
mTOR Dysregulation, Insulin Resistance, and Hypertension

 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. mTOR Pathway Overview
3. Insulin Resistance Overview
4. mTOR Dysregulation and Insulin Resistance
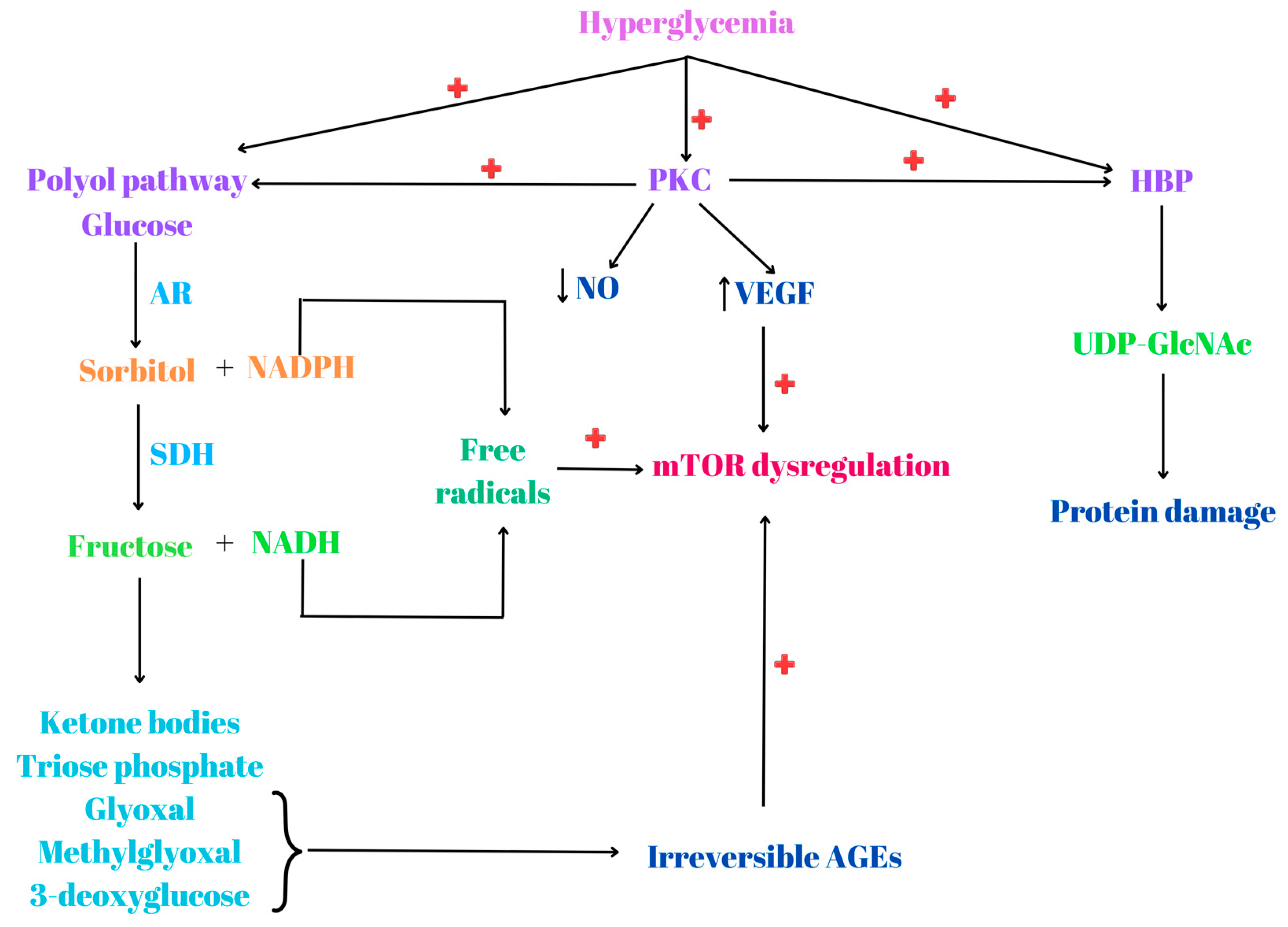
5. Hyperglycemia, mTOR Dysregulation and Stress
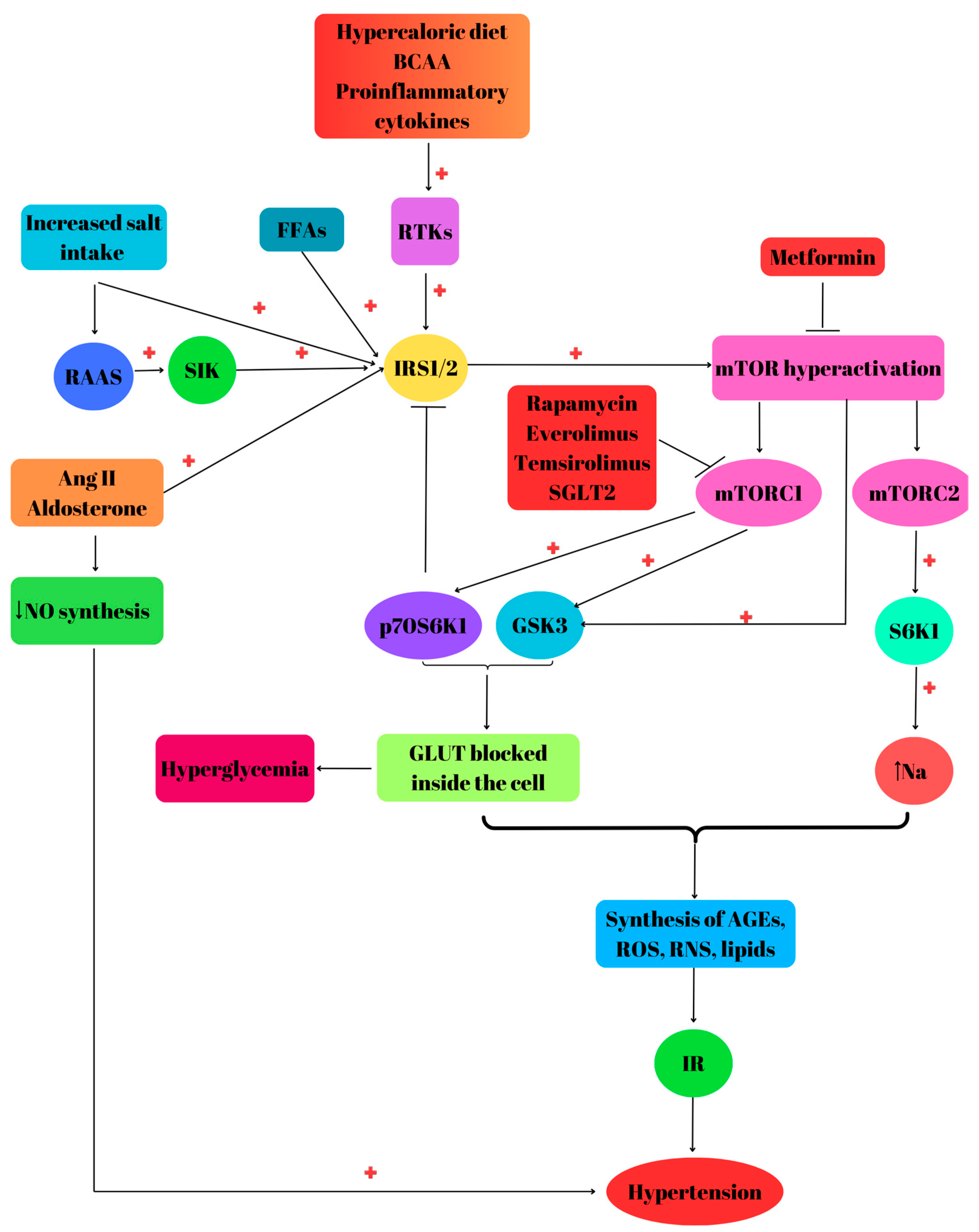
6. mTOR Dysregulation, Insulin Resistance and Hypertension
7. Metformin and mTOR Inhibitors
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hwangbo, Y.; Lee, E.K. Acute hyperglycemia associated with anti-cancer medication. Endocrinol. Metab. 2017, 32, 23–29. [Google Scholar] [CrossRef]
- Pulakat, L.; Aroor, A.R.; Gul, R.; Sowers, J.R. Cardiac insulin resistance and microRNA modulators. J. Diabetes Res. 2012, 2012, 654904. [Google Scholar] [CrossRef] [PubMed]
- da Silva Rosa, S.C.; Nayak, N.; Caymo, A.M.; Gordon, J.W. Mechanisms of muscle insulin resistance and the cross-talk with liver and adipose tissue. Physiol. Rep. 2020, 8, e14607. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, A.; Flores, R.R.; Robbins, P.D.; Niedernhofer, L.J. Role of cellular senescence in type II diabetes. Endocrinology 2021, 162, bqab136. [Google Scholar] [CrossRef]
- Akhaphong, B.; Baumann, D.C.; Beetch, M.; Lockridge, A.D.; Jo, S.; Wong, A.; Zemanovic, T.; Mohan, R.; Fondevilla, D.L.; Sia, M. Placental mTOR complex 1 regulates fetal programming of obesity and insulin resistance in mice. JCI Insight 2021, 6, e149271. [Google Scholar] [CrossRef]
- Thomas, M.S.; Calle, M.; Fernandez, M.L. Healthy plant-based diets improve dyslipidemias, insulin resistance, and inflammation in metabolic syndrome. A narrative review. Adv. Nutr. 2023, 14, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Han, L.; Hu, D. Fasting insulin, insulin resistance and risk of hypertension in the general population: A meta-analysis. Clin. Chim. Acta 2017, 464, 57–63. [Google Scholar] [CrossRef]
- Zheng, Z.-G.; Zhou, Y.-P.; Zhang, X.; Thu, P.M.; Xie, Z.-S.; Lu, C.; Pang, T.; Xue, B.; Xu, D.-Q.; Chen, Y. Anhydroicaritin improves diet-induced obesity and hyperlipidemia and alleviates insulin resistance by suppressing SREBPs activation. Biochem. Pharmacol. 2016, 122, 42–61. [Google Scholar] [CrossRef] [PubMed]
- Natali, A.; Ferrannini, E. Hypertension, insulin resistance, and the metabolic syndrome. Endocrinol. Metab. Clin. 2004, 33, 417–429. [Google Scholar] [CrossRef]
- Roberts, C.K.; Hevener, A.L.; Barnard, R.J. Metabolic syndrome and insulin resistance: Underlying causes and modification by exercise training. Compr. Physiol. 2013, 3, 1. [Google Scholar]
- Lai, Y.; Zhao, A.; Tan, M.; Yang, M.; Lin, Y.; Li, S.; Song, J.; Zheng, H.; Zhu, Z.; Liu, D. DOCK5 regulates energy balance and hepatic insulin sensitivity by targeting mTORC1 signaling. EMBO Rep. 2020, 21, e49473. [Google Scholar] [CrossRef]
- Hall, J.E.; do Carmo, J.M.; da Silva, A.A.; Wang, Z.; Hall, M.E. Obesity-induced hypertension: Interaction of neurohumoral and renal mechanisms. Circ. Res. 2015, 116, 991–1006. [Google Scholar] [CrossRef] [PubMed]
- Arabi, T.; Shafqat, A.; Sabbah, B.N.; Fawzy, N.A.; Shah, H.; Abdulkader, H.; Razak, A.; Sabbah, A.N.; Arabi, Z. Obesity-related kidney disease: Beyond hypertension and insulin-resistance. Front. Endocrinol. 2023, 13, 1095211. [Google Scholar] [CrossRef] [PubMed]
- Sivasubrmanian, S. Fathoming the role of mTOR in diabetes mellitus and its complications. Curr. Mol. Pharmacol. 2023, 16, 520–529. [Google Scholar]
- Ganesan, H.; Balasubramanian, V.; Iyer, M.; Venugopal, A.; Subramaniam, M.D.; Cho, S.-G.; Vellingiri, B. mTOR signalling pathway-A root cause for idiopathic autism? BMB Rep. 2019, 52, 424. [Google Scholar] [CrossRef] [PubMed]
- Vergès, B. mTOR and cardiovascular diseases: Diabetes mellitus. Transplantation 2018, 102, S47–S49. [Google Scholar] [CrossRef] [PubMed]
- Noori, T.; Sureda, A.; Shirooie, S. Role of natural mTOR inhibitors in treatment of diabetes mellitus. Fundam. Clin. Pharmacol. 2023, 37, 461–479. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Burillo, J.; Marqués, P.; Jiménez, B.; González-Blanco, C.; Benito, M.; Guillén, C. Insulin resistance and diabetes mellitus in Alzheimer’s disease. Cells 2021, 10, 1236. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Du, R.; Liu, W.; Huang, G.; Dong, Z.; Li, X. PI3K/Akt/mTOR signaling pathway: Role in esophageal squamous cell carcinoma, regulatory mechanisms and opportunities for targeted therapy. Front. Oncol. 2022, 12, 852383. [Google Scholar]
- Gao, Y.; Tian, T. mTOR signaling pathway and gut microbiota in various disorders: Mechanisms and potential drugs in pharmacotherapy. Int. J. Mol. Sci. 2023, 24, 11811. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef]
- Glaviano, A.; Foo, A.S.; Lam, H.Y.; Yap, K.C.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef]
- Mallela, K.; Kumar, A. Role of TSC1 in physiology and diseases. Mol. Cell. Biochem. 2021, 476, 2269–2282. [Google Scholar] [CrossRef] [PubMed]
