschart, E.; Miller, H.; Salzberg, D.; Campbell, C.; Beebe, K.; Schwalbach, C.; Adams, R.H.; Ngwube, A. Efficacy of Azacitidine and Prophylactic Donor Lymphocyte Infusion after HSCT in Pediatric Patients with Acute Myelogenous Leukemia: A Retrospective Pre-Post Study. Biol. Blood Marrow Transplant. 2023, 29, 330.e1–330.e7. [Google Scholar] [CrossRef]
- Locke, F.L.; Miklos, D.B.; Jacobson, C.A.; Perales, M.A.; Kersten, M.J.; Oluwole, O.O.; Ghobadi, A.; Rapoport, A.P.; McGuirk, J.; Pagel, J.M.; et al. All ZUMA-7 Investigators and Contributing Kite Members. Axicabtagene Ciloleucel as Second-Line Therapy for Large B-Cell Lymphoma. N. Engl. J. Med. 2022, 386, 640–654. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, C.A.; Chavez, J.C.; Sehgal, A.R.; William, B.M.; Munoz, J.; Salles, G.; Munshi, P.N.; Casulo, C.; Maloney, D.G.; de Vos, S.; et al. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): A single-arm, multicentre, phase 2 trial. Lancet Oncol. 2022, 23, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Dickinson, M.; Munoz, J.; Ulrickson, M.L.; Thieblemont, C.; Oluwole, O.O.; Herrera, A.F.; Ujjani, C.S.; Lin, Y.; Riedell, P.A.; et al. Axicabtagene ciloleucel as first-line therapy in high-risk large B-cell lymphoma: The phase 2 ZUMA-12 trial. Nat. Med. 2022, 28, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.D.; Ghobadi, A.; Oluwole, O.O.; Logan, A.C.; Boissel, N.; Cassaday, R.D.; Leguay, T.; Bishop, M.R.; Topp, M.S.; Tzachanis, D.; et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: Phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet 2021, 398, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Cheadle, E.J.; Gornall, H.; Baldan, V.; Hanson, V.; Hawkins, R.E.; Gilham, D.E. CAR T cells: Driving the road from the laboratory to the clinic. Immunol. Rev. 2014, 257, 91–106. [Google Scholar] [CrossRef]
- Smith, A.J.; Oertle, J.; Warren, D.; Prato, D. Chimeric antigen receptor (Car) T cell therapy for malignant cancers: Summary and perspective. J. Cell. Immunother. 2016, 2, 59–68. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2017, 15, 47–62. [Google Scholar] [CrossRef]
- Fried, S.; Avigdor, A.; Bielorai, B.; Meir, A.; Besser, M.J.; Schachter, J.; Shimoni, A.; Nagler, A.; Toren, A.; Jacoby, E. Earlyandlate hematologic toxicity following CD19 CAR-T cells. Bone Marrow Transplant. 2019, 54, 1643–1650. [Google Scholar] [CrossRef] [PubMed]
- Tambaro, F.P.; Singh, H.; Jones, E.; Rytting, M.; Mahadeo, K.M.; Thompson, P.; Daver, N.; DiNardo, C.; Kadia, T.; Garcia-Manero, G.; et al. Autologous CD33-CAR-T cells for treatment of relapsed/refractory acute myelogenous leukemia. Leukemia 2021, 35, 3282–3286. [Google Scholar] [CrossRef] [PubMed]
- Molica, M.; Perrone, S.; Mazzone, C.; Niscola, P.; Cesini, L.; Abruzzese, E.; de Fabritiis, P. CD33 expression and gentuzumab ozogamicin in acute myeloid leukemia: Two sides of the same coin. Cancers 2021, 13, 3214. [Google Scholar] [CrossRef] [PubMed]
