
Exploring the Role of Hormones and Cytokines in Osteoporosis Development

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Pathomechanism of Osteoporosis
2.1. Roles of Hormones and Cytokines in Osteoporosis
2.1.1. Hormones
Growth Hormone (GH)
Glucocorticoids
PTH
Estrogen Hormone
Testosterone Hormone
Calcitonin Hormone
2.1.2. Cytokines
Pro-Inflammatory Cytokines
- TNF-alpha (TNF-α)
- IL-1
- IL-6
- IL-17
- IL-8
- IL-18
- IL-23
- IL-33
Anti-Inflammatory Cytokines
- IL-10
- IL-4
- IL-13
- IL-35

{kind=link}
{kind=link}
{kind=link}
| Cytokine | Family | Effects on Osteoblast | Effects on Osteoclasts | Effects on Osteocytes | Increase | Decrease | References |
|---|---|---|---|---|---|---|---|
| TNF- | TNF- | Osteoblasts are stimulated to produce RANKL and M-CSF. At low concentrations, these molecules enhance the differentiation of mesenchymal precursor cells into osteoblasts, whereas at high concentrations, they impede osteoblast function and halt bone formation. Additionally, the suppression of IGF-1 and Runt-related transcription factor 2 (RUNX2) expressions inhibits osteoblast differentiation. | Stimulates osteoclast development and enhances RANK expression in osteoclast precursors. Supports osteoclast formation via RANKL signaling. Increases c-Fos expression in osteoclast precursors. | TNF-α is known to directly increase RANKL expression in osteocytes and induce osteoclast formation both in vitro and in vivo. Additionally, TNF-α has been observed to upregulate sclerostin expression in osteocytes, which subsequently leads to an increase in RANKL expression. | Bone resorption, osteoclast differentiation and activation | It reduces bone formation by inhibiting osteoblast function. | [131,132,236,237] |
| IL-1 | IL-1 | - | Promotes osteoclastogenesis. | IL-1α promotes osteoclast formation by increasing RANKL expression in osteocytes. Additionally, IL-1α supports the survival of osteocytes and plays a significant role in osteogenesis. IL-1α interacts with osteocytes through complex pathways such as Ca and NO signaling, affecting the balance of bone formation and resorption. | M-CSF, PGE2, bone loss, osteoclast. | OPG | [11,146,238,239,240,241] |
| IL-1 | IL-1 | Induces bone resorption in osteoblasts by activating p38 MAPK. Inhibits human osteoblast migration. | Activates osteoclasts and promotes osteoclast maturation, facilitating multinucleation and viability. | IL-1β influences bone metabolism by increasing sclerostin secretion in osteocytes and inducing osteocyte apoptosis. Furthermore, IL-1β has been reported to enhance osteocyte-mediated osteoclastogenesis, an effect that can be mitigated by mechanical loading. IL-1β promotes the differentiation and mineralization of mesenchymal stem cells into osteoblasts, can increase inflammation in the bone microenvironment, and stimulates the production of FGF23. | RANKL, bone loss, osteoclast. | Bone formation rate. | [149,153,241,242,243,244,245,246] |
| IL-4 | Th2 | - | Suppresses osteoclast formation both directly and indirectly, inhibiting the bone resorption activity of mature, differentiated osteoclasts. | IL-4 and IL-10 are key regulators in the interactions between osteocytes and bone health. IL-4, together with IL-10, promotes the osteogenic differentiation of osteoblasts, induces an anti-inflammatory phenotype in macrophages, and inhibits osteoclastogenesis. These cytokines play crucial roles in supporting bone formation and maintaining the balance between bone formation and resorption. | OPG | RANKL, osteoclastogenesis, Th2 cells produced in the presence of IL-4 prevent bone loss. | [179,219,220,221,245] |
| IL-6 | IL-12 | Inhibits osteoblast differentiation. | Directly and indirectly supports osteoclast development while restricting the differentiation of osteoclast progenitors into osteoclasts. | IL-6 plays a critical role in the interaction between osteocytes and bone metabolism. IL-6, secreted by osteocytes in response to mechanical stress, regulates bone remodeling, osteoclastogenesis, and osteoblast activity. It has been shown that IL-6 enhances osteocyte-mediated osteoclastic differentiation via the JAK2/STAT3 pathway and is important in the modulation of bone mass. | Osteogenic capacity, bone resorption, osteoclast differentiation and activation, osteoclastogenesis, rheumatoid arthritis, cancer metastasis, atherosclerosis, and type 2 diabetes. | Osteoblast differentiation, bone trabecular volume. | [129,154,243,247,248,249] |
| IL-8 | IL-8 | - | Promotes RANKL-induced osteoclastogenesis. | Osteocytes, embedded within the bone matrix, play a crucial role in bone remodeling and are sensitive to various signaling molecules, particularly inflammatory cytokines like IL-8. These cells communicate with neighboring osteoblasts and osteoclasts through canaliculi, thereby regulating bone homeostasis. It is possible that IL-8 may affect osteocyte function and potentially contribute to bone remodeling processes. | Bone resorption, neutrophil RANKL expression. | Bone density | [11,129,184,250,251] |
| IL-10 | IL-10 | Suppresses osteogenic activity in the bone marrow. | Inhibits the differentiation of osteoclast progenitors into osteoclast precursors and suppresses RANK-induced osteoclast formation. | IL-10 plays a significant role in bone metabolism and regulates osteoclastogenesis. IL-10 inhibits bone resorption by upregulating the expression of OPG while downregulating the expression of RANKL and colony-stimulating factor-1 (CSF-1). Additionally, IL-10, in conjunction with IL-4, promotes the osteogenic differentiation of osteoblasts and induces an anti-inflammatory phenotype in macrophages. This indicates that IL-10 both inhibits bone resorption and supports bone formation. | Bone formation, bone fragility. | Inhibits osteoclast differentiation, reduces bone resorption, bone loss, reduced osteoclast production, decreased bone mass. It suppresses osteogenic differentiation by inhibiting bone mineralization and synthesis of bone proteins. | [211,245,252,253] |
| IL-13 | Th2 | - | Inhibits osteoclast formation and bone resorption. | Osteocytes play a crucial role in bone remodeling and are sensitive to cytokines. IL-13, known for its anti-inflammatory properties, may affect osteocyte function and bone metabolism. Although the specific effects of IL-13 on osteocytes are not well-detailed, it is possible that IL-13, like IL-10 and IL-4, modulates osteocyte activity and bone homeostasis. | Bone formation, bone resorption control, and bone mass preservation, bone tissue strength. | Bone loss | [217,224,225] |
| IL-17 | IL-17 | Enhances the expression of cytokines such as TNF-α, IL-6, and RANKL that promote osteoclasts in osteoblasts. Supports osteoblast differentiation while preventing osteoblast calcification. | Initiates osteoclast formation. At low concentrations, it enhances autophagy and osteoclast formation in osteoclast precursors, while at high concentrations, it inhibits the differentiation of osteoclast precursors into osteoclasts. | IL-17, a proinflammatory cytokine, has significant effects on osteocytes. IL-17 promotes bone resorption by regulating RANKL production in osteocytes. Additionally, IL-17 receptor signaling mediates PTH-induced bone loss and increases osteocytic RANKL production. IL-17 also promotes bone resorption and fluid shear stress regulation through osteocyte-specific signaling pathways. | Bone loss, RANKL, pro-osteoclastogenic cytokine. | Bone resorption | [11,147,174,177,178,179,180,254,255,256] |
| IL-18 | IL-1 | - | Inhibits TNF-α-induced osteoclastogenesis through apoptosis via Fas/FasL and NO in myeloid cells. Indirectly inhibits osteoclast formation through IFN-γ and GM-CSF. | Since osteocytes are sensitive to various signaling molecules, it is possible that IL-18 could affect osteocyte function and bone metabolism. Further research is needed to understand the specific effects of IL-18 on osteocytes. | In the presence of TNF-α, IL-12 and IL-18 synergistically increase NO production. An increase in IL-18 level causes a decrease in IL-18BP level. TNF-α, IL-1β, and IL-6 increase osteoclast differentiation and activation. | Osteoclast differentiation and activation. | [186,187,188,189,190,191] |