- Larsen, L.J.; Møller, L.B. Crosstalk of hedgehog and mTORC1 pathways. Cells 2020, 9, 2316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bajraszewski, N.; Wu, E.; Wang, H.; Moseman, A.P.; Dabora, S.L.; Griffin, J.D.; Kwiatkowski, D.J. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. J. Clin. Investig. 2007, 117, 730–738. [Google Scholar] [CrossRef]
- Ching, C.B.; Hansel, D.E. Expanding therapeutic targets in bladder cancer: The PI3K/Akt/mTOR pathway. Lab. Investig. 2010, 90, 1406–1414. [Google Scholar] [CrossRef]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR signaling in cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- Jurca, C.M.; Kozma, K.; Petchesi, C.D.; Zaha, D.C.; Magyar, I.; Munteanu, M.; Faur, L.; Jurca, A.; Bembea, D.; Severin, E. Tuberous sclerosis, type II diabetes mellitus and the PI3K/AKT/mTOR signaling pathways—Case report and literature review. Genes 2023, 14, 433. [Google Scholar] [CrossRef] [PubMed]
- Lotfimehr, H.; Mardi, N.; Narimani, S.; Nasrabadi, H.T.; Karimipour, M.; Sokullu, E.; Rahbarghazi, R. mTOR signalling pathway in stem cell bioactivities and angiogenesis potential. Cell Prolif. 2023, 56, e13499. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.-Y.; Lv, M.; Luo, B.-H.; Zhao, S.-Z.; Mo, Z.-C.; Xie, Y.-J. The role of the PI3K/AKT/mTOR signalling pathway in male reproduction. Curr. Mol. Med. 2021, 21, 539–548. [Google Scholar] [PubMed]
- Tewari, D.; Patni, P.; Bishayee, A.; Sah, A.N.; Bishayee, A. Natural products targeting the PI3K-Akt-mTOR signaling pathway in cancer: A novel therapeutic strategy. Semin. Cancer Biol. 2022, 80, 1–17. [Google Scholar] [CrossRef]
- Abdo Qaid, E.Y.; Zulkipli, N.N.; Zakaria, R.; Ahmad, A.H.; Othman, Z.; Muthuraju, S.; Sasongko, T.H. The role of mTOR signalling pathway in hypoxia-induced cognitive impairment. Int. J. Neurosci. 2021, 131, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Chao, B.; Xu, J.; Liu, Z.; Tao, Y.; He, J.; Wang, J.; Yang, H.; Luo, X.; Qi, H. CPT1A modulates PI3K/Akt/mTOR pathway to promote preeclampsia. Placenta 2023, 133, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Sanvee, G.M.; Panajatovic, M.V.; Bouitbir, J.; Krähenbühl, S. Mechanisms of insulin resistance by simvastatin in C2C12 myotubes and in mouse skeletal muscle. Biochem. Pharmacol. 2019, 164, 23–33. [Google Scholar] [CrossRef]
- Nagini, S.; Sophia, J.; Mishra, R. Glycogen synthase kinases: Moonlighting proteins with theranostic potential in cancer. Semin. Cancer Biol. 2019, 56, 25–36. [Google Scholar] [CrossRef]
- Dai, D.-F.; Kang, P.; Bai, H. The mTOR signaling pathway in cardiac aging. J. Cardiovasc. Aging 2023, 3, 24. [Google Scholar] [CrossRef]
- Bodur, C.; Kazyken, D.; Huang, K.; Tooley, A.S.; Cho, K.W.; Barnes, T.M.; Lumeng, C.N.; Myers, M.G., Jr.; Fingar, D.C. TBK1-mTOR signaling attenuates obesity-linked hyperglycemia and insulin resistance. Diabetes 2022, 71, 2297–2312. [Google Scholar] [CrossRef] [PubMed]
- Kido, K.; Sase, K.; Yokokawa, T.; Fujita, S. Enhanced skeletal muscle insulin sensitivity after acute resistance-type exercise is upregulated by rapamycin-sensitive mTOR complex 1 inhibition. Sci. Rep. 2020, 10, 8509. [Google Scholar] [CrossRef] [PubMed]
- Mir, S.A.; Dar, A.; Alshehri, S.A.; Wahab, S.; Hamid, L.; Almoyad, M.A.A.; Ali, T.; Bader, G.N. Exploring the mTOR signalling pathway and its inhibitory scope in cancer. Pharmaceuticals 2023, 16, 1004. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, Y.; Wang, D.; Xu, Y.; Dong, R.; Yang, Y.; Lv, Q.; Chen, X.; Zhang, Z. The upstream pathway of mTOR-mediated autophagy in liver diseases. Cells 2019, 8, 1597. [Google Scholar] [CrossRef] [PubMed]
- Melanis, K.; Stefanou, M.-I.; Themistoklis, K.M.; Papasilekas, T. mTOR pathway–a potential therapeutic target in stroke. Ther. Adv. Neurol. Disord. 2023, 16, 17562864231187770. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, P.; Kandror, K.V. The role of mTOR in lipid homeostasis and diabetes progression. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Chen, Q. mTOR pathway: A potential therapeutic target for spinal cord injury. Biomed. Pharmacother. 2022, 145, 112430. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, Z.; Wang, D.; Jiang, Y.; Liu, Y. Targeting mTOR signaling in type 2 diabetes mellitus and diabetes complications. Curr. Drug Targets 2022, 23, 692–710. [Google Scholar] [CrossRef] [PubMed]
- Fattahi, S.; Khalifehzadeh-Esfahani, Z.; Mohammad-Rezaei, M.; Mafi, S.; Jafarinia, M. PI3K/Akt/mTOR pathway: A potential target for anti-SARS-CoV-2 therapy. Immunol. Res. 2022, 70, 269–275. [Google Scholar] [CrossRef]
- Ali, M.; Bukhari, S.A.; Lee, H.-W. Upstream signalling of mTORC1 and its hyperactivation in type 2 diabetes (T2D). BMB Rep. 2017, 50, 601. [Google Scholar] [CrossRef]
- Rubie, C.; Zimmer, J.; Lammert, F.; Gross, J.C.; Weber, S.N.; Kruse, B.; Halajda, B.; Wagner, M.; Wagenpfeil, S.; Glanemann, M. MicroRNA-496 and mechanistic target of rapamycin expression are associated with type 2 diabetes mellitus and obesity in elderly people. Ann. Nutr. Metab. 2019, 74, 279–286. [Google Scholar] [CrossRef]