- O’Hear, C.; Heiber, J.F.; Schubert, I.; Fey, G.; Geiger, T.L. Anti-CD33 chimeric antigen receptor targeting of acute myeloid leukemia. Haematologica 2014, 100, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Kenderian, S.S.; Ruella, M.; Shestova, O.; Klichinsky, M.; Aikawa, V.; Morrissette, J.J.; Scholler, J.; Song, D.; Porter, D.L.; Carroll, M.; et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia 2015, 29, 1637–1647. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tao, Z.; Xu, Y.; Liu, J.; An, N.; Wang, Y.; Xing, H.; Tian, Z.; Tang, K.; Liao, X.; et al. CD33-Specific Chimeric Antigen Receptor T Cells with Different Co-Stimulators Showed Potent Anti-Leukemia Efficacy and Different Phenotype. Hum. Gene Ther. 2018, 29, 626–639. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Yang, L.; Chukinas, J.A.; Shah, N.N.; Tarun, S.; Pouzolles, M.; Chien, C.D.; Niswander, L.M.; Welch, A.R.; Taylor, N.A.; et al. Systematic preclinical evaluation of CD33-directed chimeric antigen receptor T cell immunotherapy for acute myeloid leukemia defines optimized construct design. J. Immunother. Cancer 2021, 9, e003149, Erratum in J. Immunother. Cancer 2021, 9, e003149. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M. The UniCAR system: A modular CAR T cell approach to improve the safety of CAR T cells. Immunol. Lett. 2019, 211, 13–22. [Google Scholar] [CrossRef]
- Wang, Q.S.; Wang, Y.; Lv, H.Y.; Han, Q.W.; Fan, H.; Guo, B.; Wang, L.-L.; Han, W.-D. Treatment of Cd33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol. Ther. 2015, 23, 184–191. [Google Scholar] [CrossRef]
- Tasian, S.K.; Kenderian, S.S.; Shen, F.; Ruella, M.; Shestova, O.; Kozlowski, M.; Li, Y.; Schrank-Hacker, A.; Morrissette, J.J.; Carroll, M.; et al. Optimized depletion of chimeric antigen receptor T cells in murine xenograft models of human acute myeloid leukemia. Blood 2017, 129, 2395–2407. [Google Scholar] [CrossRef]
- Petrov, J.C.; Wada, M.; Pinz, K.G.; Yan, L.E.; Chen, K.H.; Shuai, X.; Liu, H.; Chen, X.; Leung, L.H.; Salman, H.; et al. Compound CAR T-cells as a double-pronged approach for treating acute myeloid leukemia. Leukemia 2018, 32, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, A.; Hoffmann, A.; Bergmann, R.; Koristka, S.; Berndt, N.; Arndt, C.; Rodrigues Loureiro, L.; Kittel-Boselli, E.; Mitwasi, N.; Kegler, A.; et al. Versatile chimeric antigen receptor platform for controllable and combinatorial T cell therapy. Oncoimmunology 2020, 9, 1785608. [Google Scholar] [CrossRef] [PubMed]
- Kittel-Boselli, E.; Soto, K.E.G.; Loureiro, L.R.; Hoffmann, A.; Bergmann, R.; Arndt, C.; Koristka, S.; Mitwasi, N.; Kegler, A.; Bartsch, T.; et al. Targeting Acute Myeloid Leukemia Using the RevCAR Platform: A Programmable, Switchable and Combinatorial Strategy. Cancers 2021, 13, 4785. [Google Scholar] [CrossRef] [PubMed]
- Van Rhenen, A.; van Dongen, G.A.; Kelder, A.; Rombouts, E.J.; Feller, N.; Moshaver, B.; Stigter-van Walsum, M.; Zweegman, S.; Ossenkoppele, G.J.; Jan Schuurhuis, G. The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood 2007, 7, 2659–2666. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Singh, S.; Pardoux, C.; Zhao, J.; His, E.D.; Abo, A.; Korver, W. Targeting C-type lectin-like molecule-1 for antibody-mediated immunotherapy in acute myeloid leukemia. Haematologica 2010, 95, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Zhang, Y.; Yu, L.; Wu, D. Cytotoxic effect of CLL-1 CAR-T cell immunotherapy with PD-1 silencing on relapsed/refractory acute myeloid leukemia. Mol. Med. Rep. 2021, 23, 208. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.J.; Dai, H.P.; Cui, Q.Y.; Cui, W.; Zhu, W.J.; Qu, C.J.; Kang, L.Q.; Zhu, M.Q.; Zhu, X.M.; Liu, D.D.; et al. Successful application of PD-1 knockdown CLL-1 CAR-T therapy in two AML patients with post-transplant relapse and failure of anti-CD38 CAR-T cell treatment. Am. J. Cancer Res. 2022, 12, 615–621. [Google Scholar] [PubMed]