| IL-23 | IL-12 | - | Participates in T-cell-mediated osteoclast formation, regulates osteoclast differentiation, and indirectly inhibits osteoclast formation. | It is possible that IL-23 could affect osteocyte function and bone metabolism. Given its role in inflammation and immune responses, IL-23 may regulate bone remodeling and cell communication. Further research is needed to understand the specific effects of IL-23 on osteocytes. | It transforms CD4(+) cells into Th17 cells, thus increasing the production of IL-17 by Th17 cells. Osteoclast differentiation and activation. | Osteoblast apoptosis. | [177,178,199,200,201,202] |
| IL-33 | IL-1 | Stimulates osteoblast function, promotes matrix mineral deposition, and reduces sclerostin mRNA. | Terminates the osteoclast formation initiated by RANKL and suppresses the gene expression associated with osteoblasts. It induces apoptosis in osteoclasts while preventing osteoclast formation and bone resorption induced by TNF. | IL-33 has been shown to induce IL-6 expression and regulate osteocyte function by interacting with ST2L receptors on the plasma membrane and activating specific signaling pathways. These findings suggest that IL-33 may play a significant role in regulating osteocyte function and bone metabolism, highlighting a potential connection between IL-33 and osteocytes. | IL-33 increases during inflammation, increasing osteoblast activity. | Inhibiting osteoclast formation. | [11,13,205,206,207,208,257] |
| IL-35 | IL-12 | Promote the differentiation of mesenchymal stem cells into osteoblasts. | Prevent TNF-induced osteoclast formation and promote apoptosis, support the formation of functional osteoclasts, and increase the expression of osteoclast differentiation factors. | The connection between IL-35 and osteocytes has not been directly addressed in the literature. However, based on the effects of cytokines on bone cells, it can be hypothesized that IL-35 may influence osteocyte function and bone metabolism. Further research into the role of IL-35 in bone remodeling, osteoclastogenesis, and osteoblast activity could provide insights into this connection. | JAK1/STAT1, β-catenin and Axin2, bone formation and resorption. | Osteoclast formation, TNF-induced osteoclastogenesis and bone resorption, NF-κB, MAPK, NFATc1, c-Fos, and TRAP, apoptosis and adipogenic differentiation, adipogenesis. | [11,228,230,231,232,233,258] |
2.2. GFs
2.2.1. IGFs
2.2.2. TGFs
2.2.3. FGFs
FGF-2
FGF-19
FGF-21
FGF-23
2.2.4. EGF
2.2.5. HGF
2.2.6. VEGF
2.2.7. PDGF
2.2.8. NGF
3. Conclusions and Future Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Rinonapoli, G.; Ruggiero, C.; Meccariello, L.; Bisaccia, M.; Ceccarini, P.; Caraffa, A. Osteoporosis in Men: A Review of an Underestimated Bone Condition. Int. J. Mol. Sci. 2021, 22, 2105. [Google Scholar] [CrossRef]
- Adejuyigbe, B.; Kallini, J.; Chiou, D.; Kallini, J.R. Osteoporosis: Molecular Pathology, Diagnostics, and Therapeutics. Int. J. Mol. Sci. 2023, 24, 14583. [Google Scholar] [CrossRef] [PubMed]
- Akkawi, I.; Zmerly, H. Osteoporosis: Current Concepts. Joints 2018, 6, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Xiao, P.L.; Cui, A.Y.; Hsu, C.J.; Peng, R.; Jiang, N.; Xu, X.H.; Ma, Y.G.; Liu, D.; Lu, H.D. Global, Regional Prevalence, and Risk Factors of Osteoporosis According to the World Health Organization Diagnostic Criteria: A Systematic Review and Meta-Analysis. Osteoporos. Int. 2022, 33, 2137–2153. [Google Scholar] [CrossRef] [PubMed]
- Adami, G.; Fassio, A.; Gatti, D.; Viapiana, O.; Benini, C.; Danila, M.I.; Saag, K.G.; Rossini, M. Osteoporosis in 10 Years Time: A Glimpse into the Future of Osteoporosis. Ther. Adv. Musculoskelet. Dis. 2022, 14, 1759720X2210835. [Google Scholar] [CrossRef] [PubMed]
- Malutan, A.M.; Dan, M.; Nicolae, C.; Carmen, M. Proinflammatory and Anti-Inflammatory Cytokine Changes Related to Menopause. Menopausal Rev. 2014, 3, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Najem, M.Y.; Couturaud, F.; Lemarié, C.A. Cytokine and Chemokine Regulation of Venous Thromboembolism. J. Thromb. Haemost. 2020, 18, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Leppkes, M.; Neurath, M.F. Cytokines in Inflammatory Bowel Diseases—Update 2020. Pharmacol. Res. 2020, 158, 104835. [Google Scholar] [CrossRef]
- Lan, T.; Chen, L.; Wei, X. Inflammatory Cytokines in Cancer: Comprehensive Understanding and Clinical Progress in Gene Therapy. Cells 2021, 10, 100. [Google Scholar] [CrossRef]
- Liu, C.; Chu, D.; Kalantar-Zadeh, K.; George, J.; Young, H.A.; Liu, G. Cytokines: From Clinical Significance to Quantification. Adv. Sci. 2021, 8, 2004433. [Google Scholar] [CrossRef]
- Xu, J.; Yu, L.; Liu, F.; Wan, L.; Deng, Z. The Effect of Cytokines on Osteoblasts and Osteoclasts in Bone Remodeling in Osteoporosis: A Review. Front. Immunol. 2023, 14, 1222129. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.M.; Yin, Y.; Liu, X.Q.; Du, L.J.; Wang, L.Q.; Tan, L. Study and Analysis on the Mechanisms of Action of Cytokines on Osteoclasts in Osteoporosis. TMR Theory Hypothesis 2020, 3, 393–397. [Google Scholar] [CrossRef]
- Ginaldi, L.; De Martinis, M.; Saitta, S.; Sirufo, M.M.; Mannucci, C.; Casciaro, M.; Ciccarelli, F.; Gangemi, S. Interleukin-33 Serum Levels in Postmenopausal Women with Osteoporosis. Sci. Rep. 2019, 9, 3786. [Google Scholar] [CrossRef]
- Yu, X.; Xia, Y.; Jia, J.; Yuan, G. The Role of Fibroblast Growth Factor 19 Subfamily in Different Populations Suffering from Osteoporosis. Front. Endocrinol. 2022, 13, 830022. [Google Scholar] [CrossRef] [PubMed]
- Okman-Kilic, T. Estrogen Deficiency and Osteoporosis. In Advances in Osteoporosis; InTech: London, UK, 2015. [Google Scholar]
- Augustine, M.; Horwitz, M.J. Parathyroid Hormone and Parathyroid Hormone-Related Protein Analogs as Therapies for Osteoporosis. Curr. Osteoporos. Rep. 2013, 11, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Gaffney, C.D.; Pagano, M.J.; Kuker, A.P.; Stember, D.S.; Stahl, P.J. Osteoporosis and Low Bone Mineral Density in Men with Testosterone Deficiency Syndrome. Sex. Med. Rev. 2015, 3, 298–315. [Google Scholar] [CrossRef] [PubMed]
- Chamouni, A.; Oury, F. Reciprocal Interaction between Bone and Gonads. Arch. Biochem. Biophys. 2014, 561, 147–153. [Google Scholar] [CrossRef]
- Srinivasan, A.; Wong, F.K.; Karponis, D. Calcitonin: A Useful Old Friend. J. Musculoskelet. Neuronal Interact. 2020, 20, 600–609. [Google Scholar]
- Florencio-Silva, R.; Sasso, G.R.D.S.; Sasso-Cerri, E.; Simões, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. Biomed Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef]
- Ashton, N. Physiology of Red and White Blood Cells. Anaesth. Intensive Care Med. 2007, 8, 203–208. [Google Scholar] [CrossRef]
- Weatherholt, A.M.; Fuchs, R.K.; Warden, S.J. Specialized Connective Tissue: Bone, the Structural Framework of the Upper Extremity. J. Hand Ther. 2012, 25, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Luers, J.C.; Hüttenbrink, K. Surgical Anatomy and Pathology of the Middle Ear. J. Anat. 2016, 228, 338–353. [Google Scholar] [CrossRef] [PubMed]
- Dempster, D.W.; Cosman, F.; Parisien, M.; Shen, V.; Lindsay, R. Anabolic Actions of Parathyroid Hormone on Bone. Endocr. Rev. 1993, 14, 690–709. [Google Scholar] [CrossRef] [PubMed]
- Swarthout, J.T.; D’Alonzo, R.C.; Selvamurugan, N.; Partridge, N.C. Parathyroid Hormone-Dependent Signaling Pathways Regulating Genes in Bone Cells. Gene 2002, 282, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.B.; Stein, G.S.; Stein, J.L.; Van Wijnen, A.J. Regulated Expression of the Bone-Specific Osteocalcin Gene by Vitamins and Hormones. In Vitamins & Hormones; Elsevier: Amsterdam, The Netherlands, 1998; pp. 443–509. [Google Scholar]