- Lee, S.-H.; Park, S.-Y.; Choi, C.S. Insulin resistance: From mechanisms to therapeutic strategies. Diabetes Metab. J. 2022, 46, 15–37. [Google Scholar] [CrossRef] [PubMed]
- Tsatsoulis, A.; Mantzaris, M.D.; Bellou, S.; Andrikoula, M. Insulin resistance: An adaptive mechanism becomes maladaptive in the current environment—An evolutionary perspective. Metabolism 2013, 62, 622–633. [Google Scholar] [CrossRef]
- Onyango, A.N. Cellular stresses and stress responses in the pathogenesis of insulin resistance. Oxidative Med. Cell. Longev. 2018, 2018, 4321714. [Google Scholar] [CrossRef]
- Dimitriadis, G.; Mitrou, P.; Lambadiari, V.; Maratou, E.; Raptis, S.A. Insulin effects in muscle and adipose tissue. Diabetes Res. Clin. Pract. 2011, 93, S52–S59. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Xu, G.; Tsai, S.-Y.A.; Freed, W.J.; Lee, C.-T. Transcriptional profiles of type 2 diabetes in human skeletal muscle reveal insulin resistance, metabolic defects, apoptosis, and molecular signatures of immune activation in response to infections. Biochem. Biophys. Res. Commun. 2017, 482, 282–288. [Google Scholar] [CrossRef]
- Myers, J.; Kokkinos, P.; Nyelin, E. Physical activity, cardiorespiratory fitness, and the metabolic syndrome. Nutrients 2019, 11, 1652. [Google Scholar] [CrossRef] [PubMed]
- Mirabelli, M.; Chiefari, E.; Arcidiacono, B.; Corigliano, D.M.; Brunetti, F.S.; Maggisano, V.; Russo, D.; Foti, D.P.; Brunetti, A. Mediterranean diet nutrients to turn the tide against insulin resistance and related diseases. Nutrients 2020, 12, 1066. [Google Scholar] [CrossRef]
- Tong, C.; Wu, Y.; Zhang, L.; Yu, Y. Insulin resistance, autophagy and apoptosis in patients with polycystic ovary syndrome: Association with PI3K signaling pathway. Front. Endocrinol. 2022, 13, 1091147. [Google Scholar] [CrossRef] [PubMed]
- De Cosmo, S.; Menzaghi, C.; Prudente, S.; Trischitta, V. Role of insulin resistance in kidney dysfunction: Insights into the mechanism and epidemiological evidence. Nephrol. Dial. Transplant. 2013, 28, 29–36. [Google Scholar] [CrossRef]
- Tahapary, D.L.; Pratisthita, L.B.; Fitri, N.A.; Marcella, C.; Wafa, S.; Kurniawan, F.; Rizka, A.; Tarigan, T.J.E.; Harbuwono, D.S.; Purnamasari, D. Challenges in the diagnosis of insulin resistance: Focusing on the role of HOMA-IR and Tryglyceride/glucose index. Diabetes Metab. Syndr. Clin. Res. Rev. 2022, 16, 102581. [Google Scholar] [CrossRef] [PubMed]
- Kosmas, C.E.; Bousvarou, M.D.; Kostara, C.E.; Papakonstantinou, E.J.; Salamou, E.; Guzman, E. Insulin resistance and cardiovascular disease. J. Int. Med. Res. 2023, 51, 03000605231164548. [Google Scholar] [CrossRef] [PubMed]
- Dayi, T.; Ozgoren, M. Effects of the Mediterranean diet on the components of metabolic syndrome. J. Prev. Med. Hyg. 2022, 63, E56. [Google Scholar] [PubMed]
- Perseghin, G.; Petersen, K.; Shulman, G. Cellular mechanism of insulin resistance: Potential links with inflammation. Int. J. Obes. 2003, 27, S6–S11. [Google Scholar] [CrossRef] [PubMed]
- Manaserh, I.H.; Bledzka, K.M.; Junker, A.; Grondolsky, J.; Schumacher, S.M. A cardiac amino-terminal GRK2 peptide inhibits maladaptive adipocyte hypertrophy and insulin resistance during diet-induced obesity. Basic Transl. Sci. 2022, 7, 563–579. [Google Scholar] [CrossRef] [PubMed]
- da Silva, A.A.; do Carmo, J.M.; Li, X.; Wang, Z.; Mouton, A.J.; Hall, J.E. Role of hyperinsulinemia and insulin resistance in hypertension: Metabolic syndrome revisited. Can. J. Cardiol. 2020, 36, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Bar-Tana, J. Type 2 diabetes–unmet need, unresolved pathogenesis, mTORC1-centric paradigm. Rev. Endocr. Metab. Disord. 2020, 21, 613–629. [Google Scholar] [CrossRef] [PubMed]
- Lamounier-Zepter, V.; Ehrhart-Bornstein, M.; Bornstein, S.R. Insulin resistance in hypertension and cardiovascular disease. Best Pract. Res. Clin. Endocrinol. Metab. 2006, 20, 355–367. [Google Scholar] [CrossRef]
- Perrotta, F.; Nigro, E.; Mollica, M.; Costigliola, A.; D’Agnano, V.; Daniele, A.; Bianco, A.; Guerra, G. Pulmonary hypertension and obesity: Focus on adiponectin. Int. J. Mol. Sci. 2019, 20, 912. [Google Scholar] [CrossRef]
- Lee, D.-F.; Kuo, H.-P.; Chen, C.-T.; Wei, Y.; Chou, C.-K.; Hung, J.-Y.; Yen, C.-J.; Hung, M.-C. IKKβ suppression of TSC1 function links the mTOR pathway with insulin resistance. Int. J. Mol. Med. 2008, 22, 633–638. [Google Scholar]
- Ye, J. Mechanism of insulin resistance in obesity: A role of ATP. Front. Med. 2021, 15, 372–382. [Google Scholar] [CrossRef] [PubMed]
