Abstract
Prion diseases are neurodegenerative disorders caused by misfolded prion proteins. Although rare, the said diseases are always fatal; they commonly cause death within months of developing clinical symptoms, and their diagnosis is exceptionally difficult pre-mortem. There are no known cures or treatments other than symptomatic care. Given the aggressiveness of prion diseases on onset, therapies after disease onset could be challenging. Prevention to reduce the incidence or to delay the disease onset has been suggested to be a more feasible approach. In this perspective article, we summarize our current understandings of the origin, risk factors, and clinical manifestations of prion diseases. We propose a PCR testing of the blood to identify PRNP gene polymorphisms at codons 129 and 127 in individuals with familial PRNP mutations to assess the risk. We further present the CRISPR/Cas9 gene editing strategy as a perspective preventative approach for these high-risk individuals to induce a polymorphic change at codon 127 of the PRNP gene, granting immunity to prion diseases in selected high-risk individuals, in particular, in individuals with familial PRNP mutations.
1. Introduction
Misfolded prions are unique infectious agents composed entirely of protein [1]. Unlike other pathogens, prions lack nucleic acids and consist solely of misfolded protein forms. The term “prion” is derived from “proteinaceous infectious particles” [2]. Normal mammalian tissues ubiquitously express the non-pathogenic form of the prion protein (PrPC), with particularly high expression in the central nervous system (CNS) [3]. PrPC is expressed on the cell surface and is implicated in key cellular processes including cell signaling and adhesion, neurogenesis, and neuronal homeostasis [4,5,6,7,8]. Prion diseases, also known as transmissible spongiform encephalopathies (TSEs), are subacute and fatal disorders that result in the neurodegeneration of the central nervous system (CNS) [5]. Common TSEs include Creutzfeldt–Jakob disease (CJD) and kuru in humans, scrapie in sheep, and bovine spongiform encephalopathy (BSE) in cows [9]. Here, we provide a concise review of our current understanding of the molecular genesis of TSEs and propose a perspective preventative approach to eliminate or reduce the prevalence of prion diseases in high-risk individuals.
TSEs are caused by a misfolded isoform of PrPC known as the scrapie prion protein (PrPSc), although the exact mechanisms behind prion misfolding are not entirely understood and are an active subject of ongoing research [10]. PrPSc is highly prone to aggregation and can induce the misfolding of additional PrPC molecules, leading to the formation of neurotoxic amyloid plaques and fibrils in the brain [11,12]. This pathogenic feature of PrPSc gave these disorders the name “Prion Diseases”. Structural comparisons demonstrated that the normal prion protein PrPC has a pronounced α-helical structure, while the pathogenic prion protein PrPSc has an exceedingly high abundance of, nearly all, β-sheets [13,14,15] (Figure 1). The conformational changes in PrPSc make it resistant to proteolytic degradation, further promoting plaque and fibril formation [16]. In addition to their protease-resistant properties, TSEs are characterized by their inability to elicit a detectable immune response [17,18]. Unlike most diseases, the infectious agent PrPSc is a protein peptide that has an identical amino acid sequence to the normal cellular protein isoform PrPC [19]. This feature prevents the immune system from eliciting active immune responses against the misfolded PrPSc particles [4,5,20,21,22]. Therapeutic approaches, such as targeting PrPSc-encoding nucleic acids or monoclonal antibodies targeting PrPSc are challenged by its identical peptide sequence to PrPC and the critical role of PrPC in normal cellular physiology [23]. Moreover, the lack of effective treatments and early diagnostic markers often results in a non-definitive diagnosis at an advanced stage of the disease, further complicating efforts to manage or cure TSEs [24]. With the lessons from the kuru epidemic and our understanding of the human PRNP gene polymorphisms at codon positions 127 and 129 [25], we propose a perspective preventive approach utilizing CRISPR/Cas9 technology to induce PRNP gene polymorphism at codon positions 127 and 129 to provide resistance to prion diseases in high-risk individuals. Given the infancy stage of the CRISPR technology, we also discuss the current challenges of utilizing this technology in humans.
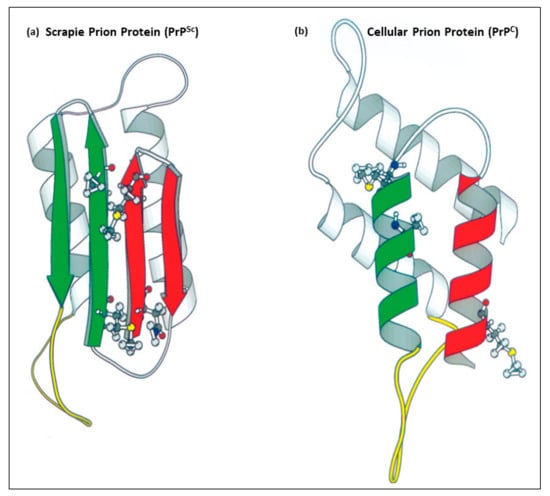
Figure 1.
Images of the tertiary structure of human prion proteins. (a) is the pathogenic protease-resistant misfolded scrapie prion isoform (PrPSc) with a high abundance of β-pleated sheets. (b) is the normal cellular prion protein (PrPC) with an α-helical structure. (a,b) are adapted from Huang et al. [26].
2. Current Understandings of Human TSEs
Prion diseases can be sporadic, genetic, or acquired [5]. Sporadic TSEs make up approximately 85% of prion diseases in humans, identified by the spontaneous conversion of PrPC into PrPSc with unknown cellular drivers [5,25]. Genetic TSEs are suspected to occur when the autosomal dominant mutation in the PRNP gene is inherited by children of whom at least one parent is a carrier [27,28]. Genetic TSEs contribute to 9–15% of human cases [29]. Acquired prion diseases have foodborne transmission and can be spread either zoonotically or between humans [30]. Other sources of acquired TSEs are the iatrogenic inoculation from prion-contaminated medical devices, such as surgical instruments and electrodes, and blood transfusions [31].
Human TSEs can present as kuru, Creutzfeldt–Jakob disease (CJD), Gerstmann–Sträussler–Scheinker syndrome (GSS), and fatal insomnia (FI), with the majority being CJD [32,33]. The most notable forms of CJD are sporadic CJD (sCJD) and variant CJD (vCJD), though they can also be transmitted genetically as familial CJD (fCJD) or iatrogenically (iCJD) [32,33,34]. sCJD contributes to approximately 85% of human prion diseases [5]. The precise drivers or risk factors for sCJD are not clear. However, methionine homozygosity at codon 129 (Met129/Met129) in the PRNP gene is one of the recognized high-risk factors for sCJD [35]. Data from three series studies in nine countries revealed that a mutation of Met129 contributes to at least 70% of the sCJD cases [35,36,37]. Age is also a risk factor for the Met129 mutation, with advanced ages being more susceptible [38]. vCJD is a zoonotic form that is contracted through the consumption of cattle affected by bovine spongiform encephalopathy (BSE), a prion disease that affects cows commonly referred to as “Mad Cow Disease” [39,40].
sCJD primarily exhibits clinical symptoms later in life, appearing in individuals between 55 and 75 years old [41,42]. Although the subclinical phase can be years to decades, the disease rapidly progresses on onset and is always fatal, with approximately 90% of patients dying within a year after the non-specific onset of symptoms [41,42]. While symptoms are highly variable, patients may initially exhibit vertigo, fatigue, insomnia, and headache [43]. These symptoms can be accompanied by memory loss, behavioral changes (depression, irritability, apathy, mood swings), and sensory changes (incoordination, visual impairment) [43]. The late stages of the disease have the potential to cause cerebellar ataxia, myoclonus, and more pronounced dementia and disorientation [43]. Extrapyramidal symptoms include bradykinesia, dystonia, rigidity, and possibly blindness [43]. As the disease continually develops, patients can gradually lose speech and mobility leading up to their death [43]. Secondary infections, such as pneumonia, are potential causes of death in TSEs [43]. vCJD, unlike sCJD, usually occurs in younger individuals, with the median age of death being 28 years old [43]. vCJD also has a longer subclinical phase than sCJD of 13–14 months [44]. The initial symptoms are similar to those of sCJD, but the most prominent early signs are painful sensory symptoms including dysesthesia and paresthesia [44,45,46].