- Pei, K.; Xu, H.; Wang, P.; Gan, W.; Hu, Z.; Su, X.; Zhang, H.; He, Y. Anti-CLL1-based CAR T-cells with 4-1-BB or CD28/CD27 stimulatory domains in treating childhood refractory/relapsed acute myeloid leukemia. Cancer Med. 2023, 12, 9655–9661. [Google Scholar] [CrossRef] [PubMed]
- Nitika Wei, J.; Hui, A.M. Role of Biomarkers in FLT3 AML. Cancers 2022, 14, 1164. [Google Scholar] [CrossRef]
- Pedersen, M.G.; Møller, B.K.; Bak, R.O. Recent Advances in the Development of Anti-FLT3 CAR T-Cell Therapies for Treatment of AML. Biomedicines 2022, 10, 2441. [Google Scholar] [CrossRef]
- Chen, L.; Mao, H.; Zhang, J.; Chu, J.; Devine, S.; Caligiuri, M.A.; Yu, J. Targeting FLT3 by chimeric antigen receptor T cells for the treatment of acute myeloid leukemia. Leukemia 2017, 31, 1830–1834. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Y.; Li, S.; Liu, J.; Xing, Y.; Xing, H.; Tian, Z.; Tang, K.; Rao, Q.; Wang, M.; et al. Targeting FLT3 in acute myeloid leukemia using ligand-based chimeric antigen receptor-engineered T cells. J. Hematol. Oncol. 2018, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Jetani, H.; Garcia-Cadenas, I.; Nerreter, T.; Thomas, S.; Rydzek, J.; Meijide, J.B.; Bonig, H.; Herr, W.; Sierra, J.; Einsele, H.; et al. CAR T-cells targeting FLT3 have potent activity against FLT3−ITD+ AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia 2018, 32, 1168–1179. [Google Scholar] [CrossRef]
- Li, K.X.; Wu, H.Y.; Pan, W.Y.; Guo, M.Q.; Qiu, D.Z.; He, Y.J.; Li, Y.H.; Yang, D.H.; Huang, Y.X. A novel approach for relapsed/refractory FLT3mut+ acute myeloid leukaemia: Synergistic effect of the combination of bispecific FLT3scFv/NKG2D-CAR T cells and gilteritinib. Mol. Cancer 2022, 21, 66, Erratum in Mol. Cancer 2022, 21, 134. [Google Scholar] [CrossRef]
- Sommer, C.; Cheng, H.Y.; Nguyen, D.; Dettling, D.; Yeung, Y.A.; Sutton, J.; Hamze, M.; Valton, J.; Smith, J.; Djuretic, I.; et al. Allogeneic FLT3 CAR T Cells with an Off-Switch Exhibit Potent Activity against AML and Can Be Depleted to Expedite Bone Marrow Recovery. Mol. Ther. 2020, 28, 2237–2251. [Google Scholar] [CrossRef] [PubMed]
- El Khawanky, N.; Hughes, A.; Yu, W.; Myburgh, R.; Matschulla, T.; Taromi, S.; Aumann, K.; Clarson, J.; Vinnakota, J.M.; Shoumariyeh, K.; et al. Demethylating therapy increases anti-CD123 CAR T cell cytotoxicity against acute myeloid leukemia. Nat. Commun. 2021, 12, 6436. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Amill, L.; Armand-Ugon, M.; Peña, S.; Casals, M.V.; Santos, C.; Frigola, G.; Minguela, A.; Torralba, B.; Casanovas, B.; Álamo, J.; et al. CD84: A novel target for CAR T-cell therapy for acute myeloid leukemia. Blood 2022, 140, 7379–7381. [Google Scholar] [CrossRef]