- van Niekerk, G.; Christowitz, C.; Engelbrecht, A.-M. Insulin-mediated immune dysfunction in the development of preeclampsia. J. Mol. Med. 2021, 99, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Czech, M.P. Mechanisms of insulin resistance related to white, beige, and brown adipocytes. Mol. Metab. 2020, 34, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Feng, Q. Pharmacological effects and molecular protective mechanisms of Astragalus polysaccharides on nonalcoholic fatty liver disease. Front. Pharmacol. 2022, 13, 854674. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Liu, C.; Wang, X.; Zhao, M.; Zhang, Z.; Zhang, X.; Wang, C.; Song, G. Resveratrol improves palmitic acid-induced insulin resistance via the DDIT4/mTOR pathway in C2C12 cells. Mol. Med. Rep. 2023, 28, 181. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yang, Y.; Liu, S.; Niu, Y.; Shao, H.; Fu, L. Aerobic exercise ameliorates insulin resistance in C57BL/6 J mice via activating Sestrin3. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2023, 1869, 166568. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Asai, A.; Fukunishi, S.; Nishiguchi, S.; Higuchi, K. Metabolic Syndrome and Sarcopenia. Nutrients 2021, 13, 3519. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Kim, Y.-B. Molecular mechanism of insulin resistance in obesity and type 2 diabetes. Korean J. Intern. Med. 2010, 25, 119. [Google Scholar] [CrossRef]
- Szymczak-Pajor, I.; Drzewoski, J.; Śliwińska, A. The molecular mechanisms by which vitamin D prevents insulin resistance and associated disorders. Int. J. Mol. Sci. 2020, 21, 6644. [Google Scholar] [CrossRef]
- Søndergaard, E.; Jensen, M.D. Quantification of adipose tissue insulin sensitivity. J. Investig. Med. 2016, 64, 989–991. [Google Scholar] [CrossRef]
- Lebovitz, H. Insulin resistance: Definition and consequences. Exp. Clin. Endocrinol. Diabetes 2001, 109, S135–S148. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.-H.; Chen, B.; Zhang, J.-P. Intracellular insulin and impaired autophagy in a zebrafish model and a cell model of type 2 diabetes. Int. J. Biol. Sci. 2017, 13, 985. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Christensen, S.M.; Duong, N.; Tran, Q.-A.; Xiong, H.M.; Huang, J.; James, S.; Vallabh, D.; Talbott, G.; Rose, M. Sirt3 pharmacologically promotes insulin sensitivity through PI3/AKT/mTOR and their downstream pathway in adipocytes. Int. J. Mol. Sci. 2022, 23, 3740. [Google Scholar] [CrossRef] [PubMed]
- Guo, S. Insulin signaling, resistance, and the metabolic syndrome: Insights from mouse models to disease mechanisms. J. Endocrinol. 2014, 220, T1. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, C.L.; Uranga, S.; Fluckey, J.D. Culprits or consequences: Understanding the metabolic dysregulation of muscle in diabetes. World J. Biol. Chem. 2021, 12, 70. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Wang, P. Role of insulin receptor substance-1 modulating PI3K/Akt insulin signaling pathway in Alzheimer’s disease. 3 Biotech 2021, 11, 179. [Google Scholar] [CrossRef] [PubMed]
- Copps, K.; White, M. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [PubMed]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef]
- Bloomgarden, Z. Diabetes and branched-chain amino acids: What is the link? J. Diabetes 2018, 10, 350–352. [Google Scholar] [CrossRef]
- Pomytkin, I.; Krasil’nikova, I.; Bakaeva, Z.; Surin, A.; Pinelis, V. Excitotoxic glutamate causes neuronal insulin resistance by inhibiting insulin receptor/Akt/mTOR pathway. Mol. Brain 2019, 12, 112. [Google Scholar] [CrossRef]
- Morita, M.; Gravel, S.-P.; Chenard, V.; Sikström, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef]
- Ong, P.S.; Wang, L.Z.; Dai, X.; Tseng, S.H.; Loo, S.J.; Sethi, G. Judicious toggling of mTOR activity to combat insulin resistance and cancer: Current evidence and perspectives. Front. Pharmacol. 2016, 7, 395. [Google Scholar] [CrossRef] [PubMed]
- Aziz, A.U.R.; Farid, S.; Qin, K.; Wang, H.; Liu, B. Regulation of insulin resistance and glucose metabolism by interaction of PIM kinases and insulin receptor substrates. Arch. Physiol. Biochem. 2020, 126, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Granata, S.; Mercuri, S.; Troise, D.; Gesualdo, L.; Stallone, G.; Zaza, G. mTOR-inhibitors and post-transplant diabetes mellitus: A link still debated in kidney transplantation. Front. Med. 2023, 10, 1168967. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Fang, X.; Green, C.D.; Das, A. mTORC1 and SGLT2 Inhibitors—A Therapeutic Perspective for Diabetic Cardiomyopathy. Int. J. Mol. Sci. 2023, 24, 15078. [Google Scholar] [CrossRef] [PubMed]
- Den Hartogh, D.J.; Vlavcheski, F.; Giacca, A.; Tsiani, E. Attenuation of free fatty acid (FFA)-induced skeletal muscle cell insulin resistance by resveratrol is linked to activation of AMPK and inhibition of mTOR and p70 S6K. Int. J. Mol. Sci. 2020, 21, 4900. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.-H.; Kim, K.-H.; Park, S.-Y.; Kim, Y.-W.; Kim, J.-Y. PPARδ attenuates alcohol-mediated insulin resistance by enhancing fatty acid-induced mitochondrial uncoupling and antioxidant defense in skeletal muscle. Front. Physiol. 2020, 11, 749. [Google Scholar] [CrossRef]