- Turner, R.T.; Riggs, B.L.; Spelsberg, T.C. Skeletal Effects of Estrogen. Endocr. Rev. 1994, 15, 275–300. [Google Scholar] [CrossRef] [PubMed]
- Eastell, R.; O’Neill, T.W.; Hofbauer, L.C.; Langdahl, B.; Reid, I.R.; Gold, D.T.; Cummings, S.R. Postmenopausal Osteoporosis. Nat. Rev. Dis. Primers 2016, 2, 16069. [Google Scholar] [CrossRef] [PubMed]
- Chotiyarnwong, P.; McCloskey, E.V. Pathogenesis of Glucocorticoid-Induced Osteoporosis and Options for Treatment. Nat. Rev. Endocrinol. 2020, 16, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.K.; Sung, Y.K. Update on Glucocorticoid Induced Osteoporosis. Endocrinol. Metab. 2021, 36, 536–543. [Google Scholar] [CrossRef]
- Abou Neel, E.; Aljabo, A.; Strange, A.; Ibrahim, S.; Coathup, M.; Young, A.; Bozec, L.; Mudera, V. Demineralization–Remineralization Dynamics in Teeth and Bone. Int. J. Nanomed. 2016, 11, 4743–4763. [Google Scholar] [CrossRef]
- Stock, S.R. The Mineral–Collagen Interface in Bone. Calcif. Tissue Int. 2015, 97, 262–280. [Google Scholar] [CrossRef]
- Song, L. Calcium and Bone Metabolism Indices. In Advances in Clinical Chemistry; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1–46. [Google Scholar]
- Kuo, T.R.; Chen, C.H. Bone Biomarker for the Clinical Assessment of Osteoporosis: Recent Developments and Future Perspectives. Biomark. Res. 2017, 5, 18. [Google Scholar] [CrossRef]
- Bradshaw, A.D.; Sage, E.H. SPARC, a Matricellular Protein That Functions in Cellular Differentiation and Tissue Response to Injury. J. Clin. Investig. 2001, 107, 1049–1054. [Google Scholar] [CrossRef]
- Vancea, A. Relationship between Osteopontin and Bone Mineral Density. Acta Endocrinol. 2021, 17, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.J.; Li, Y.S.; Zhang, F.J. Osteopontin, a Bridge Links Osteoarthritis and Osteoporosis. Front. Endocrinol. 2022, 13, 1012508. [Google Scholar] [CrossRef]
- Detsch, R.; Boccaccini, A.R. The Role of Osteoclasts in Bone Tissue Engineering. J. Tissue Eng. Regen. Med. 2015, 9, 1133–1149. [Google Scholar] [CrossRef] [PubMed]
- Prideaux, M.; Findlay, D.M.; Atkins, G.J. Osteocytes: The Master Cells in Bone Remodelling. Curr. Opin. Pharmacol. 2016, 28, 24–30. [Google Scholar] [CrossRef]
- Kini, U.; Nandeesh, B.N. Physiology of Bone Formation, Remodeling, and Metabolism. In Radionuclide and Hybrid Bone Imaging; Springer: Berlin/Heidelberg, Germany, 2012; pp. 29–57. [Google Scholar]
- Pant, A.; Paul, E.; Niebur, G.L.; Vahdati, A. Integration of Mechanics and Biology in Computer Simulation of Bone Remodeling. Prog. Biophys. Mol. Biol. 2021, 164, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Metzger, C.E.; Narayanan, S.A. The Role of Osteocytes in Inflammatory Bone Loss. Front. Endocrinol. 2019, 10, 285. [Google Scholar] [CrossRef]
- Blanchard, F.; Duplomb, L.; Baud’huin, M.; Brounais, B. The Dual Role of IL-6-Type Cytokines on Bone Remodeling and Bone Tumors. Cytokine Growth Factor Rev. 2009, 20, 19–28. [Google Scholar] [CrossRef]
- Wang, T.; Yu, X.; He, C. Pro-Inflammatory Cytokines: Cellular and Molecular Drug Targets for Glucocorticoid-Induced-Osteoporosis via Osteocyte. Curr. Drug Targets 2018, 20, 1–15. [Google Scholar] [CrossRef]
- Zhou, M.; Li, S.; Pathak, J.L. Pro-Inflammatory Cytokines and Osteocytes. Curr. Osteoporos. Rep. 2019, 17, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Mohamad, S.F.; Gunawan, A.; Blosser, R.; Childress, P.; Aguilar-Perez, A.; Ghosh, J.; Hong, J.M.; Liu, J.; Kanagasabapathy, D.; Kacena, M.A.; et al. Neonatal Osteomacs and Bone Marrow Macrophages Differ in Phenotypic Marker Expression and Function. J. Bone Miner. Res. 2020, 36, 1580–1593. [Google Scholar] [CrossRef] [PubMed]
- Miron, R.J.; Bosshardt, D.D. OsteoMacs: Key Players around Bone Biomaterials. Biomaterials 2016, 82, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Batoon, L.; Millard, S.M.; Raggatt, L.J.; Pettit, A.R. Osteomacs and Bone Regeneration. Curr. Osteoporos. Rep. 2017, 15, 385–395. [Google Scholar] [CrossRef]
- Yin, Y.; Tang, Q.; Xie, M.; Hu, L.; Chen, L. Insights into the Mechanism of Vascular Endothelial Cells on Bone Biology. Biosci. Rep. 2021, 41, BSR20203258. [Google Scholar] [CrossRef] [PubMed]
- Sivan, U.; De Angelis, J.; Kusumbe, A.P. Role of Angiocrine Signals in Bone Development, Homeostasis and Disease. Open Biol. 2019, 9, 190144. [Google Scholar] [CrossRef]
- Song, S.; Guo, Y.; Yang, Y.; Fu, D. Advances in Pathogenesis and Therapeutic Strategies for Osteoporosis. Pharmacol. Ther. 2022, 237, 108168. [Google Scholar] [CrossRef]
- Sobh, M.M.; Abdalbary, M.; Elnagar, S.; Nagy, E.; Elshabrawy, N.; Abdelsalam, M.; Asadipooya, K.; El-Husseini, A. Secondary Osteoporosis and Metabolic Bone Diseases. J. Clin. Med. 2022, 11, 2382. [Google Scholar] [CrossRef]
- Yu, B.; Wang, C. Osteoporosis and Periodontal Diseases—An Update on Their Association and Mechanistic Links. Periodontology 2000 2022, 89, 99–113. [Google Scholar] [CrossRef]
- Cannarella, R.; Barbagallo, F.; Condorelli, R.A.; Aversa, A.; La Vignera, S.; Calogero, A.E. Osteoporosis from an Endocrine Perspective: The Role of Hormonal Changes in the Elderly. J. Clin. Med. 2019, 8, 1564. [Google Scholar] [CrossRef]
- Cheng, C.H.; Chen, L.R.; Chen, K.H. Osteoporosis Due to Hormone Imbalance: An Overview of the Effects of Estrogen Deficiency and Glucocorticoid Overuse on Bone Turnover. Int. J. Mol. Sci. 2022, 23, 1376. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, J.S.; Brockwell, S.E.; Mehta, V.; Greendale, G.A.; Sowers, M.R.; Ettinger, B.; Lo, J.C.; Johnston, J.M.; Cauley, J.A.; Danielson, M.E.; et al. Bone Mineral Density Changes during the Menopause Transition in a Multiethnic Cohort of Women. J. Clin. Endocrinol. Metab. 2008, 93, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Golds, G.; Houdek, D.; Arnason, T. Male Hypogonadism and Osteoporosis: The Effects, Clinical Consequences, and Treatment of Testosterone Deficiency in Bone Health. Int. J. Endocrinol. 2017, 2017, 4602129. [Google Scholar] [CrossRef] [PubMed]
- Delitala, A.P.; Scuteri, A.; Doria, C. Thyroid Hormone Diseases and Osteoporosis. J. Clin. Med. 2020, 9, 1034. [Google Scholar] [CrossRef] [PubMed]
- Bouillon, R. Growth Hormone and Bone. Horm. Res. 1991, 36, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Gharahdaghi, N.; Phillips, B.E.; Szewczyk, N.J.; Smith, K.; Wilkinson, D.J.; Atherton, P.J. Links Between Testosterone, Oestrogen, and the Growth Hormone/Insulin-Like Growth Factor Axis and Resistance Exercise Muscle Adaptations. Front. Physiol. 2021, 11, 621226. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.; Muñoz, M.; Shibli-Rahhal, A. Anorexia Nervosa and Osteoporosis. Calcif. Tissue Int. 2022, 110, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Mazziotti, G.; Lania, A.G.; Canalis, E. Skeletal Disorders Associated with the Growth Hormone–Insulin-like Growth Factor 1 Axis. Nat. Rev. Endocrinol. 2022, 18, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Gillberg, P.; Mallmin, H.; Petrén-Mallmin, M.; Ljunghall, S.; Nilsson, A.G. Two Years of Treatment with Recombinant Human Growth Hormone Increases Bone Mineral Density in Men with Idiopathic Osteoporosis. J. Clin. Endocrinol. Metab. 2002, 87, 4900–4906. [Google Scholar] [CrossRef]
- Krantz, E.; Trimpou, P.; Landin-Wilhelmsen, K. Effect of Growth Hormone Treatment on Fractures and Quality of Life in Postmenopausal Osteoporosis: A 10-Year Follow-Up Study. J. Clin. Endocrinol. Metab. 2015, 100, 3251–3259. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, R.; Gu, Z.; Dong, C.; Guo, G.; Li, L. Effects of Glucocorticoids on Osteoporosis in Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Osteoporos. Int. 2020, 31, 1401–1409. [Google Scholar] [CrossRef]