- Mandal, K.; Wicaksono, G.; Yu, C.; Adams, J.J.; Hoopmann, M.R.; Temple, W.C.; Izgutdina, A.; Escobar, B.P.; Gorelik, M.; Ihling, C.H.; et al. Structural surfaceomics reveals an AML-specific conformation of integrin β2 as a CAR T cellular therapy target. Nat. Cancer 2023, 4, 1592–1609. [Google Scholar] [CrossRef]
- Ghamari, A.; Pakzad, P.; Majd, A.; Ebrahimi, M.; Hamidieh, A.A. Design and production an effective bispecific tandem chimeric antigen receptor on T cells against CD123 and folate receptor ß towards B-acute myeloid leukaemia blasts. Cell J. 2021, 23, 650–657. [Google Scholar]
- Scherer, L.; Tat, C.; Sauer, T.; Tashiro, H.; Naik, S.; Velasquez, M.P.; Gottschalk, S.; Rooney, C.M.; Brenner, M.K.; Omer, B. LigandCD70.CAR as a platform for dual-targeting CAR T cells for acute myeloid leukemia. Blood 2022, 140, 7396–7397. [Google Scholar] [CrossRef]
- Haubner, S.; Mansilla-Soto, J.; Nataraj, S.; Kogel, F.; Chang, Q.; de Stanchina, E.; Lopez, M.; Ng, M.R.; Fraser, K.; Subklewe, M.; et al. Cooperative CAR targeting to selectively eliminate AML and minimize escape. Cancer Cell 2023, 41, 1871–1891. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhou, G.; Zhang, L.; Zhao, Q. Building potent chimeric antigen receptor T cells with CRISPR genome editing. Front. Immunol. 2019, 10, 456. [Google Scholar] [CrossRef]
- Xavier-Ferrucio, J.; Luo, C.; Angelini, G.; Krishnamurthy, S.; Patel, N.; Pettiglio, M.; Collingsworth, T.; Isik, M.; Ghodssi, A.; Halfond, A.; et al. P1429: Multiplex deletion of myeloid antigens CD33 and CLL-1 BY CRISPR/CAS9 in human hematopoietic stem cells highlights the potential of next- generation transplants for aml treatment. Hemasphere 2022, 6, 1312–1313. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CART cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Poirot, L.; Philip, B.; Schiffer-Mannioui, C.; Le Clerre, D.; Chion-Sotinel, I.; Derniame, S.; Potrel, P.; Bas, C.; Lemaire, L.; Galetto, R.; et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res. 2015, 75, 3853–3864. [Google Scholar] [CrossRef]
- Celichowski, P.; Turi, M.; Charvátová, S.; Radhakrishnan, D.; Feizi, N.; Chyra, Z.; Šimíček, M.; Jelínek, T.; Bago, J.R.; Hájek, R.; et al. Tuning CARs: Recent advances in modulating chimeric antigen receptor (CAR) T cell activity for improved safety, efficacy, and flexibility. J. Transl. Med. 2023, 21, 197. [Google Scholar] [CrossRef]
- Berdien, B.; Mock, U.; Atanackovic, D.; Fehse, B. TALEN-mediated editing of endogenous T-cell receptors facilitates efficient reprogramming of T lymphocytes by lentiviral gene transfer. Gene Ther. 2014, 21, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; DeAngelo, D.J.; Pemmaraju, N.; Dinner, S.; Gill, S.; Olin, R.L.; Wang, E.S.; Konopleva, M.; Stark, E.; Korngold, A.; et al. AMELI-01: A Phase I Trial of UCART123v1.2, an Anti-CD123 Allogeneic CAR-T Cell Product, in Adult Patients with Relapsed or Refractory (R/R) CD123+ Acute Myeloid Leukemia (AML). Blood 2022, 140 (Suppl. S1), 2371–2373. [Google Scholar] [CrossRef]
- Loff, S.; Dietrich, J.; Meyer, J.-E.; Riewaldt, J.; Spehr, J.; von Bonin, M.; Gründer, C.; Swayampakula, M.; Franke, K.; Feldmann, A.; et al. Rapidly switchable universal CAR-T cells for treatment of cd123-positive leukemia. Mol. Ther. Oncol. 2023, 17, 408–420. [Google Scholar] [CrossRef]