- Lee, H.-K.; Kwon, B.; Lemere, C.A.; De La Monte, S.; Itamura, K.; Ha, A.Y.; Querfurth, H.W. mTORC2 (Rictor) in Alzheimer’s disease and reversal of amyloid-β expression-induced insulin resistance and toxicity in rat primary cortical neurons. J. Alzheimer’s Dis. 2017, 56, 1015–1036. [Google Scholar] [CrossRef] [PubMed]
- den Hartigh, L.J.; Goodspeed, L.; Wang, S.A.; Kenerson, H.L.; Omer, M.; O’Brien, K.D.; Ladiges, W.; Yeung, R.; Subramanian, S. Chronic oral rapamycin decreases adiposity, hepatic triglycerides and insulin resistance in male mice fed a diet high in sucrose and saturated fat. Exp. Physiol. 2018, 103, 1469–1480. [Google Scholar] [CrossRef]
- Sharma, A.; Chetty, V. Obesity, hypertension and insulin resistance. Acta Diabetol. 2005, 42, s3–s8. [Google Scholar] [CrossRef]
- Boyer, F.; Vidot, J.B.; Dubourg, A.G.; Rondeau, P.; Essop, M.F.; Bourdon, E. Oxidative stress and adipocyte biology: Focus on the role of AGEs. Oxidative Med. Cell. Longev. 2015, 2015, 534873. [Google Scholar] [CrossRef] [PubMed]
- Kang, Q.; Yang, C. Oxidative stress and diabetic retinopathy: Molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol. 2020, 37, 101799. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Jacobs, K.; Haucke, E.; Santos, A.N.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Baver, S.B.; Hope, K.; Guyot, S.; Bjørbaek, C.; Kaczorowski, C.; O’Connell, K.M. Leptin modulates the intrinsic excitability of AgRP/NPY neurons in the arcuate nucleus of the hypothalamus. J. Neurosci. 2014, 34, 5486–5496. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.-C.; Wu, S.-C.; Xu, K.-D.; Liao, B.-C.; Wu, J.-F.; Cheng, A.-S. Scopoletin protects against methylglyoxal-induced hyperglycemia and insulin resistance mediated by suppression of advanced glycation endproducts (AGEs) generation and anti-glycation. Molecules 2015, 20, 2786–2801. [Google Scholar] [CrossRef] [PubMed]
- González, P.; Lozano, P.; Ros, G.; Solano, F. Hyperglycemia and oxidative stress: An integral, updated and critical overview of their metabolic interconnections. Int. J. Mol. Sci. 2023, 24, 9352. [Google Scholar] [CrossRef] [PubMed]
- Stefano, G.B.; Challenger, S.; Kream, R.M. Hyperglycemia-associated alterations in cellular signaling and dysregulated mitochondrial bioenergetics in human metabolic disorders. Eur. J. Nutr. 2016, 55, 2339–2345. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Bhadada, S.K. AGEs accumulation with vascular complications, glycemic control and metabolic syndrome: A narrative review. Bone 2023, 176, 116884. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Eshwaran, R.; Beck, S.C.; Hammes, H.-P.; Wieland, T.; Feng, Y. Contribution of the hexosamine biosynthetic pathway in the hyperglycemia-dependent and-independent breakdown of the retinal neurovascular unit. Mol. Metab. 2023, 73, 101736. [Google Scholar] [CrossRef]
- Chatterjee, A.; Eshwaran, R.; Poschet, G.; Lomada, S.; Halawa, M.; Wilhelm, K.; Schmidt, M.; Hammes, H.-P.; Wieland, T.; Feng, Y. Involvement of NDPK-B in glucose metabolism-mediated endothelial damage via activation of the hexosamine biosynthesis pathway and suppression of O-GlcNAcase activity. Cells 2020, 9, 2324. [Google Scholar] [CrossRef]
- Sage, A.T.; Walter, L.A.; Shi, Y.; Khan, M.I.; Kaneto, H.; Capretta, A.; Werstuck, G.H. Hexosamine biosynthesis pathway flux promotes endoplasmic reticulum stress, lipid accumulation, and inflammatory gene expression in hepatic cells. Am. J. Physiol.-Endocrinol. Metab. 2010, 298, E499–E511. [Google Scholar] [CrossRef]
- Vasconcelos-dos-Santos, A.; de Queiroz, R.M.; da Costa Rodrigues, B.; Todeschini, A.R.; Dias, W.B. Hyperglycemia and aberrant O-GlcNAc ylation: Contributions to tumor progression. J. Bioenerg. Biomembr. 2018, 50, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Wronka, M.; Krzemińska, J.; Młynarska, E.; Rysz, J.; Franczyk, B. The influence of lifestyle and treatment on oxidative stress and inflammation in diabetes. Int. J. Mol. Sci. 2022, 23, 15743. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Xu, B.-T.; Wan, S.-R.; Ma, X.-M.; Long, Y.; Xu, Y.; Jiang, Z.-Z. The role of oxidative stress in diabetes mellitus-induced vascular endothelial dysfunction. Cardiovasc. Diabetol. 2023, 22, 237. [Google Scholar] [CrossRef] [PubMed]
- Luc, K.; Schramm-Luc, A.; Guzik, T.; Mikolajczyk, T. Oxidative stress and inflammatory markers in prediabetes and diabetes. J. Physiol. Pharmacol. 2019, 70, 809–824. [Google Scholar]
- Lima, J.E.; Moreira, N.C.; Sakamoto-Hojo, E.T. Mechanisms underlying the pathophysiology of type 2 diabetes: From risk factors to oxidative stress, metabolic dysfunction, and hyperglycemia. Mutat. Res./Genet. Toxicol. Environ. Mutagen. 2022, 874, 503437. [Google Scholar] [CrossRef]
- Jubaidi, F.F.; Zainalabidin, S.; Taib, I.S.; Abdul Hamid, Z.; Mohamad Anuar, N.N.; Jalil, J.; Mohd Nor, N.A.; Budin, S.B. The role of PKC-MAPK signalling pathways in the development of hyperglycemia-induced cardiovascular complications. Int. J. Mol. Sci. 2022, 23, 8582. [Google Scholar] [CrossRef] [PubMed]