- Urquiaga, M.; Saag, K.G. Risk for Osteoporosis and Fracture with Glucocorticoids. Best Pract. Res. Clin. Rheumatol. 2022, 36, 101793. [Google Scholar] [CrossRef]
- English, K.A.; Lines, K.E.; Thakker, R.V. Genetics of Hereditary Forms of Primary Hyperparathyroidism. Hormones 2024, 23, 3–14. [Google Scholar] [CrossRef]
- Magagnoli, L.; Ciceri, P.; Cozzolino, M. Secondary Hyperparathyroidism in Chronic Kidney Disease: Pathophysiology, Current Treatments and Investigational Drugs. Expert Opin. Investig. Drugs 2024, 33, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Xu, Y.; Fu, Q.; He, M. Parathyroid Hormone Regulates Osteoblast Differentiation in a Wnt/β-Catenin-Dependent Manner. Mol. Cell Biochem. 2011, 355, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Rejnmark, L.; Ejlsmark-Svensson, H. Effects of PTH and PTH Hypersecretion on Bone: A Clinical Perspective. Curr. Osteoporos. Rep. 2020, 18, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Vilardaga, J.-P.; Romero, G.; Friedman, P.A.; Gardella, T.J. Molecular Basis of Parathyroid Hormone Receptor Signaling and Trafficking: A Family B GPCR Paradigm. Cell. Mol. Life Sci. 2011, 68, 1–13. [Google Scholar] [CrossRef]
- Zhao, L.; Yuan, Q.; Dai, A.; He, X.; Chen, C.; Zhang, C.; Xu, Y.; Zhou, Y.; Wang, M.; Yang, D.; et al. Molecular Recognition of Two Endogenous Hormones by the Human Parathyroid Hormone Receptor-1. Acta Pharmacol. Sin. 2023, 44, 1227–1237. [Google Scholar] [CrossRef]
- Liu, H.; Liu, L.; Rosen, C.J. PTH and the Regulation of Mesenchymal Cells within the Bone Marrow Niche. Cells 2024, 13, 406. [Google Scholar] [CrossRef]
- De Freitas, P.H.L.; Li, M.; Ninomiya, T.; Nakamura, M.; Ubaidus, S.; Oda, K.; Udagawa, N.; Maeda, T.; Takagi, R.; Amizuka, N. Intermittent PTH Administration Stimulates Pre-Osteoblastic Proliferation without Leading to Enhanced Bone Formation in Osteoclast-Less c-Fos−/− Mice. J. Bone Miner. Res. 2009, 24, 1586–1597. [Google Scholar] [CrossRef]
- Lotinun, S.; Sibonga, J.D.; Turner, R.T. Differential Effects of Intermittent and Continuous Administration of Parathyroid Hormone on Bone Histomorphometry and Gene Expression. Endocrine 2002, 17, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, V.; Cox, D.R. Dynamics of Bone Cell Interactions and Differential Responses to PTH and Antibody-Based Therapies. Bull. Math. Biol. 2019, 81, 3575–3622. [Google Scholar] [CrossRef]
- Li, G.; Liu, S.; Xu, H.; Chen, Y.; Deng, J.; Xiong, A.; Wang, D.; Weng, J.; Yu, F.; Gao, L.; et al. Potential Effects of Teriparatide (PTH (1–34)) on Osteoarthritis: A Systematic Review. Arthritis Res. Ther. 2023, 25, 3. [Google Scholar] [CrossRef] [PubMed]
- Jilka, R.L.; Weinstein, R.S.; Bellido, T.; Parfitt, A.M.; Manolagas, S.C. Osteoblast Programmed Cell Death (Apoptosis): Modulation by Growth Factors and Cytokines. J. Bone Miner. Res. 1998, 13, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.L.; Cain, R.L.; Halladay, D.L.; Yang, X.; Zeng, Q.; Miles, R.R.; Chandrasekhar, S.; Martin, T.J.; Onyia, J.E. Catabolic Effects of Continuous Human PTH (1–38) in Vivo Is Associated with Sustained Stimulation of RANKL and Inhibition of Osteoprotegerin and Gene-Associated Bone Formation. Endocrinology 2001, 142, 4047–4054. [Google Scholar] [CrossRef] [PubMed]
- Abu-Amer, Y. NF-ΚB Signaling and Bone Resorption. Osteoporos. Int. 2013, 24, 2377–2386. [Google Scholar] [CrossRef] [PubMed]
- Bradford, P.G.; Gerace, K.V.; Roland, R.L.; Chrzan, B.G. Estrogen Regulation of Apoptosis in Osteoblasts. Physiol. Behav. 2010, 99, 181–185. [Google Scholar] [CrossRef]
- Almeida, M.; Martin-Millan, M.; Ambrogini, E.; Bradsher, R.; Han, L.; Chen, X.D.; Roberson, P.K.; Weinstein, R.S.; O’Brien, C.A.; Jilka, R.L.; et al. Estrogens Attenuate Oxidative Stress and the Differentiation and Apoptosis of Osteoblasts by DNA-Binding-Independent Actions of the ERα. J. Bone Miner. Res. 2010, 25, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Zilberman, Y.; Wassermann, K.; Bain, S.D.; Sadovsky, Y.; Gazit, D. Estrogen Modulates Estrogen Receptor and Expression, Osteogenic Activity, and Apoptosis in Mesenchymal Stem Cells (MSCs) of Osteoporotic Mice. J. Cell Biochem. 2001, 81, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Tella, S.H.; Gallagher, J.C. Bazedoxifene + Conjugated Estrogens in HT for the Prevention of Osteoporosis and Treatment of Vasomotor Symptoms Associated with the Menopause. Expert Opin. Pharmacother. 2013, 14, 2407–2420. [Google Scholar] [CrossRef]
- Wang, L.T.; Chen, L.R.; Chen, K.H. Hormone-Related and Drug-Induced Osteoporosis: A Cellular and Molecular Overview. Int. J. Mol. Sci. 2023, 24, 5814. [Google Scholar] [CrossRef] [PubMed]
- Gambacciani, M.; Levancini, M. Featured Editorial Hormone Replacement Therapy and the Prevention of Postmenopausal Osteoporosis. Menopausal Rev. 2014, 4, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, J. Prevention and Treatment of Osteoporosis in Women. Post Reprod. Health 2023, 29, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Abu, E.O.; Horner, A.; Kusec, V.; Triffitt, J.T.; Compston, J.E. The Localization of Androgen Receptors in Human Bone. J. Clin. Endocrinol. Metab. 1997, 82, 3493–3497. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bloom, I.; Dennison, E.M.; Ward, K.A.; Robinson, S.M.; Barker, M.; Cooper, C.; Lawrence, W. Understanding Influences on Physical Activity Participation by Older Adults: A Qualitative Study of Community-Dwelling Older Adults from the Hertfordshire Cohort Study, UK. PLoS ONE 2022, 17, e0263050. [Google Scholar] [CrossRef] [PubMed]
- Notelovitz, M. Androgen Effects on Bone and Muscle. Fertil. Steril. 2002, 77, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ominsky, M.S.; Stolina, M.; Warmington, K.S.; Geng, Z.; Niu, Q.T.; Asuncion, F.J.; Tan, H.L.; Grisanti, M.; Dwyer, D.; et al. Increased RANK Ligand in Bone Marrow of Orchiectomized Rats and Prevention of Their Bone Loss by the RANK Ligand Inhibitor Osteoprotegerin. Bone 2009, 45, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.Y.; Ima-Nirwana, S. The Effects of Orchidectomy and Supraphysiological Testosterone Administration on Trabecular Bone Structure and Gene Expression in Rats. Aging Male 2015, 18, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Bellido, T.; Jilka, R.L.; Boyce, B.F.; Girasole, G.; Broxmeyer, H.; Dalrymple, S.A.; Murray, R.; Manolagas, S.C. Regulation of Interleukin-6, Osteoclastogenesis, and Bone Mass by Androgens. The Role of the Androgen Receptor. J. Clin. Investig. 1995, 95, 2886–2895. [Google Scholar] [CrossRef]
- Gill, R.K.; Turner, R.T.; Wronski, T.J.; Bell, N.H. Orchiectomy Markedly Reduces the Concentration of the Three Isoforms of Transforming Growth Factor β in Rat Bone, and Reduction Is Prevented by Testosterone. Endocrinology 1998, 139, 546–550. [Google Scholar] [CrossRef]
- Mohamad, N.V.; Soelaiman, I.N.; Chin, K.Y. A Concise Review of Testosterone and Bone Health. Clin. Interv. Aging 2016, 11, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- Ng Tang Fui, M.; Hoermann, R.; Bracken, K.; Handelsman, D.J.; Inder, W.J.; Stuckey, B.G.A.; Yeap, B.B.; Ghasem-Zadeh, A.; Robledo, K.P.; Jesudason, D.; et al. Effect of Testosterone Treatment on Bone Microarchitecture and Bone Mineral Density in Men: A 2-Year RCT. J. Clin. Endocrinol. Metab. 2021, 106, e3143–e3158. [Google Scholar] [CrossRef] [PubMed]
- Corona, G.; Vena, W.; Pizzocaro, A.; Giagulli, V.A.; Francomano, D.; Rastrelli, G.; Mazziotti, G.; Aversa, A.; Isidori, A.M.; Pivonello, R.; et al. Testosterone Supplementation and Bone Parameters: A Systematic Review and Meta-Analysis Study. J. Endocrinol. Investig. 2022, 45, 911–926. [Google Scholar] [CrossRef] [PubMed]