PrPSc, due to its tertiary and/or quaternary structure, is characterized by its distinctive properties, especially the ability to maintain its integrity under conditions that would denature most other proteins [16]. This stability allows prions to accumulate in cells. The scrapie isoform is protease resistant and is highly heat resistant. Cooking meat infected with PrPSc may reduce, but will not completely eliminate, its pathogenicity [16,47]. It was shown experimentally that heating PrPSc at 600 °C and 1000 °C can achieve lower infectivity and destroy prion infectivity, respectively [48]. In laboratory practices, heating to 115 °C or autoclaving was shown to be effective in reducing the infectivity of prions causing BSEs [47]. Certain proteases can degrade PrPSc, but the optimum conditions for degradation are a high pH of 10–12 and a temperature of 50–60 °C [48,49]. This provides a solution for destroying prions in the environment but is not feasible in vivo or during food processing. Based on a series of experiments conducted by the CDC, autoclaving PrPSc-contaminated instruments in sodium hydroxide is sufficient to eliminate the risk to the operator [50]. The CDC has developed a guideline that is incorporated in “Chemical and Autoclave Sterilization Methods Outlined in Annex III of the WHO Infection Control Guidelines for TSEs”.
The mechanisms by which PrPSc infects and degrades the CNS are not well understood. Notwithstanding, enough information is present to draw plausible inferences about acquired PrPSc. It is believed that orally acquired PrPSc exploits the gut-associated lymphoid tissue (GALT) in the small intestine as a means to spread to the CNS [51]. The GALT typically functions with the mesenteric lymph nodes (MLNs) to prevent infections by triggering the production of immune cells [51]. Intestinal M cells, a type of intestinal epithelial cell (IEC) located in the same region as GALT lymphoid follicles, usually function to translocate antigens to the GALT to elicit an immune response [52]. In the case of PrPSc infections, the M cells uptake the PrPSc protein into the GALT and spread into follicular dendritic cells (FDCs), specialized antigen-presenting cells in the lymphoid follicles [53]. PrPC is a cell-surface glycoprotein attached to the plasma membrane by a glycosylphosphatidylinositol (GPI) anchor found in high levels in lymphoid cells in addition to the CNS in which it is prominent. Thus, the protein is also largely expressed in the plasma membrane of FDCs [54,55,56,57,58]. As FDCs capture and retain PrPSc, the high concentrations of PrPC enable the replication and accumulation of PrPSc by inducing the misfolding of PrPC [53,56]. When the concentration is sufficient for neuroinvasion, PrPSc spreads, possibly via the peripheral nervous system, to infect the CNS [53]. The hematogenous spread of prions to the CNS may also be possible [53]. Whether non-acquired CJD utilizes the same neuroinvasion pathways or directly occurs in the CNS is not clear. Once PrPSc occurs in the CNS, it is suspected to spread between cells by means including direct cell contact by tunneling nanotubes (TNTs), exosome-mediated transfer, cell-to-cell contact, or from membrane budding and transport in vesicles [5]. After dissemination, PrPSc induces abnormal conformational changes in the already-formed PrPC of surrounding cells, resulting in an exponential rate of PrPSc replication and aggregation [5]. PrPSc accumulation causes spongiform degeneration, neuroinflammation, neuronal apoptosis, and synaptic changes [59] (Figure 2). How prion protein aggregates cause neurodegeneration is not well understood [59].
Prion diseases are exceedingly difficult to diagnose, as they have incubation periods of years to decades and exhibit clinical symptoms that are indistinguishably similar to other neurological disorders [24]. Furthermore, all definite diagnosis methods require a sample of brain tissue that can only be extracted postmortem [24]. Testing methods include the processing of tissue using immunohistochemistry or Western blotting for PrPSc detection [24]. Antemortem testing using EEGs, MRIs, and examining cerebrospinal fluid for elevated 14-3-3 protein levels can be conducted but is limited to a non-definitive diagnosis [60,61,62]. Real-time quaking-induced conversion (RT-QuIC) assays have made a considerable impact on clinical diagnosis and have become the standard tool for sCJD diagnosis [63,64,65,66]. By exploiting the ability of PrPSc found in cerebrospinal fluid (CSF) to induce the conversion of PrPC to PrPSc and the consequent aggregation, the RT-QuIC assay can detect the formation of the aggregated PrPC in real time using fluorescent dyes [63,64,65,66]. The method can reach a specificity of 99–100% and sensitivities of up to 97%, depending on the alleles being detected of MM, MV, or VV and specific study sites [63].

Figure 2.
Images of uninfected and prion-infected brain tissues. (A,B) are MRI scans of uninfected (A) and prion-infected (B) brains. Arrows and circles in (B) indicate the gross images of spongiform degeneration in two regions of the brain. (C,D) are H&E stains of uninfected (C) and prion-infected (D) brain sections. Arrows in (D) indicate spongiform degeneration at the cellular level. (A,B) are adapted from Zeidler et al. [67]. (C) is from Practical Surgical Neuropathology: A Diagnostic Approach (Second Edition by Daniel J. Brat), Chapter 2, Normal Brain Histopathology [68]. (D) is adapted from Britannica, T. Editors of Encyclopaedia [69].
Certain individuals have a higher risk of developing sCJD, and the relative risk factors can be determined by examining the polymorphisms at codon 129 in the PRNP gene with PCR [70]. The common alleles at this codon are methionine 129 (Met129) and valine 129 (Val129) [70]. Individuals with homozygotes of the allele Met129 (Met129/Met129) have a higher risk of developing sCJD than those with the heterozygous allele (Met129/Val129) [70]. Whether individuals with homozygous valine 129 (Val129/Val129) have a significantly higher risk than those with the heterozygous allele (Met129/Val129) is not clear, as studies from different historical periods and different geographic locations reported inconsistent outcomes [38,71]. The exact mechanism behind prion protein misfolding in sCJD is still not well understood. Family history also plays a significant role in determining the risk of a patient developing fCJD. Being an autosomal dominant mutation, it is very probable that the children of an fCJD carrier may exhibit clinical symptoms later in life [28].
3. Lessons from the Kuru Epidemic
Kuru, a TSE acquired from consuming prion-infected human brain and nervous tissue, was a prominent disease of the Eastern Highlands Province (EHP) of Papua New Guinea in the 1950s before the country conformed to Western influence [72,73,74,75,76,77,78]. A common practice by the Fore tribe, inhabitants of the area, was the consumption of deceased relatives at mortuary feasts to free the spirit of the dead (endocannibalism). It was believed by the Fore tribe that the deceased being eaten by their loved ones was preferable to being consumed by insects. This practice enabled kuru to widely spread among the Fore people, peaking with mortality rates of up to 3.5% of the population during the 1940s and 1950s [79]. The disease disproportionately affected more women and children than men, as it was customary for the women and children to consume the brain while the men preferred muscle tissue. It is noteworthy that 60% of kuru cases were in adult females while only 2% were found in adult males, with the remainder being in children [73,79]. By 1960, the practice of endocannibalism mostly ceased; subsequently, kuru cases gradually disappeared [73].
During the kuru epidemic in the EHP, G127V, a new PRNP polymorphism that substitutes glycine (Gly127) with Val127 arose [80]. The combination of the G127V mutation with homozygous methionine 129 (Gly127Met129/Val127Met129) provides complete resistance to kuru and sCJD but not vCJD [80]. With this knowledge, Asante et al. generated transgenic mice that were homozygotes or heterozygotes of human PrP Val127Met129 (HuPrP Val127Met129/Val127Met129 or Gly127Met129/Val127Met129) and tested their resistance to human TSEs, including kuru, sCJD, and iCJD. The Val127Met129/Val127Met129 mice were completely resistant to all four of the tested human TSE types (types 1–4) by molecular classification [80,81], with no clinical symptoms or subclinical infection [80]. That study also found that mice with the G127V heterozygosity Gly127Met129/Val127Met129 were only resistant to human TSE types 1–3 but not to type 4. Whether Val127Met129/Val127Met129 or Gly127Met129/Val127Met129 provide resistance to other types of human TSEs is not known from this study. X-ray crystallography and NMR spectroscopy demonstrated that the G127V polymorphism induces structural changes that constrain the conformation of PrPC in regions associated with prion disease, preventing β-sheet formation [15,82]. The mutation increases the intermolecular hydrogen bonding between PrPC dimers, reducing the instability and the likelihood of protein misfolding [15,82].
4. Current Development in Treating Prion Diseases
The current treatment for prion diseases is limited to supportive care, as no treatments with disease-modifying effects are currently available. With the understanding that PrPC protein misfolding is the cause of prion diseases, various strategies are in development to prevent PrPC misfolding or to target misfolded PrPSc aggregates. These strategies are often coupled with treatments that directly reduce the production of PrPC [83,84]. However, the development of effective treatments for prion diseases is particularly challenging compared to the more common, slowly progressive neurodegenerative disorders due to the rapid progression and lack of definitive screening biological markers [85]. Furthermore, designing formal clinical trials with an adequate patient population for prion diseases has proven to be extremely difficult, largely because of the rarity of prion diseases. Much of the clinical evidence regarding treatment effects is based on anecdotal observations rather than on rigorous scientific data. Consequently, only six treatments for prion diseases were advanced to the stage of clinical evaluations: flupirtine, quinacrine, doxycycline, pentosan polysulfate, anti-PrPSc monoclonal antibody PRN100, and a PRNP antisense oligonucleotide (ASO) [25,85].