- Adua, E. Decoding the mechanism of hypertension through multiomics profiling. J. Hum. Hypertens. 2023, 37, 253–264. [Google Scholar] [CrossRef]
- Te Riet, L.; van Esch, J.H.; Roks, A.J.; van den Meiracker, A.H.; Danser, A.J. Hypertension: Renin–angiotensin–aldosterone system alterations. Circ. Res. 2015, 116, 960–975. [Google Scholar] [CrossRef]
- Rossier, B.C.; Bochud, M.; Devuyst, O. The hypertension pandemic: An evolutionary perspective. Physiology 2017, 32, 112–125. [Google Scholar] [CrossRef]
- Deedwania, P. Hypertension, dyslipidemia, and insulin resistance in patients with diabetes mellitus or the cardiometabolic syndrome: Benefits of vasodilating β-blockers. J. Clin. Hypertens. 2011, 13, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, W.A.; Wyne, K. Renin-angiotensin-aldosterone system in diabetes and hypertension. J. Clin. Hypertens. 2011, 13, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Sorn, S.R.; Lee, Y.; Kang, I. Salt induces adipogenesis/lipogenesis and inflammatory adipocytokines secretion in adipocytes. Int. J. Mol. Sci. 2019, 20, 160. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Sowers, J.R. Hypertension in diabetes: An update of basic mechanisms and clinical disease. Hypertension 2021, 78, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.A.; Yang, Y.; Zhang, L.; Sun, Z.; Jia, G.; Parrish, A.R.; Sowers, J.R. Insulin resistance, cardiovascular stiffening and cardiovascular disease. Metabolism 2021, 119, 154766. [Google Scholar] [CrossRef] [PubMed]
- Tagi, V.M.; Mainieri, F.; Chiarelli, F. Hypertension in patients with insulin resistance: Etiopathogenesis and management in children. Int. J. Mol. Sci. 2022, 23, 5814. [Google Scholar] [CrossRef] [PubMed]
- Pulakat, L.; DeMarco, V.G.; Whaley-Connell, A.; Sowers, J.R. The impact of overnutrition on insulin metabolic signaling in the heart and the kidney. Cardiorenal Med. 2011, 1, 102–112. [Google Scholar] [CrossRef]
- Hwang, H.J.; Kim, N.; Herman, A.B.; Gorospe, M.; Lee, J.-S. Factors and pathways modulating endothelial cell senescence in vascular aging. Int. J. Mol. Sci. 2022, 23, 10135. [Google Scholar] [CrossRef]
- Gleason, C.E.; Frindt, G.; Cheng, C.-J.; Ng, M.; Kidwai, A.; Rashmi, P.; Lang, F.; Baum, M.; Palmer, L.G.; Pearce, D. mTORC2 regulates renal tubule sodium uptake by promoting ENaC activity. J. Clin. Investig. 2015, 125, 117–128. [Google Scholar] [CrossRef]
- Mansley, M.K.; Wilson, S.M. Dysregulation of epithelial Na+ absorption induced by inhibition of the kinases TORC1 and TORC2. Br. J. Pharmacol. 2010, 161, 1778–1792. [Google Scholar] [CrossRef]
- Kumar, V.; Evans, L.C.; Kurth, T.; Yang, C.; Wollner, C.; Nasci, V.; Zheleznova, N.N.; Bukowy, J.; Dayton, A.; Cowley, A.W., Jr. Therapeutic suppression of mTOR (mammalian target of rapamycin) signaling prevents and reverses salt-induced hypertension and kidney injury in Dahl salt-sensitive rats. Hypertension 2019, 73, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Kundu, B.K.; Zhong, M.; Sen, S.; Davogustto, G.; Keller, S.R.; Taegtmeyer, H. Remodeling of glucose metabolism precedes pressure overload-induced left ventricular hypertrophy: Review of a hypothesis. Cardiology 2015, 130, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Birajdar, S.V.; Mazahir, F.; Alam, M.I.; Kumar, A.; Yadav, A.K. Repurposing and clinical attributes of antidiabetic drugs for the treatment of neurodegenerative disorders. Eur. J. Pharmacol. 2023, 961, 176117. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, H.; Yang, Y.; Zhang, S.; Wang, J.; Zhang, D.; Yu, H. Metformin attenuates UVA-induced skin photoaging by suppressing mitophagy and the PI3K/AKT/mTOR pathway. Int. J. Mol. Sci. 2022, 23, 6960. [Google Scholar] [CrossRef] [PubMed]
- Ampuero, J.; Romero-Gomez, M. Prevention of hepatocellular carcinoma by correction of metabolic abnormalities: Role of statins and metformin. World J. Hepatol. 2015, 7, 1105. [Google Scholar] [CrossRef] [PubMed]
- Citi, V.; Barresi, E.; Piragine, E.; Spezzini, J.; Testai, L.; Da Settimo, F.; Martelli, A.; Taliani, S.; Calderone, V. Anti-Proliferative Properties of the Novel Hybrid Drug Met-ITC, Composed of the Native Drug Metformin with the Addition of an Isothiocyanate H2S Donor Moiety, in Different Cancer Cell Lines. Int. J. Mol. Sci. 2023, 24, 16131. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Esteva, F.; Ensor, J.; Hortobagyi, G.; Lee, M.-H.; Yeung, S.-C. Metformin and thiazolidinediones are associated with improved breast cancer-specific survival of diabetic women with HER2+ breast cancer. Ann. Oncol. 2012, 23, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Pusceddu, S.; Buzzoni, R.; Vernieri, C.; Concas, L.; Marceglia, S.; Giacomelli, L.; Milione, M.; Leuzzi, L.; Femia, D.; Formisano, B. Metformin with everolimus and octreotide in pancreatic neuroendocrine tumor patients with diabetes. Future Oncol. 2016, 12, 1251–1260. [Google Scholar] [CrossRef]