- Fuggle, N.R.; Beaudart, C.; Bruyère, O.; Abrahamsen, B.; Al-Daghri, N.; Burlet, N.; Chandran, M.; Rosa, M.M.; Cortet, B.; Demonceau, C.; et al. Evidence-Based Guideline for the Management of Osteoporosis in Men. Nat. Rev. Rheumatol. 2024, 20, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Summers, R.; Macnab, R. Thyroid, Parathyroid Hormones and Calcium Homeostasis. Anaesth. Intensive Care Med. 2017, 18, 522–526. [Google Scholar] [CrossRef]
- Copp, D.H.; Cheney, B. Calcitonin—A Hormone from the Parathyroid Which Lowers the Calcium-Level of the Blood. Nature 1962, 193, 381–382. [Google Scholar] [CrossRef] [PubMed]
- Hurley, D.L.; Tiegs, R.D.; Wahner, H.W.; Heath, H. Axial and Appendicular Bone Mineral Density in Patients with Long-Term Deficiency or Excess of Calcitonin. N. Engl. J. Med. 1987, 317, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Wüster, C.; Raue, F.; Meyer, C.; Bergmann, M.; Ziegler, R. Long-Term Excess of Endogenous Calcitonin in Patients with Medullary Thyroid Carcinoma Does Not Affect Bone Mineral Density. J. Endocrinol. 1992, 134, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, P.F.; Baruch, H. Is Calcitonin an Important Physiological Substance? Int. J. Basic Clin. Endocrinol. 2003, 21, 201–208. [Google Scholar] [CrossRef]
- Miller, S. Calcitonin—Guardian of the Mammalian Skeleton or Is It Just a Fish Story? Endocrinology 2006, 147, 4007–4009. [Google Scholar] [CrossRef]
- Davey, R.A.; Findlay, D.M. Calcitonin: Physiology or Fantasy? J. Bone Miner. Res. 2013, 28, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.E.; Singer, F.R.; Gorn, A.H.; Hofer, D.P.; Nimni, M.E. Calcitonin Stimulates Bone Formation When Administered Prior to Initiation of Osteogenesis. J. Clin. Investig. 1981, 68, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Farley, J.R.; Tarbaux, N.M.; Hall, S.L.; Linkhart, T.A.; Baylink, D.J. The Anti-Bone-Resorptive Agent Calcitonin Also Acts In Vitro to Directly Increase Bone Formation and Bone Cell Proliferation. Endocrinology 1988, 123, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Villa, I.; Dal Fiume, C.; Maestroni, A.; Rubinacci, A.; Ravasi, F.; Guidobono, F. Human Osteoblast-like Cell Proliferation Induced by Calcitonin-Related Peptides Involves PKC Activity. Am. J. Physiol.-Endocrinol. Metab. 2003, 284, E627–E633. [Google Scholar] [CrossRef] [PubMed]
- Cornish, J.; Callon, K.E.; Gasser, J.A.; Bava, U.; Gardiner, E.M.; Coy, D.H.; Cooper, G.J.S.; Reid, I.R. Systemic Administration of a Novel Octapeptide, Amylin-(1—8), Increases Bone Volume in Male Mice. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E730–E735. [Google Scholar] [CrossRef] [PubMed]
- Cornish, J.; Callon, K.E.; Cooper, G.J.S.; Reid, I.R. Amylin Stimulates Osteoblast Proliferation and Increases Mineralized Bone Volume in Adult Mice. Biochem. Biophys. Res. Commun. 1995, 207, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Bigazzi, P.E.; Yoshida, T. Similarities of T Cell Function in Cell-Mediated Immunity and Antibody Production. Cell Immunol. 1974, 12, 150–159. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Gadina, M.; Siegel, R.M. Cytokines and Cytokine Receptors. In Clinical Immunology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 127–155.e1. [Google Scholar]
- Akdoğan, M.; Yöntem, M. Sitokinler. Online Türk. Sağlık Bilim. Derg. 2018, 3, 36–45. [Google Scholar] [CrossRef]
- Zhang, J.M.; An, J. Cytokines, Inflammation, and Pain. Int. Anesthesiol. Clin. 2007, 45, 27–37. [Google Scholar] [CrossRef]
- Sprague, A.H.; Khalil, R.A. Inflammatory Cytokines in Vascular Dysfunction and Vascular Disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef]
- Camacho, V.; Kuznetsova, V.; Welner, R.S. Inflammatory Cytokines Shape an Altered Immune Response during Myeloid Malignancies. Front. Immunol. 2021, 12, 772408. [Google Scholar] [CrossRef] [PubMed]
- McCusker, R.H.; Kelley, K.W. Immune–Neural Connections: How the Immune System’s Response to Infectious Agents Influences Behavior. J. Exp. Biol. 2013, 216, 84–98. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, A.; Pohler, E.; Moraga, I. Molecular and Cellular Factors Determining the Functional Pleiotropy of Cytokines. FEBS J. 2023, 290, 2525–2552. [Google Scholar] [CrossRef]
- Murakami, M.; Kamimura, D.; Hirano, T. Pleiotropy and Specificity: Insights from the Interleukin 6 Family of Cytokines. Immunity 2019, 50, 812–831. [Google Scholar] [CrossRef]
- Hopkins, S.J. The Pathophysiological Role of Cytokines. Leg. Med. 2003, 5, S45–S57. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Proinflammatory Cytokines. Chest 2000, 118, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.P.; Fahmi, H. Role of Proinflammatory Cytokines in the Pathophysiology of Osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Opal, S.M.; DePalo, V.A. Anti-Inflammatory Cytokines. Chest 2000, 117, 1162–1172. [Google Scholar] [CrossRef] [PubMed]
- Schett, G. Effects of Inflammatory and Anti-inflammatory Cytokines on the Bone. Eur. J. Clin. Investig. 2011, 41, 1361–1366. [Google Scholar] [CrossRef] [PubMed]
- Bolamperti, S.; Villa, I.; Rubinacci, A. Bone Remodeling: An Operational Process Ensuring Survival and Bone Mechanical Competence. Bone Res. 2022, 10, 48. [Google Scholar] [CrossRef]
- Mulvihill, B.M.; McNamara, L.M.; Prendergast, P.J. Loss of Trabeculae by Mechano-Biological Means May Explain Rapid Bone Loss in Osteoporosis. J. R. Soc. Interface 2008, 5, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Lerner, U.H. Bone Remodeling in Post-Menopausal Osteoporosis. J. Dent. Res. 2006, 85, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Šromová, V.; Sobola, D.; Kaspar, P. A Brief Review of Bone Cell Function and Importance. Cells 2023, 12, 2576. [Google Scholar] [CrossRef] [PubMed]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef] [PubMed]
- Carswell, E.A.; Old, L.J.; Kassel, R.L.; Green, S.; Fiore, N.; Williamson, B. An Endotoxin-Induced Serum Factor That Causes Necrosis of Tumors. Proc. Natl. Acad. Sci. USA 1975, 72, 3666–3670. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Yao, Z.; Li, F.; Zhang, Q.; Badell, I.R.; Schwarz, E.M.; Takeshita, S.; Wagner, E.F.; Noda, M.; Matsuo, K.; et al. NF-κB P50 and P52 Regulate Receptor Activator of NF-κB Ligand (RANKL) and Tumor Necrosis Factor-Induced Osteoclast Precursor Differentiation by Activating c-Fos and NFATc1. J. Biol. Chem. 2007, 282, 18245–18253. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Galson, D.L.; Zhao, C.; Peng, L.; Laplace, C.; Wang, K.Z.Q.; Bachler, M.A.; Amano, H.; Aburatani, H.; Ishikawa, H.; et al. Nuclear Factor of Activated T-Cells (NFAT) Rescues Osteoclastogenesis in Precursors Lacking c-Fos. J. Biol. Chem. 2004, 279, 26475–26480. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, J.; Akhavan, N.S.; Mullins, A.P.; Arjmandi, B.H. Macrophage Polarization and Osteoporosis: A Review. Nutrients 2020, 12, 2999. [Google Scholar] [CrossRef] [PubMed]
- Parameswaran, N.; Patial, S. Tumor Necrosis Factor-α Signaling in Macrophages. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 87–103. [Google Scholar] [CrossRef]
- Faustman, D.L.; Davis, M. TNF Receptor 2 and Disease: Autoimmunity and Regenerative Medicine. Front. Immunol. 2013, 4, 478. [Google Scholar] [CrossRef]
- Osta, B.; Benedetti, G.; Miossec, P. Classical and Paradoxical Effects of TNF-α on Bone Homeostasis. Front. Immunol. 2014, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Murad, R.; Shezad, Z.; Ahmed, S.; Ashraf, M.; Qadir, M.; Rehman, R. Serum Tumour Necrosis Factor Alpha in Osteopenic and Osteoporotic Postmenopausal Females: A Cross-Sectional Study in Pakistan. J. Pak. Med. Assoc. 2018, 68, 428–431. [Google Scholar] [PubMed]