Flupirtine is a triaminopyridine that exhibits anti-apoptotic activity against PrPSc and amyloid beta (Aβ) peptides [86]. It was the first anti-prion drug to enter clinical trials in which patients with sCJD were treated. While improved cognitive performance was observed, there was no significant improvement in patient survival times [87]. Alternately, quinacrine is an antimalarial drug that was thought to inhibit the conversion of PrPC to PrPSc [88]. Quinacrine was tested in multiple clinical trials, but the outcomes were inconclusive [89,90,91]. At best, a pilot clinical trial demonstrated that quinacrine induced a marginal improvement in alleviating symptoms [92]. However, expanded clinical trials with larger patient populations did not demonstrate the same benefit of quinacrine [93,94].
With the positive outcome of antibody-based therapy in treating certain neurological disorders such as Alzheimer’s disease [95,96], the use of a monoclonal antibody to target PrPSc had been pursued. However, clinical trials with the PrPSc-targeting monoclonal antibody PRN100 failed to slow down prion disease progression, despite a reduction in PrPSc deposits that was found in one patient in a postmortem autopsy [97]. Achieving a plausible clinical benefit with PrPSc-targeting monoclonal antibodies remains challenging.
Tetracyclines, such as doxycycline, have been shown to mitigate PrPSc aggregation and neurotoxicity and prolong the survival of animals with prion diseases [98,99,100]. Doxycycline has been tested in multiple clinical trials, but conflicting results were observed. In one study, doxycycline was used to treat CJD patients and showed an improvement in patient survival by approximately 80% [101]. Conversely, a different clinical trial showed no significant changes to patient survival and disease progression [102]. The anti-PRNP ASO, ION717, was recently developed. ION717 was shown to induce a significantly prolonged survival of mice with scrapie [103]. ION717 recently entered a phase I/II clinical trial (NCT06153966).
5. Perspective Prevention Strategies
Although passive and active immunization strategies and RNAi gene therapies to target PrPC have been proposed and are currently in development [25,104,105,106,107,108,109,110,111], given that PrPC is important for normal cellular function, the potential off-target unwanted toxicity could be a concern [112,113,114,115,116]. In another critical aspect, due to the rapid progression of prion diseases once the symptoms begin, achieving clinical benefit within this therapeutic window can be extremely challenging. Thus, preventing or delaying prion disease onset has been suggested as one of the most promising goals in carriers of pathogenic mutations in the PRNP gene [25,111].
With the lessons from the kuru epidemic and the outcome from the transgenic animal study by Asante et al. [80], we propose that CRISPR/Cas9 may be utilized to prevent disease onset in high-risk individuals suspected of being carriers of prion diseases (Figure 3). This proposed approach could leverage the future success of safer viral-vector-based or non-viral delivery systems into the somatic cells of individuals confirmed as being PRNP high-risk mutation carriers [117,118,119,120,121,122]. Candidates would be those who have a family history of fCJD and potentially a family history of sCJD with known pathogenic mutations in the PRNP gene. Specifically, those with homozygosity for Met129 (Gly127Met129/Gly127Met129) would be reasonable candidates through PCR screening. The strategy would be introducing one- or two-nucleotide substitution(s) in the chromosome of the 20th pair to induce the desired polymorphic change to Gly127Met129/Val127Met129 or Val127Met129/Val127Met129 in the PRNP gene. gRNA designed to target a region of the PRNP gene that includes the 127th codon position functions with the Cas9 enzyme in the Cas9-gRNA complex to induce a double-strand break (DSB) in the targeted area. Noteworthily, the success of CRISPR/Cas9 in preventing TSEs relies on the homology-directed repair (HDR) pathway of the DSB using a donor DNA template with a nearly identical nucleotide sequence. PCR can be used as verification that the mutation has been correctly incorporated after the DSB is repaired. It should be emphasized that the proposed strategy represents a perspective concept or direction in developing feasible strategies for preventing TSEs in high-risk individuals. Acknowledging the potential and the infancy stage of using CRISPR technology in treating human diseases, the feasibility and safety of this proposed strategy need to be rigorously tested in relevant pre-clinical models. Encouragingly, the utilization of CRISP-Cas9 technology to treat human diseases is under active investigation in various pre-clinical animal models of human diseases [123,124,125,126,127,128,129]. Recent clinical trials have demonstrated excellent safety profiles using in vivo CRISPR/Cas9 editing of the KLKB1 gene to treat patients with hereditary angioedema and editing the HSV-1 gene to treat patients with herpetic stromal keratitis [130,131]. The FDA approval and later successes of using CRISPR/Cas9 ex vivo genome-edited hematological stem cells to treat sickle-cell disease also shed light on the viability of the proposed approaches [132].
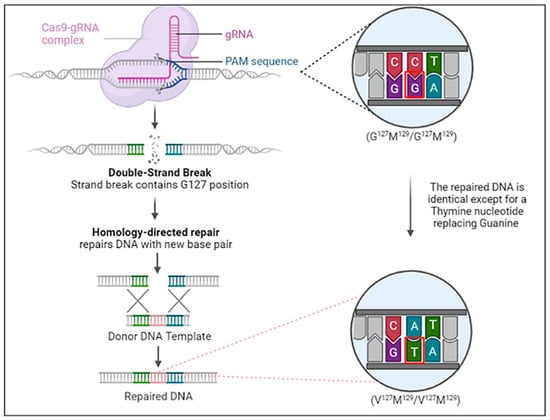
Figure 3.
The proposed strategy of CRISPR/Cas9 to introduce the Gly127 polymorphism in PRNP gene to prevent prion diseases during early life of high-risk individuals, in particular of individuals with familial PRNP mutations. Only a single or double base pair substitution (outlined in red) is necessary to induce the Gly127 polymorphism into the PRNP gene.
6. Current Limitations and Challenges of Using CRISPR Gene Editing for Therapy
Given the recent history of CRISPR/Cas9 technology, its application in treating human diseases is still in its infancy, pending the resolution of the following challenges. The first challenge is potential non-specific integration. At present, CRISPR/Cas9 has the potential to induce nucleotide base mismatches between the gRNA and non-target sequences that may lead to off-target effects, primarily being unknown mutations [133,134,135]. This is caused by a duplex conformation that forms upon mismatched base interactions under strong force [136,137]. Many active investigations are being conducted to overcome these challenges, potentially through approaches to control Cas9 activation. For example, the multiple high-fidelity mutants of Cas9, such as HypaCas9, Cas9-HF1, and SuperFi-Cas9, have exhibited improved accuracy, although at the cost of reduced efficiency [138,139,140,141]. While these mutants are not without limitations, they suggest that utilizing Cas9 variants represents a promising approach for mitigating the off-target effects of CRISPR/Cas9.
The second challenge is the dependency of Cas9 activity on a suitable PAM site to perform base changes [142,143]. This limits the applicability of CRISPR/Cas9 as a therapeutic approach to certain diseases. However, Walton et al. mutated the amino acid sites of Cas9 to produce an SpCas9 variant (SpRY) that is less dependent on PAM restrictions [144]. When inducing the same mutations in the Cas9-HF1 variant (SpRY-HF1), almost all off-target effects were eliminated [145]. Further, modifying Nme2Cas9, a Cas9 variant derived from Neisseria meningitidis, has shown promises [146,147]. Nme2Cas9 has strong gene-editing activity in mammalian cells and exhibits enhanced capabilities to target a greater abundance of potential target sites [146,147]. The smaller size of Nme2Cas9 also provides a greater potential for targeted delivery [146,147]. These approaches, although still under active investigations, suggest that developing PAM-independent Cas9 may increase the potential for precise genome editing across a wider variety of genomic sequences.
The third challenge is the potential to induce chromosomal disorganization when repairing DSBs. Activation of the NHEJ repair pathway is the commonly triggered response to DSBs. While the probability of chromosomal disorganization during DSB repair is extremely low, base pair deletions or chromosomal structural translocations can potentially lead to malignancy [148,149,150,151,152,153]. It was shown that the recurrent cleavage of target genes by CRISPR/Cas9 is a significant contributor to the potential occurrence of chromosomal translocations and deletions [148]. It is noteworthy that Yin et al. combined the structural domain of the exonuclease TREX2 with Cas9 to create Cas9TX, which was shown to be highly effective in reducing chromosomal translocations [148]. Collectively, these studies suggest that modifications to Cas9 have the potential to mitigate adverse effects while ensuring genome editing efficiency.