- Shen, Z.; Xue, D.; Wang, K.; Zhang, F.; Shi, J.; Jia, B.; Yang, D.; Zhang, Q.; Zhang, S.; Jiang, H. Metformin exerts an antitumor effect by inhibiting bladder cancer cell migration and growth, and promoting apoptosis through the PI3K/AKT/mTOR pathway. BMC Urol. 2022, 22, 79. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Pauli, C.; Du, X.; Wang, D.G.; Li, X.; Wu, D.; Amadiume, S.C.; Goncalves, M.D.; Hodakoski, C.; Lundquist, M.R. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018, 560, 499–503. [Google Scholar] [CrossRef]
- Cheung, Y.-M.M.; McDonnell, M.; Hamnvik, O.-P.R. A targeted approach to phosphoinositide-3-kinase/Akt/mammalian target of rapamycin-induced hyperglycemia. Curr. Probl. Cancer 2022, 46, 100776. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.D.; Baker, B.C.; Scott, E.M.; Forbes, K. Interaction between metformin, folate and vitamin B12 and the potential impact on fetal growth and long-term metabolic health in diabetic pregnancies. Int. J. Mol. Sci. 2021, 22, 5759. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Yu, H.; Jin, Y.; Mo, J.; Sui, J.; Qian, X.; Chen, T. Metformin facilitates osteoblastic differentiation and M2 macrophage polarization by PI3K/AKT/mTOR pathway in human umbilical cord mesenchymal stem cells. Stem Cells Int. 2022, 2022, 9498876. [Google Scholar] [CrossRef]
- Singh, S.K.; Apata, T.; Singh, S.; McFadden, M.; Singh, R. Clinical implication of metformin in relation to diabetes mellitus and ovarian cancer. Biomedicines 2021, 9, 1020. [Google Scholar] [CrossRef] [PubMed]
- Triggle, C.R.; Mohammed, I.; Bshesh, K.; Marei, I.; Ye, K.; Ding, H.; MacDonald, R.; Hollenberg, M.D.; Hill, M.A. Metformin: Is it a drug for all reasons and diseases? Metabolism 2022, 133, 155223. [Google Scholar] [CrossRef] [PubMed]
- Ala, M.; Ala, M. Metformin for cardiovascular protection, inflammatory bowel disease, osteoporosis, periodontitis, polycystic ovarian syndrome, neurodegeneration, cancer, inflammation and senescence: What is next? ACS Pharmacol. Transl. Sci. 2021, 4, 1747–1770. [Google Scholar] [CrossRef] [PubMed]
- Pusceddu, S.; Vernieri, C.; Di Maio, M.; Marconcini, R.; Spada, F.; Massironi, S.; Ibrahim, T.; Brizzi, M.P.; Campana, D.; Faggiano, A. Metformin use is associated with longer progression-free survival of patients with diabetes and pancreatic neuroendocrine tumors receiving everolimus and/or somatostatin analogues. Gastroenterology 2018, 155, 479–489.e7. [Google Scholar] [CrossRef] [PubMed]
- Wynn, A.; Vacheron, A.; Zuber, J.; Solomon, S.S. Metformin associated with increased survival in type 2 diabetes patients with pancreatic cancer and lymphoma. Am. J. Med. Sci. 2019, 358, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Deblon, N.; Bourgoin, L.; Veyrat-Durebex, C.; Peyrou, M.; Vinciguerra, M.; Caillon, A.; Maeder, C.; Fournier, M.; Montet, X.; Rohner-Jeanrenaud, F. Chronic mTOR inhibition by rapamycin induces muscle insulin resistance despite weight loss in rats. Br. J. Pharmacol. 2012, 165, 2325–2340. [Google Scholar] [CrossRef]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.M.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef]
- D’Elia, J.A.; Weinrauch, L.A. Hyperglycemia and hyperlipidemia with kidney or liver transplantation: A review. Biology 2023, 12, 1185. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Once again on rapamycin-induced insulin resistance and longevity: Despite of or owing to. Aging 2012, 4, 350. [Google Scholar] [CrossRef]
- Kezic, A.; Popovic, L.; Lalic, K. mTOR inhibitor therapy and metabolic consequences: Where do we stand? Oxidative Med. Cell. Longev. 2018, 2018, 2640342. [Google Scholar] [CrossRef]
- Wang, T.; Kusudo, T.; Takeuchi, T.; Yamashita, Y.; Kontani, Y.; Okamatsu, Y.; Saito, M.; Mori, N.; Yamashita, H. Evodiamine inhibits insulin-stimulated mTOR-S6K activation and IRS1 serine phosphorylation in adipocytes and improves glucose tolerance in obese/diabetic mice. PLoS ONE 2013, 8, e83264. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.J.; Palming, J.; Rizell, M.; Aureliano, M.; Carvalho, E.; Svensson, M.K.; Eriksson, J.W. mTOR inhibition with rapamycin causes impaired insulin signalling and glucose uptake in human subcutaneous and omental adipocytes. Mol. Cell. Endocrinol. 2012, 355, 96–105. [Google Scholar] [CrossRef]
- Zhang, N.; Ma, S. Research progress of 70 kDa ribosomal protein S6 kinase (P70S6K) inhibitors as effective therapeutic tools for obesity, type II diabetes and cancer. Curr. Med. Chem. 2020, 27, 4699–4719. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, G.; Capone, D. Inhibition of the mTOR pathway: A possible protective role in coronary artery disease. Ann. Med. 2013, 45, 348–356. [Google Scholar] [CrossRef]
- Das, A.; Durrant, D.; Koka, S.; Salloum, F.N.; Xi, L.; Kukreja, R.C. Mammalian target of rapamycin (mTOR) inhibition with rapamycin improves cardiac function in type 2 diabetic mice: Potential role of attenuated oxidative stress and altered contractile protein expression. J. Biol. Chem. 2014, 289, 4145–4160. [Google Scholar] [CrossRef]