- Zha, L.; He, L.; Liang, Y.; Qin, H.; Yu, B.; Chang, L.; Xue, L. TNF-α Contributes to Postmenopausal Osteoporosis by Synergistically Promoting RANKL-Induced Osteoclast Formation. Biomed. Pharmacother. 2018, 102, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Lange, U.; Teichmann, J.; Müller-Ladner, U.; Strunk, J. Increase in Bone Mineral Density of Patients with Rheumatoid Arthritis Treated with Anti-TNF-α Antibody: A Prospective Open-Label Pilot Study. Rheumatology 2005, 44, 1546–1548. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage Plasticity, Polarization, and Function in Health and Disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Lim, D.H.; Oh, J.S.; Kim, Y.G.; Lee, C.K.; Yoo, B.; Hong, S. Effect of TNF Inhibitors on Bone Mineral Density in Rheumatoid Arthritis Patients Receiving Bisphosphonate: A Retrospective Cohort Study. Rheumatol. Int. 2020, 40, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J. TNF-mediated Inflammatory Disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef]
- Nakamura, I.; Jimi, E. Regulation of Osteoclast Differentiation and Function by Interleukin-1. In Vitamins & Hormones; Elsevier: Amsterdam, The Netherlands, 2006; pp. 357–370. [Google Scholar]
- Lee, S.K.; Gardner, A.E.; Kalinowski, J.F.; Jastrzebski, S.L.; Lorenzo, J.A. RANKL-Stimulated Osteoclast-like Cell Formation In Vitro Is Partially Dependent on Endogenous Interleukin-1 Production. Bone 2006, 38, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Kitaura, H.; Zhou, P.; Ross, F.P.; Teitelbaum, S.L. IL-1 Mediates TNF-Induced Osteoclastogenesis. J. Clin. Investig. 2005, 115, 282–290. [Google Scholar] [CrossRef]
- Tanabe, N.; Maeno, M.; Suzuki, N.; Fujisaki, K.; Tanaka, H.; Ogiso, B.; Ito, K. IL-1α Stimulates the Formation of Osteoclast-like Cells by Increasing M-CSF and PGE2 Production and Decreasing OPG Production by Osteoblasts. Life Sci. 2005, 77, 615–626. [Google Scholar] [CrossRef]
- Fischer, V.; Haffner-Luntzer, M. Interaction between Bone and Immune Cells: Implications for Postmenopausal Osteoporosis. Semin. Cell Dev. Biol. 2022, 123, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Romas, E.; Martin, T.J. Cytokines in the Pathogenesis of Osteoporosis. Osteoporos. Int. 1997, 7, 47–53. [Google Scholar] [CrossRef]
- Pacifici, R.; Rifas, L.; McCracken, R.; Vered, I.; McMurtry, C.; Avioli, L.V.; Peck, W.A. Ovarian Steroid Treatment Blocks a Postmenopausal Increase in Blood Monocyte Interleukin 1 Release. Proc. Natl. Acad. Sci. USA 1989, 86, 2398–2402. [Google Scholar] [CrossRef] [PubMed]
- Pacifici, R. Estrogen, Cytokines, and Pathogenesis of Postmenopausal Osteoporosis. J. Bone Miner. Res. 1996, 11, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Harrell, C.R.; Markovic, B.S.; Fellabaum, C.; Arsenijevic, N.; Djonov, V.; Volarevic, V. The Role of Interleukin 1 Receptor Antagonist in Mesenchymal Stem Cell-based Tissue Repair and Regeneration. BioFactors 2020, 46, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Chang, J.; Li, J.; Li, Z.; Li, Z.; Zhang, H.; Liu, Q. Protective Effects of Oridonin against Osteoporosis by Regulating Immunity and Activating the Wnt3a/β-Catenin/VEGF Pathway in Ovariectomized Mice. Int. Immunopharmacol. 2023, 118, 110011. [Google Scholar] [CrossRef] [PubMed]
- Rogers, A.; Eastell, R. Effects of Estrogen Therapy of Postmenopausal Women on Cytokines Measured in Peripheral Blood. J. Bone Miner. Res. 1998, 13, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. Interleukin-6 Family Cytokines. Cold Spring Harb. Perspect. Biol. 2018, 10, a028415. [Google Scholar] [CrossRef] [PubMed]
- Briso, E.M.; Dienz, O.; Rincon, M. Cutting Edge: Soluble IL-6R Is Produced by IL-6R Ectodomain Shedding in Activated CD4 T Cells. J. Immunol. 2008, 180, 7102–7106. [Google Scholar] [CrossRef]
- Modur, V.; Li, Y.; Zimmerman, G.A.; Prescott, S.M.; McIntyre, T.M. Retrograde Inflammatory Signaling from Neutrophils to Endothelial Cells by Soluble Interleukin-6 Receptor Alpha. J. Clin. Investig. 1997, 100, 2752–2756. [Google Scholar] [CrossRef]
- Scheller, J.; Garbers, C.; Rose-John, S. Interleukin-6: From Basic Biology to Selective Blockade of pro-Inflammatory Activities. Semin. Immunol. 2014, 26, 2–12. [Google Scholar] [CrossRef]
- De Benedetti, F.; Rucci, N.; Del Fattore, A.; Peruzzi, B.; Paro, R.; Longo, M.; Vivarelli, M.; Muratori, F.; Berni, S.; Ballanti, P.; et al. Impaired Skeletal Development in Interleukin-6–Transgenic Mice: A Model for the Impact of Chronic Inflammation on the Growing Skeletal System. Arthritis Rheum. 2006, 54, 3551–3563. [Google Scholar] [CrossRef]
- Kudo, O.; Sabokbar, A.; Pocock, A.; Itonaga, I.; Fujikawa, Y.; Athanasou, N.A. Interleukin-6 and Interleukin-11 Support Human Osteoclast Formation by a RANKL-Independent Mechanism. Bone 2003, 32, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Axmann, R.; Böhm, C.; Krönke, G.; Zwerina, J.; Smolen, J.; Schett, G. Inhibition of Interleukin-6 Receptor Directly Blocks Osteoclast Formation In Vitro and In Vivo. Arthritis Rheum. 2009, 60, 2747–2756. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Tang, S.; Ye, G.; Wang, P.; Li, J.; Liu, W.; Li, M.; Wang, S.; Wu, X.; Cen, S.; et al. Interleukin-6/Interleukin-6 Receptor Complex Promotes Osteogenic Differentiation of Bone Marrow-Derived Mesenchymal Stem Cells. Stem Cell Res. Ther. 2018, 9, 13. [Google Scholar] [CrossRef]
- Kaneshiro, S.; Ebina, K.; Shi, K.; Higuchi, C.; Hirao, M.; Okamoto, M.; Koizumi, K.; Morimoto, T.; Yoshikawa, H.; Hashimoto, J. IL-6 Negatively Regulates Osteoblast Differentiation through the SHP2/MEK2 and SHP2/Akt2 Pathways In Vitro. J. Bone Miner. Metab. 2014, 32, 378–392. [Google Scholar] [CrossRef] [PubMed]
- Joseph, C.; Kenny, A.M.; Taxel, P.; Lorenzo, J.A.; Duque, G.; Kuchel, G.A. Role of Endocrine-Immune Dysregulation in Osteoporosis, Sarcopenia, Frailty and Fracture Risk. Mol. Aspects Med. 2005, 26, 181–201. [Google Scholar] [CrossRef]
- Jilka, R.L.; Hangoc, G.; Girasole, G.; Passeri, G.; Williams, D.C.; Abrams, J.S.; Boyce, B.; Broxmeyer, H.; Manolagas, S.C. Increased Osteoclast Development after Estrogen Loss: Mediation by Interleukin-6. Science 1992, 257, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Li, H. Association of IL-6 174G/C (Rs1800795) and 572C/G (Rs1800796) Polymorphisms with Risk of Osteoporosis: A Meta-Analysis. BMC Musculoskelet. Disord. 2020, 21, 330. [Google Scholar] [CrossRef]
- Sims, N.A. Influences of the IL-6 Cytokine Family on Bone Structure and Function. Cytokine 2021, 146, 155655. [Google Scholar] [CrossRef]
- Wang, T.; He, C. TNF-α and IL-6: The Link between Immune and Bone System. Curr. Drug Targets 2020, 21, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Coates, B.A.; McKenzie, J.A.; Yoneda, S.; Silva, M.J. Interleukin-6 (IL-6) Deficiency Enhances Intramembranous Osteogenesis Following Stress Fracture in Mice. Bone 2021, 143, 115737. [Google Scholar] [CrossRef]
- Wang, S.Y.; Jiang, J.H.; Liu, S.Y.; Zhang, J.; Gao, X.; Liu, H.; Ke, K.X.; Jiang, Y.; Liu, L.; He, B.C. Interleukin 6 Promotes BMP9-Induced Osteoblastic Differentiation through Stat3/MTORC1 in Mouse Embryonic Fibroblasts. Aging 2023, 15, 718–733. [Google Scholar] [CrossRef]
- Kassem, M.; Harris, S.A.; Spelsberg, T.C.; Riggs, B.L. Estrogen Inhibits Interleukin-6 Production and Gene Expression in a Human Osteoblastic Cell Line with High Levels of Estrogen Receptors. J. Bone Miner. Res. 1996, 11, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Song, X.; Chen, X.; Jiang, R.; Peng, K.; Tang, X.; Liu, Z. Antiosteoporotic Effect of Hesperidin against Ovariectomy-induced Osteoporosis in Rats via Reduction of Oxidative Stress and Inflammation. J. Biochem. Mol. Toxicol. 2021, 35, e22832. [Google Scholar] [CrossRef]
- Tyagi, A.M.; Srivastava, K.; Mansoori, M.N.; Trivedi, R.; Chattopadhyay, N.; Singh, D. Estrogen Deficiency Induces the Differentiation of IL-17 Secreting Th17 Cells: A New Candidate in the Pathogenesis of Osteoporosis. PLoS ONE 2012, 7, e44552. [Google Scholar] [CrossRef]
- Tyagi, A.M.; Mansoori, M.N.; Srivastava, K.; Khan, M.P.; Kureel, J.; Dixit, M.; Shukla, P.; Trivedi, R.; Chattopadhyay, N.; Singh, D. Enhanced Immunoprotective Effects by Anti-IL-17 Antibody Translates to Improved Skeletal Parameters under Estrogen Deficiency Compared with Anti-RANKL and Anti-TNF-α Antibodies. J. Bone Miner. Res. 2014, 29, 1981–1992. [Google Scholar] [CrossRef]