Lastly, the proper delivery systems for clinical use remain an actively investigated area in gene therapy. Both viral and non-viral vectors have been used as the delivery system in clinical studies [154,155]. Improving the specificity of target cell delivery, the efficiency of delivery, and reducing immunogenicity are among the current enduring efforts to improve the delivery system for CRISPR/Cas9 [156,157,158,159,160]. In addition, confounders such as sex, age, and genetic ethnicity should also be considered as crucially important variables that may impact the efficacy of CRISPR/Cas9-based interventions [161]. Notwithstanding, the recent successes proving the safety of using CRISPR/Cas9 in clinical trials, along with the current active investigations to improve the technology, strongly suggest that the future of using this technology to prevent prion diseases in high-risk individuals holds great promise.
Author Contributions
Conceptualization, M.M.M.; Investigation, M.M.M.; Project Administration, M.M.M. and Q.C.; Visualization, M.M.M.; Writing—Original Draft, M.M.M.; Writing—Review and Editing, M.M.M. and Q.C.; Supervision, Q.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wille, H.; Requena, J.R. The Structure of PrP(Sc) Prions. Pathogens 2018, 7, 20. [Google Scholar] [CrossRef]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef]
- Grassmann, A.; Wolf, H.; Hofmann, J.; Graham, J.; Vorberg, I. Cellular aspects of prion replication in vitro. Viruses 2013, 5, 374–405. [Google Scholar] [CrossRef]
- Legname, G. Elucidating the function of the prion protein. PLoS Pathog. 2017, 13, e1006458. [Google Scholar] [CrossRef]
- Sigurdson, C.J.; Bartz, J.C.; Glatzel, M. Cellular and Molecular Mechanisms of Prion Disease. Annu. Rev. Pathol. 2019, 14, 497–516. [Google Scholar] [CrossRef]
- Stahl, N.; Borchelt, D.R.; Hsiao, K.; Prusiner, S.B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987, 51, 229–240. [Google Scholar] [CrossRef]
- Haraguchi, T.; Fisher, S.; Olofsson, S.; Endo, T.; Groth, D.; Tarentino, A.; Borchelt, D.R.; Teplow, D.; Hood, L.; Burlingame, A.; et al. Asparagine-linked glycosylation of the scrapie and cellular prion proteins. Arch. Biochem. Biophys. 1989, 274, 1–13. [Google Scholar] [CrossRef]
- Linden, R. The Biological Function of the Prion Protein: A Cell Surface Scaffold of Signaling Modules. Front. Mol. Neurosci. 2017, 10, 77. [Google Scholar] [CrossRef]
- Zerr, I.; Ladogana, A.; Mead, S.; Hermann, P.; Forloni, G.; Appleby, B.S. Creutzfeldt-Jakob disease and other prion diseases. Nat. Rev. Dis. Primers 2024, 10, 14. [Google Scholar] [CrossRef]
- Jankovska, N.; Rusina, R.; Bruzova, M.; Parobkova, E.; Olejar, T.; Matej, R. Human Prion Disorders: Review of the Current Literature and a Twenty-Year Experience of the National Surveillance Center in the Czech Republic. Diagnostics 2021, 11, 1821. [Google Scholar] [CrossRef]
- Chang, S.C.; Hannaoui, S.; Arifin, M.I.; Huang, Y.; Tang, X.; Wille, H.; Gilch, S. Propagation of PrP(Sc) in mice reveals impact of aggregate composition on prion disease pathogenesis. Commun. Biol. 2023, 6, 1162. [Google Scholar] [CrossRef]
- Kupfer, L.; Hinrichs, W.; Groschup, M.H. Prion protein misfolding. Curr. Mol. Med. 2009, 9, 826–835. [Google Scholar] [CrossRef]
- Eghiaian, F. Structuring the puzzle of prion propagation. Curr. Opin. Struct. Biol. 2005, 15, 724–730. [Google Scholar] [CrossRef]
- Martins, V.R. A receptor for infectious and cellular prion protein. Braz. J. Med. Biol. Res. 1999, 32, 853–859. [Google Scholar] [CrossRef][Green Version]
- Kraus, A.; Hoyt, F.; Schwartz, C.L.; Hansen, B.; Artikis, E.; Hughson, A.G.; Raymond, G.J.; Race, B.; Baron, G.S.; Caughey, B. High-resolution structure and strain comparison of infectious mammalian prions. Mol. Cell 2021, 81, 4540–4551.e6. [Google Scholar] [CrossRef]
- Concha-Marambio, L.; Diaz-Espinoza, R.; Soto, C. The extent of protease resistance of misfolded prion protein is highly dependent on the salt concentration. J. Biol. Chem. 2014, 289, 3073–3079. [Google Scholar] [CrossRef]
- Zabel, M.D.; Avery, A.C. Prions—Not your immunologist’s pathogen. PLoS Pathog. 2015, 11, e1004624. [Google Scholar] [CrossRef]
- Porter, D.D.; Porter, H.G.; Cox, N.A. Failure to demonstrate a humoral immune response to scrapie infection in mice. J. Immunol. 1973, 111, 1407–1410. [Google Scholar] [CrossRef]
- Abalos, G.C.; Cruite, J.T.; Bellon, A.; Hemmers, S.; Akagi, J.; Mastrianni, J.A.; Williamson, R.A.; Solforosi, L. Identifying key components of the PrPC-PrPSc replicative interface. J. Biol. Chem. 2008, 283, 34021–34028. [Google Scholar] [CrossRef]
- Aucouturier, P.; Carnaud, C. The immune system and prion diseases: A relationship of complicity and blindness. J. Leukoc. Biol. 2002, 72, 1075–1083. [Google Scholar] [CrossRef]
- Aucouturier, P.; Carp, R.I.; Carnaud, C.; Wisniewski, T. Prion diseases and the immune system. Clin. Immunol. 2000, 96, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicka, A.; Deptula, W. The role of the immune system in the pathogenesis of prion diseases. Postepy Hig. Med. Dosw. 2008, 62, 166–173. [Google Scholar]
- Mabbott, N.A.; Bradford, B.M.; Pal, R.; Young, R.; Donaldson, D.S. The Effects of Immune System Modulation on Prion Disease Susceptibility and Pathogenesis. Int. J. Mol. Sci. 2020, 21, 7299. [Google Scholar] [CrossRef] [PubMed]
- Figgie, M.P., Jr.; Appleby, B.S. Clinical Use of Improved Diagnostic Testing for Detection of Prion Disease. Viruses 2021, 13, 789. [Google Scholar] [CrossRef] [PubMed]
- Baiardi, S.; Mammana, A.; Capellari, S.; Parchi, P. Human prion disease. Molecular pathogenesis, and possible therapeutic targets and strategies. Expert Opin. Ther. Targets 2023, 27, 1271–1284. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Prusiner, S.B.; Cohen, F.E. Scrapie prions: A three-dimensional model of an infectious fragment. Fold Des. 1996, 1, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G. Genetic background of human prion diseases. Ideggyogy. Szle. 2007, 60, 438–446. [Google Scholar]
- Goldman, J.S.; Vallabh, S.M. Genetic counseling for prion disease: Updates and best practices. Genet. Med. 2022, 24, 1993–2003. [Google Scholar] [CrossRef]
- Asher, D.M.; Gregori, L. Human transmissible spongiform encephalopathies: Historic view. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 153, pp. 1–17. [Google Scholar]
- Will, R.G. Acquired prion disease: Iatrogenic CJD, variant CJD, kuru. Br. Med. Bull. 2003, 66, 255–265. [Google Scholar] [CrossRef]
- Brown, P.; Preece, M.; Brandel, J.P.; Sato, T.; McShane, L.; Zerr, I.; Fletcher, A.; Will, R.G.; Pocchiari, M.; Cashman, N.R.; et al. Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology 2000, 55, 1075–1081. [Google Scholar] [CrossRef]
- Brandel, J.P.; Knight, R. Variant Creutzfeldt-Jakob disease. Handb. Clin. Neurol. 2018, 153, 191–205. [Google Scholar] [PubMed]
- Ironside, J.W. Variant Creutzfeldt-Jakob disease. Haemophilia 2010, 5 (Suppl 16), 175–180. [Google Scholar] [CrossRef] [PubMed]
- Laskowska, E.; Kuczynska-Wisnik, D.; Lipinska, B. Proteomic analysis of protein homeostasis and aggregation. J. Proteom. 2019, 198, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Palmer, M.S.; Dryden, A.J.; Hughes, J.T.; Collinge, J. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt-Jakob disease. Nature 1991, 352, 340–342. [Google Scholar] [CrossRef] [PubMed]