- Wang, J.; Fan, S.; Xiong, Q.; Niu, Y.; Zhang, X.; Qin, J.; Shi, Y.; Zhang, L. Glucagon-like peptide-1 attenuates cardiac hypertrophy via the AngII/AT1R/ACE2 and AMPK/mTOR/p70S6K pathways. Acta Biochim. Et Biophys. Sin. 2021, 53, 1189–1197. [Google Scholar] [CrossRef]
- Patel, S.M.; Kang, Y.M.; Im, K.; Neuen, B.L.; Anker, S.D.; Bhatt, D.L.; Butler, J.; Cherney, D.Z.; Claggett, B.L.; Fletcher, R.A. Sodium-Glucose Cotransporter-2 Inhibitors and Major Adverse Cardiovascular Outcomes: A SMART-C Collaborative Meta-Analysis. Circulation 2024, 149, 1789–1801. [Google Scholar] [CrossRef]
- Schaub, J.A.; AlAkwaa, F.M.; McCown, P.J.; Naik, A.S.; Nair, V.; Eddy, S.; Menon, R.; Otto, E.A.; Demeke, D.; Hartman, J. SGLT2 inhibitors mitigate kidney tubular metabolic and mTORC1 perturbations in youth-onset type 2 diabetes. J. Clin. Investig. 2023, 133, e164486. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Fasting and rapamycin: Diabetes versus benevolent glucose intolerance. Cell Death Dis. 2019, 10, 607. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ye, S. Rapamycin improves insulin resistance and hepatic steatosis in type 2 diabetes rats through activation of autophagy. Cell Biol. Int. 2018, 42, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Reifsnyder, P.C.; Flurkey, K.; Doty, R.; Calcutt, N.A.; Koza, R.A.; Harrison, D.E. Rapamycin/metformin co-treatment normalizes insulin sensitivity and reduces complications of metabolic syndrome in type 2 diabetic mice. Aging Cell 2022, 21, e13666. [Google Scholar] [CrossRef] [PubMed]
- Reifsnyder, P.C.; Flurkey, K.; Te, A.; Harrison, D.E. Rapamycin treatment benefits glucose metabolism in mouse models of type 2 diabetes. Aging 2016, 8, 3120. [Google Scholar] [CrossRef] [PubMed]
- Gadallah, S.H.; Ghanem, H.M.; Abdel-Ghaffar, A.; Metwaly, F.G.; Hanafy, L.K.; Ahmed, E.K. 4-Phenylbutyric acid and rapamycin improved diabetic status in high fat diet/streptozotocin-induced type 2 diabetes through activation of autophagy. Arch. Physiol. Biochem. 2021, 127, 235–244. [Google Scholar] [CrossRef]
- Zhao, M.; Li, X.W.; Chen, D.Z.; Hao, F.; Tao, S.X.; Yu, H.Y.; Cheng, R.; Liu, H. Neuro-protective role of metformin in patients with acute stroke and type 2 diabetes mellitus via AMPK/mammalian target of rapamycin (mTOR) signaling pathway and oxidative stress. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 2186. [Google Scholar] [CrossRef] [PubMed]
- Temiz-Resitoglu, M.; Guden, D.S.; Senol, S.P.; Vezir, O.; Sucu, N.; Kibar, D.; Yılmaz, S.N.; Tunctan, B.; Malik, K.U.; Sahan-Firat, S. Pharmacological inhibition of mammalian target of rapamycin attenuates deoxycorticosterone acetate salt–induced hypertension and related pathophysiology: Regulation of oxidative stress, inflammation, and cardiovascular hypertrophy in male rats. J. Cardiovasc. Pharmacol. 2022, 79, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yan, J.; Wang, H.; Shi, M.; Zhang, M.; Yang, W.; Peng, C.; Li, H. Rapamycin ameliorates inflammation and fibrosis in the early phase of cirrhotic portal hypertension in rats through inhibition of mTORC1 but not mTORC2. PLoS ONE 2014, 9, e83908. [Google Scholar] [CrossRef]
- Kumar, V.; Wollner, C.; Kurth, T.; Bukowy, J.D.; Cowley, A.W., Jr. Inhibition of mammalian target of rapamycin complex 1 attenuates salt-induced hypertension and kidney injury in Dahl salt-sensitive rats. Hypertension 2017, 70, 813–821. [Google Scholar] [CrossRef]
- Kurniawan, R.; Nurkolis, F.; Taslim, N.A.; Subali, D.; Surya, R.; Gunawan, W.B.; Alisaputra, D.; Mayulu, N.; Salindeho, N.; Kim, B. Carotenoids composition of green algae Caulerpa racemosa and their antidiabetic, anti-obesity, antioxidant, and anti-inflammatory properties. Molecules 2023, 28, 3267. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stanciu, S.M.; Jinga, M.; Miricescu, D.; Stefani, C.; Nica, R.I.; Stanescu-Spinu, I.-I.; Vacaroiu, I.A.; Greabu, M.; Nica, S. mTOR Dysregulation, Insulin Resistance, and Hypertension. Biomedicines 2024, 12, 1802. https://doi.org/10.3390/biomedicines12081802
Stanciu SM, Jinga M, Miricescu D, Stefani C, Nica RI, Stanescu-Spinu I-I, Vacaroiu IA, Greabu M, Nica S. mTOR Dysregulation, Insulin Resistance, and Hypertension. Biomedicines. 2024; 12(8):1802. https://doi.org/10.3390/biomedicines12081802
Chicago/Turabian StyleStanciu, Silviu Marcel, Mariana Jinga, Daniela Miricescu, Constantin Stefani, Remus Iulian Nica, Iulia-Ioana Stanescu-Spinu, Ileana Adela Vacaroiu, Maria Greabu, and Silvia Nica. 2024. "mTOR Dysregulation, Insulin Resistance, and Hypertension" Biomedicines 12, no. 8: 1802. https://doi.org/10.3390/biomedicines12081802
APA StyleStanciu, S. M., Jinga, M., Miricescu, D., Stefani, C., Nica, R. I., Stanescu-Spinu, I.-I., Vacaroiu, I. A., Greabu, M., & Nica, S. (2024). mTOR Dysregulation, Insulin Resistance, and Hypertension. Biomedicines, 12(8), 1802. https://doi.org/10.3390/biomedicines12081802