- Ciucci, T.; Ibáñez, L.; Boucoiran, A.; Birgy-Barelli, E.; Pène, J.; Abou-Ezzi, G.; Arab, N.; Rouleau, M.; Hébuterne, X.; Yssel, H.; et al. Bone Marrow Th17 TNFα Cells Induce Osteoclast Differentiation, and Link Bone Destruction to IBD. Gut 2015, 64, 1072–1081. [Google Scholar] [CrossRef]
- Kim, Y.G.; Park, J.W.; Lee, J.M.; Suh, J.Y.; Lee, J.K.; Chang, B.S.; Um, H.S.; Kim, J.Y.; Lee, Y. IL-17 Inhibits Osteoblast Differentiation and Bone Regeneration in Rat. Arch. Oral Biol. 2014, 59, 897–905. [Google Scholar] [CrossRef]
- Weitzmann, M.N. The Role of Inflammatory Cytokines, the RANKL/OPG Axis, and the Immunoskeletal Interface in Physiological Bone Turnover and Osteoporosis. Scientifica 2013, 2013, 125705. [Google Scholar] [CrossRef]
- Srivastava, R.K.; Dar, H.Y.; Mishra, P.K. Immunoporosis: Immunology of Osteoporosis—Role of T Cells. Front. Immunol. 2018, 9, 657. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Suematsu, A.; Okamoto, K.; Yamaguchi, A.; Morishita, Y.; Kadono, Y.; Tanaka, S.; Kodama, T.; Akira, S.; Iwakura, Y.; et al. Th17 Functions as an Osteoclastogenic Helper T Cell Subset That Links T Cell Activation and Bone Destruction. J. Exp. Med. 2006, 203, 2673–2682. [Google Scholar] [CrossRef] [PubMed]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T Cell Subsets and Their Signature Cytokines in Autoimmune and Inflammatory Diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Adamopoulos, I.E.; Chao, C.; Geissler, R.; Laface, D.; Blumenschein, W.; Iwakura, Y.; McClanahan, T.; Bowman, E.P. Interleukin-17A Upregulates Receptor Activator of NF-κB on Osteoclast Precursors. Arthritis Res. Ther. 2010, 12, R29. [Google Scholar] [CrossRef]
- Cruickshank, A.M.; Fraser, W.D.; Burns, H.J.G.; Van Damme, J.; Shenkin, A. Response of Serum Interleukin-6 in Patients Undergoing Elective Surgery of Varying Severity. Clin. Sci. 1990, 79, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Kopesky, P.; Tiedemann, K.; Alkekhia, D.; Zechner, C.; Millard, B.; Schoeberl, B.; Komarova, S.V. Autocrine Signaling Is a Key Regulatory Element during Osteoclastogenesis. Biol. Open 2014, 3, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Sousa, L.H.; Linhares, E.V.M.; Alexandre, J.T.; Lisboa, M.R.; Furlaneto, F.; Freitas, R.; Ribeiro, I.; Val, D.; Marques, M.; Chaves, H.V.; et al. Effects of Atorvastatin on Periodontitis of Rats Subjected to Glucocorticoid-Induced Osteoporosis. J. Periodontol. 2016, 87, 1206–1216. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Sun, Y.; Xu, W.; Lin, T.; Zeng, H. Expression of RANKL by Peripheral Neutrophils and Its Association with Bone Mineral Density in COPD. Respirology 2017, 22, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Okamura, H.; Tsutsui, H.; Komatsu, T.; Yutsudo, M.; Hakura, A.; Tanimoto, T.; Torigoe, K.; Okura, T.; Nukada, Y.; Hattori, K.; et al. Cloning of a New Cytokine That Induces IFN-γ Production by T Cells. Nature 1995, 378, 88–91. [Google Scholar] [CrossRef]
- Kitaura, H.; Tatamiya, M.; Nagata, N.; Fujimura, Y.; Eguchi, T.; Yoshida, N.; Nakayama, K. IL-18 Induces Apoptosis of Adherent Bone Marrow Cells in TNF-α Mediated Osteoclast Formation in Synergy with IL-12. Immunol. Lett. 2006, 107, 22–31. [Google Scholar] [CrossRef]
- Morita, Y.; Kitaura, H.; Yoshimatsu, M.; Fujimura, Y.; Kohara, H.; Eguchi, T.; Yoshida, N. IL-18 Inhibits TNF-α-Induced Osteoclastogenesis Possibly via a T Cell-Independent Mechanism in Synergy with IL-12 In Vivo. Calcif. Tissue Int. 2010, 86, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Kitaura, H.; Fujimura, Y.; Yoshimatsu, M.; Kohara, H.; Morita, Y.; Aonuma, T.; Fukumoto, E.; Masuyama, R.; Yoshida, N.; Takano-Yamamoto, T. IL-12- and IL-18-Mediated, Nitric Oxide-Induced Apoptosis in TNF-α-Mediated Osteoclastogenesis of Bone Marrow Cells. Calcif. Tissue Int. 2011, 89, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Horwood, N.J.; Udagawa, N.; Elliott, J.; Grail, D.; Okamura, H.; Kurimoto, M.; Dunn, A.R.; Martin, T.; Gillespie, M.T. Interleukin 18 Inhibits Osteoclast Formation via T Cell Production of Granulocyte Macrophage Colony-Stimulating Factor. J. Clin. Investig. 1998, 101, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, M.N.; Shukla, P.; Kakaji, M.; Tyagi, A.M.; Srivastava, K.; Shukla, M.; Dixit, M.; Kureel, J.; Gupta, S.; Singh, D. IL-18BP Is Decreased in Osteoporotic Women: Prevents Inflammasome Mediated IL-18 Activation and Reduces Th17 Differentiation. Sci. Rep. 2016, 6, 33680. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, N.; Horwood, N.J.; Elliott, J.; Mackay, A.; Owens, J.; Okamura, H.; Kurimoto, M.; Chambers, T.J.; Martin, T.J.; Gillespie, M.T. Interleukin-18 (Interferon-γ–Inducing Factor) Is Produced by Osteoblasts and Acts Via Granulocyte/Macrophage Colony-Stimulating Factor and Not Via Interferon-γ to Inhibit Osteoclast Formation. J. Exp. Med. 1997, 185, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.H.; Yang, M.Y. The Role of Macrophage in the Pathogenesis of Osteoporosis. Int. J. Mol. Sci. 2019, 20, 2093. [Google Scholar] [CrossRef] [PubMed]
- Yun, T.J.; Chaudhary, P.M.; Shu, G.L.; Frazer, J.K.; Ewings, M.K.; Schwartz, S.M.; Pascual, V.; Hood, L.E.; Clark3, E.A. OPG/FDCR-1, a TNF Receptor Family Member, Is Expressed in Lymphoid Cells and Is Up-Regulated by Ligating CD40. J. Immunol. 1998, 161, 6113–6121. [Google Scholar] [CrossRef] [PubMed]
- Amarasekara, D.S.; Yun, H.; Kim, S.; Lee, N.; Kim, H.; Rho, J. Regulation of Osteoclast Differentiation by Cytokine Networks. Immune Netw. 2018, 18, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, H.; Mazure, R.; Gonzalez, D.; Flores, D.; Pedreira, S.; Niveloni, S.; Smecuol, E.; Maurino, E.; Bai, J.C. Risk of Fractures in Celiac Disease Patients: A Cross-Sectional, Case-Control Study. Am. J. Gastroenterol. 2000, 95, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Collin, P.; Kaukinen, K.; Välimäki, M.; Salmi, J. Endocrinological Disorders and Celiac Disease. Endocr. Rev. 2002, 23, 464–483. [Google Scholar] [CrossRef]
- Clowes, J.A.; Riggs, B.L.; Khosla, S. The Role of the Immune System in the Pathophysiology of Osteoporosis. Immunol. Rev. 2005, 208, 207–227. [Google Scholar] [CrossRef] [PubMed]
- Taranta, A.; Fortunati, D.; Longo, M.; Rucci, N.; Iacomino, E.; Aliberti, F.; Facciuto, E.; Migliaccio, S.; Bardella, M.T.; Dubini, A.; et al. Imbalance of Osteoclastogenesis-Regulating Factors in Patients with Celiac Disease. J. Bone Miner. Res. 2004, 19, 1112–1121. [Google Scholar] [CrossRef]
- Ju, J.H.; Cho, M.L.; Moon, Y.M.; Oh, H.J.; Park, J.S.; Jhun, J.Y.; Min, S.Y.; Cho, Y.G.; Park, K.S.; Yoon, C.H.; et al. IL-23 Induces Receptor Activator of NF-κB Ligand Expression on CD4+ T Cells and Promotes Osteoclastogenesis in an Autoimmune Arthritis Model. J. Immunol. 2008, 181, 1507–1518. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.K.; Kang, Y.M.; Han, S. Osteoclasts in the Inflammatory Arthritis: Implications for Pathologic Osteolysis. Immune Netw. 2019, 19, e2. [Google Scholar] [CrossRef]
- Sims, N.A.; Green, J.R.; Glatt, M.; Schlict, S.; Martin, T.J.; Gillespie, M.T.; Romas, E. Targeting Osteoclasts with Zoledronic Acid Prevents Bone Destruction in Collagen-induced Arthritis. Arthritis Rheum. 2004, 50, 2338–2346. [Google Scholar] [CrossRef]
- Shukla, P.; Mansoori, M.N.; Kakaji, M.; Shukla, M.; Gupta, S.K.; Singh, D. Interleukin 27 (IL-27) Alleviates Bone Loss in Estrogen-Deficient Conditions by Induction of Early Growth Response-2 Gene. J. Biol. Chem. 2017, 292, 4686–4699. [Google Scholar] [CrossRef]
- De Martinis, M.; Sirufo, M.M.; Suppa, M.; Ginaldi, L. IL-33/IL-31 Axis in Osteoporosis. Int. J. Mol. Sci. 2020, 21, 1239. [Google Scholar] [CrossRef]