- Lampe, J.; Kitzler, H.; Walter, M.C.; Lochmüller, H.; Reichmann, H. Methionine homozygosity at prion gene codon 129 may predispose to sporadic inclusion-body myositis. Lancet 1999, 353, 465–466. [Google Scholar] [CrossRef] [PubMed]
- Will, R.G.; Alperovitch, A.; Poser, S.; Pocchiari, M.; Hoffman, A.; Mitrova, E.; de Silva, R.; D’Alessandro, M.; Delasnerie-Laupretre, N.; Zerr, I.; et al. Descriptive epidemiology of Creutzfeldt-Jakob disease in six European countries, 1993–1995. EU Collaborative Study Group for CJD. Ann. Neurol. 1998, 43, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Alperovitch, A.; Zerr, I.; Pocchiari, M.; Mitrova, E.; de Pedro Cuesta, J.; Hegyi, I.; Collins, S.; Kretzschmar, H.; van Duijn, C.; Will, R.G. Codon 129 prion protein genotype and sporadic Creutzfeldt-Jakob disease. Lancet 1999, 353, 1673–1674. [Google Scholar] [CrossRef] [PubMed]
- Amin, R.; Darwin, R.; Chakraborty, S.; Chandran, D.; Chopra, H.; Dhama, K. Bovine Spongiform Encephalopathy, “Mad Cow’s Disease” and Variant Creutzfeldt-Jakob Disease in Humans, A Critical Update. Arch. Med. Res. 2023, 54, 102854. [Google Scholar] [CrossRef]
- Harman, J.L.; Silva, C.J. Bovine spongiform encephalopathy. J. Am. Vet. Med. Assoc. 2009, 234, 59–72. [Google Scholar] [CrossRef]
- Uttley, L.; Carroll, C.; Wong, R.; Hilton, D.A.; Stevenson, M. Creutzfeldt-Jakob disease: A systematic review of global incidence, prevalence, infectivity, and incubation. Lancet Infect. Dis. 2020, 20, e2–e10. [Google Scholar] [CrossRef]
- Vacca, V.M. Understanding Creutzfeldt-Jakob disease. Nursing 2016, 46, 36–42, quiz 42–43. [Google Scholar] [CrossRef] [PubMed]
- Sitammagari, K.K.; Masood, W. Creutzfeldt Jakob Disease; StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Macleod, M.A.; Stewart, G.E.; Zeidler, M.; Will, R.; Knight, R. Sensory features of variant Creutzfeldt-Jakob disease. J. Neurol. 2002, 249, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Heath, C.A.; Cooper, S.A.; Murray, K.; Lowman, A.; Henry, C.; Macleod, M.A.; Stewart, G.E.; Zeidler, M.; MacKenzie, J.M.; Ironside, J.W.; et al. Validation of diagnostic criteria for variant Creutzfeldt-Jakob disease. Ann. Neurol. 2010, 67, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.; Knight, R. Clinical features of variant Creutzfeldt-Jakob disease. Rev. Med. Virol. 2002, 12, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Langeveld, J.P.M.; Balkema-Buschmann, A.; Becher, D.; Thomzig, A.; Nonno, R.; Andréoletti, O.; Davidse, A.; Di Bari, M.A.; Pirisinu, L.; Agrimi, U.; et al. Stability of BSE infectivity towards heat treatment even after proteolytic removal of prion protein. Vet. Res. 2021, 52, 59. [Google Scholar] [CrossRef] [PubMed]
- Saunders, S.E.; Bartelt-Hunt, S.L.; Bartz, J.C. Prions in the environment: Occurrence, fate and mitigation. Prion 2008, 2, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Hui, Z.; Doi, H.; Kanouchi, H.; Matsuura, Y.; Mohri, S.; Nonomura, Y.; Oka, T. Alkaline serine protease produced by Streptomyces sp. degrades PrP(Sc). Biochem. Biophys. Res. Commun. 2004, 321, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.A.; Merritt, K. Use of containment pans and lids for autoclaving caustic solutions. Am. J. Infect. Control 2003, 31, 257–260. [Google Scholar] [CrossRef]
- Donaldson, D.S.; Else, K.J.; Mabbott, N.A. The Gut-Associated Lymphoid Tissues in the Small Intestine, Not the Large Intestine, Play a Major Role in Oral Prion Disease Pathogenesis. J. Virol. 2015, 89, 9532–9547. [Google Scholar] [CrossRef]
- Ohno, H. Intestinal M cells. J. Biochem. 2016, 159, 151–160. [Google Scholar] [CrossRef]
- Donaldson, D.S.; Kobayashi, A.; Ohno, H.; Yagita, H.; Williams, I.R.; Mabbott, N.A. M cell-depletion blocks oral prion disease pathogenesis. Mucosal Immunol. 2012, 5, 216–225. [Google Scholar] [CrossRef]
- Rezk, S.A.; Nathwani, B.N.; Zhao, X.; Weiss, L.M. Follicular dendritic cells: Origin, function, and different disease-associated patterns. Hum. Pathol. 2013, 44, 937–950. [Google Scholar] [CrossRef]
- Puig, B.; Altmeppen, H.C.; Linsenmeier, L.; Chakroun, K.; Wegwitz, F.; Piontek, U.K.; Tatzelt, J.; Bate, C.; Magnus, T.; Glatzel, M. GPI-anchor signal sequence influences PrPC sorting, shedding and signalling, and impacts on different pathomechanistic aspects of prion disease in mice. PLoS Pathog. 2019, 15, e1007520. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, L.; Brown, K.L.; Bradford, B.M.; Hopkins, J.; Bailey, M.; Rajewsky, K.; Manson, J.C.; Mabbott, N.A. Follicular dendritic cell-specific prion protein (PrP) expression alone is sufficient to sustain prion infection in the spleen. PLoS Pathog. 2011, 7, e1002402. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.; Kim, M.Y. The role of cellular prion protein in immune system. BMB Rep. 2023, 56, 645–650. [Google Scholar] [CrossRef]
- Isaacs, J.D.; Jackson, G.S.; Altmann, D.M. The role of the cellular prion protein in the immune system. Clin. Exp. Immunol. 2006, 146, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Satani, N. The intricate mechanisms of neurodegeneration in prion diseases. Trends Mol. Med. 2011, 17, 14–24. [Google Scholar] [CrossRef]
- Wieser, H.G.; Schindler, K.; Zumsteg, D. EEG in Creutzfeldt-Jakob disease. Clin. Neurophysiol. 2006, 117, 935–951. [Google Scholar] [CrossRef]
- Macfarlane, R.G.; Wroe, S.J.; Collinge, J.; Yousry, T.A.; Jäger, H.R. Neuroimaging findings in human prion disease. J. Neurol. Neurosurg. Psychiatry 2007, 78, 664–670. [Google Scholar] [CrossRef]
- Zerr, I.; Bodemer, M.; Gefeller, O.; Otto, M.; Poser, S.; Wiltfang, J.; Windl, O.; Kretzschmar, H.A.; Weber, T. Detection of 14–3-3 protein in the cerebrospinal fluid supports the diagnosis of Creutzfeldt-Jakob disease. Ann. Neurol. 1998, 43, 32–40. [Google Scholar] [CrossRef]
- Atarashi, R. RT-QuIC as ultrasensitive method for prion detection. Cell Tissue Res. 2023, 392, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Brusick, D.J. Genetic risk assessment. JAPCA 1987, 37, 795. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Srivastava, A.; Alam, P.; Caughey, B. RT-QuIC and Related Assays for Detecting and Quantifying Prion-like Pathological Seeds of alpha-Synuclein. Biomolecules 2022, 12, 576. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.M.; Salamat, M.K.F.; de Wolf, C.; McCutcheon, S.; Blanco, A.R.; Manson, J.C.; Hunter, N.; Houston, E.F. Development of a sensitive real-time quaking-induced conversion (RT-QuIC) assay for application in prion-infected blood. PLoS ONE 2023, 18, e0293845. [Google Scholar] [CrossRef] [PubMed]
- Zeidler, M.; Collie, D.A.; Macleod, M.A.; Sellar, R.J.; Knight, R. FLAIR MRI in sporadic Creutzfeldt-Jakob disease. Neurology 2001, 56, 282. [Google Scholar] [CrossRef]
- Perry, A.; Bratt, D.J. Practical Surgical Neuropathology: A Diagnostic Approach, 2nd ed.; Pattern Recognition; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Britannica. Creutzfeldt–Jakob Disease. 2024. Available online: https://www.britannica.com/science/Creutzfeldt-Jakob-disease (accessed on 19 May 2024).