- Khawar, M.B.; Sun, H. CAR-NK cells: From natural basis to design for kill. Front. Immunol. 2021, 12, 707542. [Google Scholar] [CrossRef]
- Mehta, R.S.; Rezvani, K. Chimeric antigen receptor expressing natural killer cells for the immunotherapy of cancer. Front. Immunol. 2018, 9, 283. [Google Scholar] [CrossRef] [PubMed]
- Albinger, N.; Pfeifer, R.; Nitsche, M.; Mertlitz, S.; Campe, J.; Stein, K.; Kreyenberg, H.; Schubert, R.; Quadflieg, M.; Schneider, D.; et al. Primary CD33-targeting CAR-NK cells for the treatment of acute myeloid leukemia. Blood Cancer J. 2022, 12, 61. [Google Scholar] [CrossRef] [PubMed]
- Kararoudi, M.N.; Likhite, S.; Elmas, E.; Yamamoto, K.; Schwartz, M.; Sorathia, K.; Pereira, M.d.S.F.; Sezgin, Y.; Devine, R.D.; Lyberger, J.M.; et al. Optimization and validation of CAR transduction into human primary NK cells using CRISPR and AAV. Cell Rep. Methods 2022, 2, 100236. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Drug | ClinicalTrials.gov Identifier | Phase of Clinical Study | Available Online |
|---|---|---|---|
| Anti-CD33 CAR-T cells | NCT04835519 | Phase 1, Phase 2 | https://clinicaltrials.gov/ct2/show/NCT04835519, accessed on 30 April 2023 |
| SC-DARIC33 (anti-CD33 CAR-T cells) | NCT05105152 | Phase 1 | https://clinicaltrials.gov/ct2/show/NCT05105152, accessed on 30 April 2023 |
| Anti-CD123 CAR-T cells | NCT04318678 | Phase 1 | https://clinicaltrials.gov/ct2/show/NCT04318678, accessed on 30 April 2023 |
| Anti-CD123 CAR-T cells | NCT04272125 | Phase 1, Phase 2 | https://clinicaltrials.gov/ct2/show/NCT04272125, accessed on 30 April 2023 |
| UCART123v1.2 (Allogeneic Engineered T cells Expressing Anti-CD123 CAR) | NCT03190278 | Phase 1 | https://clinicaltrials.gov/ct2/show/NCT03190278, accessed on 30 April 2023 |
| Anti-FLT3 CAR-T | NCT05023707 | Phase 1, Phase 2 | https://clinicaltrials.gov/ct2/show/NCT05023707, accessed on 30 April 2023 |
| TAA05 (anti-FLT3 CAR-T cells) | NCT05432401 | Early Phase 1 | https://clinicaltrials.gov/ct2/show/NCT05432401, accessed on 30 April 2023 |
| Anti-CLL-1 CAR-T cells | NCT05252572 | Early Phase 1 | https://clinicaltrials.gov/ct2/show/NCT05252572, accessed on 30 April 2023 |
| Anti-CLL-1 CAR-T cells | NCT04219163 | Phase 1 | https://clinicaltrials.gov/ct2/show/NCT04219163, accessed on 30 April 2023 |
| CLL-1, CD33 and/or CD123-specific CAR-T cells | NCT04010877 | Phase 1, Phase 2 | https://clinicaltrials.gov/ct2/show/NCT04010877, accessed on 30 April 2023 |
| Dual CD33/CLL-1 CAR-T | NCT05248685 | Phase 1 | https://clinicaltrials.gov/ct2/show/NCT05248685, accessed on 30 April 2023 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canichella, M.; de Fabritiis, P. Cell-Based Treatment in Acute Myeloid Leukemia Relapsed after Allogeneic Stem Cell Transplantation. Biomedicines 2024, 12, 1721. https://doi.org/10.3390/biomedicines12081721
Canichella M, de Fabritiis P. Cell-Based Treatment in Acute Myeloid Leukemia Relapsed after Allogeneic Stem Cell Transplantation. Biomedicines. 2024; 12(8):1721. https://doi.org/10.3390/biomedicines12081721
Chicago/Turabian StyleCanichella, Martina, and Paolo de Fabritiis. 2024. "Cell-Based Treatment in Acute Myeloid Leukemia Relapsed after Allogeneic Stem Cell Transplantation" Biomedicines 12, no. 8: 1721. https://doi.org/10.3390/biomedicines12081721