- Karamujic-Comic, H.; Ahmad, S.; Lysen, T.S.; Heshmatollah, A.; Roshchupkin, G.V.; Vernooij, M.W.; Rozemuller, J.M.; Ikram, M.A.; Amin, N.; van Duijn, C.M. Prion protein codon 129 polymorphism in mild cognitive impairment and dementia: The Rotterdam Study. Brain Commun. 2020, 2, fcaa030. [Google Scholar] [CrossRef] [PubMed]
- Zerr, I.; Parchi, P. Sporadic Creutzfeldt-Jakob disease. Handb. Clin. Neurol. 2018, 153, 155–174. [Google Scholar]
- Mead, S.; Whitfield, J.; Poulter, M.; Shah, P.; Uphill, J.; Beck, J.; Campbell, T.; Al-Dujaily, H.; Hummerich, H.; Alpers, M.P.; et al. Genetic susceptibility, evolution and the kuru epidemic. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 3741–3746. [Google Scholar] [CrossRef]
- Mahat, S.; Asuncion, R.M.D. Kuru; StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Kothekar, H.; Chaudhary, K. Kuru Disease: Bridging the Gap Between Prion Biology and Human Health. Cureus 2024, 16, e51708. [Google Scholar] [CrossRef]
- Liberski, P.P.; Gajos, A.; Sikorska, B.; Lindenbaum, S. Kuru, the First Human Prion Disease. Viruses 2019, 11, 232. [Google Scholar] [CrossRef]
- Liberski, P.P. Kuru: A journey back in time from papua new Guinea to the neanderthals’ extinction. Pathogens 2013, 2, 472–505. [Google Scholar] [CrossRef] [PubMed]
- Sikorska, B.; Liberski, P.P. Human prion diseases: From Kuru to variant Creutzfeldt-Jakob disease. In Protein Aggregation and Fibrillogenesis in Cerebral and Systemic Amyloid Disease; Subcellular Biochemistry; Springer: Berlin/Heidelberg, Germany, 2012; Volume 65, pp. 457–496. [Google Scholar]
- Liberski, P.P.; Sikorska, B.; Brown, P. Kuru: The first prion disease. Adv. Exp. Med. Biol. 2012, 724, 143–153. [Google Scholar] [PubMed]
- Alpers, M.P. Review. The epidemiology of kuru: Monitoring the epidemic from its peak to its end. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 3707–3713. [Google Scholar] [CrossRef] [PubMed]
- Asante, E.A.; Smidak, M.; Grimshaw, A.; Houghton, R.; Tomlinson, A.; Jeelani, A.; Jakubcova, T.; Hamdan, S.; Richard-Londt, A.; Linehan, J.M.; et al. A naturally occurring variant of the human prion protein completely prevents prion disease. Nature 2015, 522, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.F.; Joiner, S.; Wadsworth, J.D.; Sidle, K.C.; Bell, J.E.; Budka, H.; Ironside, J.W.; Collinge, J. Molecular classification of sporadic Creutzfeldt-Jakob disease. Brain 2003, 126, 1333–1346. [Google Scholar] [CrossRef] [PubMed]
- Hosszu, L.L.P.; Conners, R.; Sangar, D.; Batchelor, M.; Sawyer, E.B.; Fisher, S.; Cliff, M.J.; Hounslow, A.M.; McAuley, K.; Brady, R.L.; et al. Structural effects of the highly protective V127 polymorphism on human prion protein. Commun. Biol. 2020, 3, 402. [Google Scholar] [CrossRef] [PubMed]
- Mallucci, G.; Dickinson, A.; Linehan, J.; Klöhn, P.C.; Brandner, S.; Collinge, J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 2003, 302, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Biggi, S.; Pancher, M.; Stincardini, C.; Luotti, S.; Massignan, T.; Vedove, A.D.; Astolfi, A.; Gatto, P.; Lolli, G.; Barreca, M.L.; et al. Identification of compounds inhibiting prion replication and toxicity by removing PrP(C) from the cell surface. J. Neurochem. 2020, 152, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Forloni, G.; Roiter, I.; Tagliavini, F. Clinical trials of prion disease therapeutics. Curr. Opin. Pharmacol. 2019, 44, 53–60. [Google Scholar] [CrossRef]
- Harish, S.; Bhuvana, K.; Bengalorkar, G.M.; Kumar, T. Flupirtine: Clinical pharmacology. J. Anaesthesiol. Clin. Pharmacol. 2012, 28, 172–177. [Google Scholar] [CrossRef]
- Otto, M.; Cepek, L.; Ratzka, P.; Doehlinger, S.; Boekhoff, I.; Wiltfang, J.; Irle, E.; Pergande, G.; Ellers-Lenz, B.; Windl, O.; et al. Efficacy of flupirtine on cognitive function in patients with CJD: A double-blind study. Neurology 2004, 62, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Doh-Ura, K.; Iwaki, T.; Caughey, B. Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J. Virol. 2000, 74, 4894–4897. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.J.; Lewis, V.; Brazier, M.; Hill, A.F.; Fletcher, A.; Masters, C.L. Quinacrine does not prolong survival in a murine Creutzfeldt-Jakob disease model. Ann. Neurol. 2002, 52, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Barret, A.; Tagliavini, F.; Forloni, G.; Bate, C.; Salmona, M.; Colombo, L.; Luigi, A.D.; Limido, L.; Suardi, S.; Rossi, G.; et al. Evaluation of quinacrine treatment for prion diseases. J. Virol. 2003, 77, 8462–8469. [Google Scholar] [CrossRef]
- Murakami-Kubo, I.; Doh-Ura, K.; Ishikawa, K.; Kawatake, S.; Sasaki, K.; Kira, J.I.; Ohta, S.; Iwaki, T. Quinoline derivatives are therapeutic candidates for transmissible spongiform encephalopathies. J. Virol. 2004, 78, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Yamada, T.; Kusuhara, T.; Furukawa, H.; Takahashi, M.; Yamauchi, A.; Kataoka, Y. Results of quinacrine administration to patients with Creutzfeldt-Jakob disease. Dement. Geriatr. Cogn. Disord. 2004, 17, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J.; Gorham, M.; Hudson, F.; Kennedy, A.; Keogh, G.; Pal, S.; Rossor, M.; Rudge, P.; Siddique, D.; Spyer, M.; et al. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): A patient-preference trial. Lancet Neurol. 2009, 8, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Geschwind, M.D.; Kuo, A.L.; Wong, K.S.; Haman, A.; Devereux, G.; Raudabaugh, B.J.; Johnson, D.Y.; Torres-Chae, C.C.; Finley, R.; Garcia, P.; et al. Quinacrine treatment trial for sporadic Creutzfeldt-Jakob disease. Neurology 2013, 81, 2015–2023. [Google Scholar] [CrossRef]
- Van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Sims, J.R.; Zimmer, J.A.; Evans, C.D.; Lu, M.; Ardayfio, P.; Sparks, J.; Wessels, A.M.; Shcherbinin, S.; Wang, H.; Monkul Nery, E.S.; et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA 2023, 330, 512–527. [Google Scholar] [CrossRef]
- Mead, S.; Khalili-Shirazi, A.; Potter, C.; Mok, T.; Nihat, A.; Hyare, H.; Canning, S.; Schmidt, C.; Campbell, T.; Darwent, L.; et al. Prion protein monoclonal antibody (PRN100) therapy for Creutzfeldt-Jakob disease: Evaluation of a first-in-human treatment programme. Lancet Neurol. 2022, 21, 342–354. [Google Scholar] [CrossRef]
- Tagliavini, F.; Forloni, G.; Colombo, L.; Rossi, G.; Girola, L.; Canciani, B.; Angeretti, N.; Giampaolo, L.; Peressini, E.; Awan, T.; et al. Tetracycline affects abnormal properties of synthetic PrP peptides and PrP(Sc) in vitro. J. Mol. Biol. 2000, 300, 1309–1322. [Google Scholar] [CrossRef] [PubMed]
- Forloni, G.; Iussich, S.; Awan, T.; Colombo, L.; Angeretti, N.; Girola, L.; Bertani, I.; Poli, G.; Caramelli, M.; Bruzzone, M.G.; et al. Tetracyclines affect prion infectivity. Proc. Natl. Acad. Sci. USA 2002, 99, 10849–10854. [Google Scholar] [CrossRef] [PubMed]
- De Luigi, A.; Colombo, L.; Diomede, L.; Capobianco, R.; Mangieri, M.; Miccolo, C.; Limido, L.; Forloni, G.; Tagliavini, F.; Salmona, M. The efficacy of tetracyclines in peripheral and intracerebral prion infection. PLoS ONE 2008, 3, e1888. [Google Scholar] [CrossRef]
- Forloni, G.; Salmona, M.; Marcon, G.; Tagliavini, F. Tetracyclines and prion infectivity. Infect. Disord. Drug Targets 2009, 9, 23–30. [Google Scholar] [CrossRef]
- Haik, S.; Marcon, G.; Mallet, A.; Tettamanti, M.; Welaratne, A.; Giaccone, G.; Azimi, S.; Pietrini, V.; Fabreguettes, J.R.; Imperiale, D.; et al. Doxycycline in Creutzfeldt-Jakob disease: A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014, 13, 150–158. [Google Scholar] [CrossRef]
- Raymond, G.J.; Zhao, H.T.; Race, B.; Raymond, L.D.; Williams, K.; Swayze, E.E.; Graffam, S.; Le, J.; Caron, T.; Stathopoulos, J.; et al. Antisense oligonucleotides extend survival of prion-infected mice. JCI Insight 2019, 5, e131175. [Google Scholar] [CrossRef]
- Colini Baldeschi, A.; Zattoni, M.; Vanni, S.; Nikolic, L.; Ferracin, C.; La Sala, G.; Summa, M.; Bertorelli, R.; Bertozzi, S.M.; Giachin, G.; et al. Innovative Non-PrP-Targeted Drug Strategy Designed to Enhance Prion Clearance. J. Med. Chem. 2022, 65, 8998–9010. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.R.P.; Muxfeldt, M.; Boechat, F.; Souza, M.; Silva, J.L.; de Moraes, M.C.; Rangel, L.P.; Vieira, T.C.R.G.; Batalha, P.N. Aminoquinolones and Their Benzoquinone Dimer Hybrids as Modulators of Prion Protein Conversion. Molecules 2022, 27, 7935. [Google Scholar] [CrossRef]
- Lehmann, S.; Relano-Gines, A.; Resina, S.; Brillaud, E.; Casanova, D.; Vincent, C.; Hamela, C.; Poupeau, S.; Laffont, M.; Gabelle, A.; et al. Systemic delivery of siRNA down regulates brain prion protein and ameliorates neuropathology in prion disorder. PLoS ONE 2014, 9, e88797. [Google Scholar] [CrossRef]
- McCarthy, J.M.; Franke, M.; Resenberger, U.K.; Waldron, S.; Simpson, J.C.; Tatzelt, J.; Appelhans, D.; Rogers, M.S. Anti-prion drug mPPIg5 inhibits PrP(C) conversion to PrP(Sc). PLoS ONE 2013, 8, e55282. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, A.; Eigenbrod, S.; Al-Khadra, S.; Hofmann, A.; Mitteregger, G.; Moser, M.; Bertsch, U.; Kretzschmar, H. Lentivector-mediated RNAi efficiently suppresses prion protein and prolongs survival of scrapie-infected mice. J. Clin. Investig. 2006, 116, 3204–3210. [Google Scholar] [CrossRef] [PubMed]
- Relano-Gines, A.; Gabelle, A.; Lehmann, S.; Milhavet, O.; Crozet, C. Gene and cell therapy for prion diseases. Infect. Disord. Drug Targets 2009, 9, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Rigter, A.; Langeveld, J.P.; van Zijderveld, F.G.; Bossers, A. Prion protein self-interactions: A gateway to novel therapeutic strategies? Vaccine 2010, 28, 7810–7823. [Google Scholar] [CrossRef]
- Zattoni, M.; Legname, G. Tackling prion diseases: A review of the patent landscape. Expert Opin. Ther. Pat. 2021, 31, 1097–1115. [Google Scholar] [CrossRef] [PubMed]
- Westergard, L.; Christensen, H.M.; Harris, D.A. The cellular prion protein (PrP(C)): Its physiological function and role in disease. Biochim. Biophys. Acta 2007, 1772, 629–644. [Google Scholar] [CrossRef]
- Brown, D.R.; Nicholas, R.S.; Canevari, L. Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J. Neurosci. Res. 2002, 67, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Schulz-Schaeffer, W.J.; Schmidt, B.; Kretzschmar, H.A. Prion protein-deficient cells show altered response to oxidative stress due to decreased SOD-1 activity. Exp. Neurol. 1997, 146, 104–112. [Google Scholar] [CrossRef]
- Bounhar, Y.; Zhang, Y.; Goodyer, C.G.; Leblanc, A. Prion protein protects human neurons against Bax-mediated apoptosis. J. Biol. Chem. 2001, 276, 39145–39149. [Google Scholar] [CrossRef]
- Roucou, X.; Guo, Q.; Zhang, Y.; Goodyer, C.G.; Leblanc, A.C. Cytosolic prion protein is not toxic and protects against Bax-mediated cell death in human primary neurons. J. Biol. Chem. 2003, 278, 40877–40881. [Google Scholar] [CrossRef]
- Cui, T.; Cai, B.; Tian, Y.; Liu, X.; Liang, C.; Gao, Q.; Li, B.; Ding, Y.; Li, R.; Zhou, Q.; et al. Therapeutic In Vivo Gene Editing Achieved by a Hypercompact CRISPR-Cas12f1 System Delivered with All-in-One Adeno-Associated Virus. Adv. Sci. 2024, 11, e2308095. [Google Scholar] [CrossRef]
- Dubey, A.K.; Mostafavi, E. Biomaterials-mediated CRISPR/Cas9 delivery: Recent challenges and opportunities in gene therapy. Front. Chem. 2023, 11, 1259435. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.R.; Chen, E.; Perez, B.S.; Sandoval Espinoza, C.R.; Kang, M.H.; Trinidad, M.; Ngo, W.; Doudna, J.A. In vivo human T cell engineering with enveloped delivery vehicles. Nat. Biotechnol. 2024, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sahel, D.K.; Vora, L.K.; Saraswat, A.; Sharma, S.; Monpara, J.; D’Souza, A.A.; Mishra, D.; Tryphena, K.P.; Kawakita, S.; Khan, S.; et al. CRISPR/Cas9 Genome Editing for Tissue-Specific In Vivo Targeting: Nanomaterials and Translational Perspective. Adv. Sci. 2023, 10, e2305072. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, F.; Begum, A.A.; Dai, C.C.; Toth, I.; Moyle, P.M. Recent advances in the delivery and applications of nonviral CRISPR/Cas9 gene editing. Drug Deliv. Transl. Res. 2023, 13, 1500–1519. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kelly, K.; Lee, J.; Echeverria, D.; Cooper, D.; Panwala, R.; Amrani, N.; Chen, Z.; Gaston, N.; Wagh, A.; et al. Self-delivering, chemically modified CRISPR RNAs for AAV co-delivery and genome editing in vivo. Nucleic Acids Res. 2024, 52, 977–997. [Google Scholar] [CrossRef]
- Burdo, T.H.; Chen, C.; Kaminski, R.; Sariyer, I.K.; Mancuso, P.; Donadoni, M.; Smith, M.D.; Sariyer, R.; Caocci, M.; Liao, S.; et al. Preclinical safety and biodistribution of CRISPR targeting SIV in non-human primates. Gene Ther. 2024, 31, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Deneault, E. Recent Therapeutic Gene Editing Applications to Genetic Disorders. Curr. Issues Mol. Biol. 2024, 46, 4147–4185. [Google Scholar] [CrossRef]
- Gao, M.; He, Y.; Zhu, X.; Peng, W.; Zhou, Y.; Deng, Y.; Liao, G.; Ni, W.; Li, Y.; Gao, J.; et al. One-step in vivo gene knock-out in porcine embryos using recombinant adeno-associated viruses. Front. Cell Dev. Biol. 2024, 12, 1376936. [Google Scholar] [CrossRef]
- Laurent, M.; Geoffroy, M.; Pavani, G.; Guiraud, S. CRISPR-Based Gene Therapies: From Preclinical to Clinical Treatments. Cells 2024, 13, 800. [Google Scholar] [CrossRef]
- Li, T.; Yang, Y.; Qi, H.; Cui, W.; Zhang, L.; Fu, X.; He, X.; Liu, M.; Li, P.F.; Yu, T. CRISPR/Cas9 therapeutics: Progress and prospects. Signal Transduct. Target. Ther. 2023, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Metzger, J.M.; Wang, Y.; Neuman, S.S.; Snow, K.J.; Murray, S.A.; Lutz, C.M.; Bondarenko, V.; Felton, J.; Gimse, K.; Xie, R.; et al. Efficient in vivo neuronal genome editing in the mouse brain using nanocapsules containing CRISPR-Cas9 ribonucleoproteins. Biomaterials 2023, 293, 121959. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Zheng, C.; Xu, W.; Zhang, S.; Liu, S.; Chen, X.; Yao, K. Breaking genetic shackles: The advance of base editing in genetic disorder treatment. Front. Pharmacol. 2024, 15, 1364135. [Google Scholar] [CrossRef] [PubMed]
- Longhurst, H.J.; Lindsay, K.; Petersen, R.S.; Fijen, L.M.; Gurugama, P.; Maag, D.; Butler, J.S.; Shah, M.Y.; Golden, A.; Xu, Y.; et al. CRISPR-Cas9 In Vivo Gene Editing of KLKB1 for Hereditary Angioedema. N. Engl. J. Med. 2024, 390, 432–441. [Google Scholar] [CrossRef]
- Wei, A.; Yin, D.; Zhai, Z.; Ling, S.; Le, H.; Tian, L.; Xu, J.; Paludan, S.R.; Cai, Y.; Hong, J. In vivo CRISPR gene editing in patients with herpetic stromal keratitis. Mol. Ther. 2023, 31, 3163–3175. [Google Scholar] [CrossRef] [PubMed]
- Parums, D.V. Editorial: First Regulatory Approvals for CRISPR-Cas9 Therapeutic Gene Editing for Sickle Cell Disease and Transfusion-Dependent beta-Thalassemia. Med. Sci. Monit. 2024, 30, e944204. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.F.; Chen, G.J.; Luo, Y.L.; Zhang, Y.; Zhao, G.; Lu, Z.D.; Czarna, A.; Gu, Z.; Wang, J. Rational designs of in vivo CRISPR-Cas delivery systems. Adv. Drug Deliv. Rev. 2021, 168, 3–29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Shen, J.; Li, D.; Cheng, Y. Strategies in the delivery of Cas9 ribonucleoprotein for CRISPR/Cas9 genome editing. Theranostics 2021, 11, 614–648. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef]
- Liu, M.S.; Gong, S.; Yu, H.H.; Jung, K.; Johnson, K.A.; Taylor, D.W. Engineered CRISPR/Cas9 enzymes improve discrimination by slowing DNA cleavage to allow release of off-target DNA. Nat. Commun. 2020, 11, 3576. [Google Scholar] [CrossRef]
- Singh, D.; Wang, Y.; Mallon, J.; Yang, O.; Fei, J.; Poddar, A.; Ceylan, D.; Bailey, S.; Ha, T. Mechanisms of improved specificity of engineered Cas9s revealed by single-molecule FRET analysis. Nat. Struct. Mol. Biol. 2018, 25, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Kocak, D.D.; Josephs, E.A.; Bhandarkar, V.; Adkar, S.S.; Kwon, J.B.; Gersbach, C.A. Increasing the specificity of CRISPR systems with engineered RNA secondary structures. Nat. Biotechnol. 2019, 37, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, A.; Fujii, W.; Sugiura, K.; Naito, K. High-fidelity endonuclease variant HypaCas9 facilitates accurate allele-specific gene modification in mouse zygotes. Commun. Biol. 2019, 2, 371. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef]
- Kulcsar, P.I.; Talas, A.; Ligeti, Z.; Krausz, S.L.; Welker, E. SuperFi-Cas9 exhibits remarkable fidelity but severely reduced activity yet works effectively with ABE8e. Nat. Commun. 2022, 13, 6858. [Google Scholar] [CrossRef] [PubMed]
- Collias, D.; Beisel, C.L. CRISPR technologies and the search for the PAM-free nuclease. Nat. Commun. 2021, 12, 555. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014, 513, 569–573. [Google Scholar] [CrossRef]
- Walton, R.T.; Christie, K.A.; Whittaker, M.N.; Kleinstiver, B.P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 2020, 368, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Chu, A.H.Y.; Bao, S.; Hoang, D.A.; Kebede, F.T.; Xiong, W.; Ji, M.; Shi, J.; Zheng, Z. Rationally engineered Staphylococcus aureus Cas9 nucleases with high genome-wide specificity. Proc. Natl. Acad. Sci. USA 2019, 116, 20969–20976. [Google Scholar] [CrossRef]
- Edraki, A.; Mir, A.; Ibraheim, R.; Gainetdinov, I.; Yoon, Y.; Song, C.Q.; Cao, Y.; Gallant, J.; Xue, W.; Rivera- Pérez, J.A.; et al. A Compact, High-Accuracy Cas9 with a Dinucleotide PAM for In Vivo Genome Editing. Mol. Cell 2019, 73, 714–726.e4. [Google Scholar] [CrossRef]
- Ibraheim, R.; Tai, P.W.L.; Mir, A.; Javeed, N.; Wang, J.; Rodríguez, T.C.; Namkung, S.; Nelson, S.; Khokhar, E.S.; Mintzer, E.; et al. Self-inactivating, all-in-one AAV vectors for precision Cas9 genome editing via homology-directed repair in vivo. Nat. Commun. 2021, 12, 6267. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Lu, R.; Xin, C.; Wang, Y.; Ling, X.; Li, D.; Zhang, W.; Liu, M.; Xie, W.; Kong, L.; et al. Cas9 exo-endonuclease eliminates chromosomal translocations during genome editing. Nat. Commun. 2022, 13, 1204. [Google Scholar] [CrossRef]
- Adikusuma, F.; Piltz, S.; Corbett, M.A.; Turvey, M.; McColl, S.R.; Helbig, K.J.; Beard, M.R.; Hughes, J.; Pomerantz, R.T.; Thomas, P.Q. Large deletions induced by Cas9 cleavage. Nature 2018, 560, E8–E9. [Google Scholar] [CrossRef] [PubMed]
- Nahmad, A.D.; Reuveni, E.; Goldschmidt, E.; Tenne, T.; Liberman, M.; Horovitz-Fried, M.; Khosravi, R.; Kobo, H.; Reinstein, E.; Madi, A.; et al. Frequent aneuploidy in primary human T cells after CRISPR-Cas9 cleavage. Nat. Biotechnol. 2022, 40, 1807–1813. [Google Scholar] [CrossRef]
- Tao, J.; Wang, Q.; Mendez-Dorantes, C.; Burns, K.H.; Chiarle, R. Frequency and mechanisms of LINE-1 retrotransposon insertions at CRISPR/Cas9 sites. Nat. Commun. 2022, 13, 3685. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, M.L.; Papathanasiou, S.; Doerfler, P.A.; Blaine, L.J.; Sun, L.; Yao, Y.; Zhang, C.Z.; Weiss, M.J.; Pellman, D. Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat. Genet. 2021, 53, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Zuccaro, M.V.; Xu, J.; Mitchell, C.; Marin, D.; Zimmerman, R.; Rana, B.; Weinstein, E.; King, R.T.; Palmerola, K.L.; Smith, M.E.; et al. Allele-Specific Chromosome Removal after Cas9 Cleavage in Human Embryos. Cell 2020, 183, 1650–1664.e15. [Google Scholar] [CrossRef] [PubMed]
- Duan, D. Systemic delivery of adeno-associated viral vectors. Curr. Opin. Virol. 2016, 21, 16–25. [Google Scholar] [CrossRef]
- Ablain, J.; Zon, L.I. Tissue-specific gene targeting using CRISPR/Cas9. Methods Cell Biol. 2016, 135, 189–202. [Google Scholar]
- Xu, X.; Liu, C.; Wang, Y.; Koivisto, O.; Zhou, J.; Shu, Y.; Zhang, H. Nanotechnology-based delivery of CRISPR/Cas9 for cancer treatment. Adv. Drug Deliv. Rev. 2021, 176, 113891. [Google Scholar] [CrossRef]
- Cai, W.; Luo, T.; Mao, L.; Wang, M. Spatiotemporal Delivery of CRISPR/Cas9 Genome Editing Machinery Using Stimuli-Responsive Vehicles. Angew. Chem. Int. Ed. Engl. 2021, 60, 8596–8606. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, Y.; Liu, X.; Widjaya, A.S.; Jiang, B.; Jiang, Y. Macrophage-biomimetic anti-inflammatory liposomes for homing and treating of aortic dissection. J. Control Release 2021, 337, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Jahromi, L.P.; Shahbazi, M.A.; Maleki, A.; Azadi, A.; Santos, H.A. Chemically Engineered Immune Cell-Derived Microrobots and Biomimetic Nanoparticles: Emerging Biodiagnostic and Therapeutic Tools. Adv. Sci. 2021, 8, 2002499. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Wang, Y.; Wijaya, A.; Liu, B.; Maruf, A.; Wang, J.; Xu, J.; Liao, X.; Wu, W.; Wang, G. ROS-responsive biomimetic nanoparticles for potential application in targeted anti-atherosclerosis. Regen. Biomater. 2021, 8, rbab033. [Google Scholar] [CrossRef]
- Guo, M.; Xiong, Y. Sex-biased genome-editing effects of CRISPR-Cas9 across cancer cells dependent on p53 status. iScience 2023, 26, 107529. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).