- Molofsky, A.B.; Savage, A.K.; Locksley, R.M. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity 2015, 42, 1005–1019. [Google Scholar] [CrossRef]
- Pichery, M.; Mirey, E.; Mercier, P.; Lefrancais, E.; Dujardin, A.; Ortega, N.; Girard, J.-P. Endogenous IL-33 Is Highly Expressed in Mouse Epithelial Barrier Tissues, Lymphoid Organs, Brain, Embryos, and Inflamed Tissues: In Situ Analysis Using a Novel Il-33–LacZ Gene Trap Reporter Strain. J. Immunol. 2012, 188, 3488–3495. [Google Scholar] [CrossRef] [PubMed]
- Palmer, G.; Lipsky, B.P.; Smithgall, M.D.; Meininger, D.; Siu, S.; Talabot-Ayer, D.; Gabay, C.; Smith, D.E. The IL-1 Receptor Accessory Protein (AcP) Is Required for IL-33 Signaling and Soluble AcP Enhances the Ability of Soluble ST2 to Inhibit IL-33. Cytokine 2008, 42, 358–364. [Google Scholar] [CrossRef]
- Kiyomiya, H.; Ariyoshi, W.; Okinaga, T.; Kaneuji, T.; Mitsugi, S.; Sakurai, T.; Habu, M.; Yoshioka, I.; Tominaga, K.; Nishihara, T. IL-33 Inhibits RANKL-Induced Osteoclast Formation through the Regulation of Blimp-1 and IRF-8 Expression. Biochem. Biophys. Res. Commun. 2015, 460, 320–326. [Google Scholar] [CrossRef] [PubMed]
- De Martinis, M.; Ginaldi, L.; Sirufo, M.M.; Bassino, E.M.; De Pietro, F.; Pioggia, G.; Gangemi, S. IL-33/Vitamin D Crosstalk in Psoriasis-Associated Osteoporosis. Front. Immunol. 2021, 11, 604055. [Google Scholar] [CrossRef] [PubMed]
- Neill, D.R.; Wong, S.H.; Bellosi, A.; Flynn, R.J.; Daly, M.; Langford, T.K.A.; Bucks, C.; Kane, C.M.; Fallon, P.G.; Pannell, R.; et al. Nuocytes Represent a New Innate Effector Leukocyte That Mediates Type-2 Immunity. Nature 2010, 464, 1367–1370. [Google Scholar] [CrossRef] [PubMed]
- Önel, A.U.; Yildirim, M. Sitokinler Ve Kanatlilarda Sitokinlerin Aşi Adjuvanti Olarak Kullanimi. Vet. Farmakoloji Ve Toksikoloji Derneği Bülteni 2021, 12, 21–32. [Google Scholar] [CrossRef]
- Van Vlasselaer, P.; Borremans, B.; Van Den Heuvel, R.; Van Gorp, U.; de Waal Malefyt, R. Interleukin-10 Inhibits the Osteogenic Activity of Mouse Bone Marrow. Blood 1993, 82, 2361–2370. [Google Scholar] [CrossRef] [PubMed]
- Sobieski, M.A.; Graham, J.D.; Pappas, P.S.; Tatooles, A.J.; Slaughter, M.S. Reducing the Effects of the Systemic Inflammatory Response to Cardiopulmonary Bypass: Can Single Dose Steroids Blunt Systemic Inflammatory Response Syndrome? ASAIO J. 2008, 54, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Whitlock, R.P.; Young, E.; Noora, J.; Farrokhyar, F.; Blackall, M.; Teoh, K.H. Pulse Low Dose Steroids Attenuate Post-Cardiopulmonary Bypass SIRS.; SIRS I. J. Surg. Res. 2006, 132, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Nelms, K.; Keegan, A.D.; Zamorano, J.; Ryan, J.J.; Paul, W.E. THE IL-4 RECEPTOR: Signaling Mechanisms and Biologic Functions. Annu. Rev. Immunol. 1999, 17, 701–738. [Google Scholar] [CrossRef] [PubMed]
- Stein, N.C.; Kreutzmann, C.; Zimmermann, S.P.; Niebergall, U.; Hellmeyer, L.; Goettsch, C.; Schoppet, M.; Hofbauer, L.C. Interleukin-4 and Interleukin-13 Stimulate the Osteoclast Inhibitor Osteoprotegerin by Human Endothelial Cells through the STAT6 Pathway. J. Bone Miner. Res. 2008, 23, 750–758. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Shukla, M.; Yakubenko, V.P.; Mulya, A.; Kundu, S.; Cathcart, M.K. IL-4 and IL-13 Employ Discrete Signaling Pathways for Target Gene Expression in Alternatively Activated Monocytes/Macrophages. Free Radic. Biol. Med. 2013, 54, 1–16. [Google Scholar] [CrossRef]
- Palmqvist, P.; Lundberg, P.; Persson, E.; Johansson, A.; Lundgren, I.; Lie, A.; Conaway, H.H.; Lerner, U.H. Inhibition of Hormone and Cytokine-Stimulated Osteoclastogenesis and Bone Resorption by Interleukin-4 and Interleukin-13 Is Associated with Increased Osteoprotegerin and Decreased RANKL and RANK in a STAT6-Dependent Pathway. J. Biol. Chem. 2006, 281, 2414–2429. [Google Scholar] [CrossRef]
- Lind, M.; Deleuran, B.; Yssel, H.; Fink-Eriksen, E.; Thestrup-Pedersen, K. IL-4 and IL-13, but Not IL-10, Are Chemotactic Factors for Human Osteoblasts. Cytokine 1995, 7, 78–82. [Google Scholar] [CrossRef]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of Effector CD4 T Cell Populations. Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef]
- Srivastava, R.K. Osteoimmunology The i Nexus i between Bone and Immune System. Front. Biosci. 2018, 23, 4600. [Google Scholar] [CrossRef]
- Ren, W.; Liu, G.; Chen, S.; Yin, J.; Wang, J.; Tan, B.; Wu, G.; Bazer, F.W.; Peng, Y.; Li, T.; et al. Melatonin Signaling in T Cells: Functions and Applications. J. Pineal Res. 2017, 62, e12394. [Google Scholar] [CrossRef]
- Petra, A.I.; Tsilioni, I.; Taracanova, A.; Katsarou-Katsari, A.; Theoharides, T.C. Interleukin 33 and Interleukin 4 Regulate Interleukin 31 Gene Expression and Secretion from Human Laboratory of Allergic Diseases 2 Mast Cells Stimulated by Substance P and/or Immunoglobulin E. Allergy Asthma Proc. 2018, 39, 153–160. [Google Scholar] [CrossRef]
- Silfverswärd, C.; Frost, A.; Brändström, H.; Nilsson, O.; Ljunggren, Ö. Interleukin-4 and Interleukin-13 Potentiate Interleukin-1 Induced Secretion of Interleukin-6 in Human Osteoblast-like Cells. J. Orthop. Res. 2004, 22, 1058–1062. [Google Scholar] [CrossRef]
- Harmer, D.; Falank, C.; Reagan, M.R. Interleukin-6 Interweaves the Bone Marrow Microenvironment, Bone Loss, and Multiple Myeloma. Front. Endocrinol. 2019, 9, 788. [Google Scholar] [CrossRef]
- Sun, W.; Meednu, N.; Rosenberg, A.; Rangel-Moreno, J.; Wang, V.; Glanzman, J.; Owen, T.; Zhou, X.; Zhang, H.; Boyce, B.F.; et al. B Cells Inhibit Bone Formation in Rheumatoid Arthritis by Suppressing Osteoblast Differentiation. Nat. Commun. 2018, 9, 5127. [Google Scholar] [CrossRef]
- Socha, L.A.; Gowardman, J.; Silva, D.; Correcha, M.; Petrosky, N. Elevation in Interleukin 13 Levels in Patients Diagnosed with Systemic Inflammatory Response Syndrome. Intensive Care Med. 2006, 32, 244–250. [Google Scholar] [CrossRef]
- Pacifici, R. T Cells: Critical Bone Regulators in Health and Disease. Bone 2010, 47, 461–471. [Google Scholar] [CrossRef]
- Peng, M.; Wang, Y.; Qiang, L.; Xu, Y.; Li, C.; Li, T.; Zhou, X.; Xiao, M.; Wang, J. Interleukin-35 Inhibits TNF-α-Induced Osteoclastogenesis and Promotes Apoptosis via Shifting the Activation from TNF Receptor-Associated Death Domain (TRADD)–TRAF2 to TRADD–Fas-Associated Death Domain by JAK1/STAT1. Front. Immunol. 2018, 9, 1417. [Google Scholar] [CrossRef]
- Li, J.; Chen, X.; Lu, L.; Yu, X. The Relationship between Bone Marrow Adipose Tissue and Bone Metabolism in Postmenopausal Osteoporosis. Cytokine Growth Factor. Rev. 2020, 52, 88–98. [Google Scholar] [CrossRef]
- Tencerova, M.; Kassem, M. The Bone Marrow-Derived Stromal Cells: Commitment and Regulation of Adipogenesis. Front. Endocrinol. 2016, 7, 127. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Lu, J. Interleukin-35 Promote Osteogenesis and Inhibit Adipogenesis: Role of Wnt/β-Catenin and PPARγ Signaling Pathways. Inflammation 2023, 46, 522–533. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Y.; Yuan, L.; Yao, L.; Yang, J.; Xia, L.; Shen, H.; Lu, J. Interleukin-35 Is Involved in Angiogenesis/Bone Remodeling Coupling through T Helper 17/Interleukin-17 Axis. Front. Endocrinol. 2021, 12, 642676. [Google Scholar] [CrossRef]
- Li, Y.; Yuan, L.; Jiang, S.; Liu, S.; Xia, L.; Shen, H.; Lu, J. Interleukin-35 Stimulates Tumor Necrosis Factor-α Activated Osteoblasts Differentiation through Wnt/β-Catenin Signaling Pathway in Rheumatoid Arthritis. Int. Immunopharmacol. 2019, 75, 105810. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Lu, J. Interleukin-35 Controls the Balance between Osteogenic and Adipogenic Differentiation of Progenitor Cells. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Cho, K.A.; Lee, J.K.; Kim, Y.H.; Park, M.; Woo, S.Y.; Ryu, K.H. Mesenchymal Stem Cells Ameliorate B-Cell-Mediated Immune Responses and Increase IL-10-Expressing Regulatory B Cells in an EBI3-Dependent Manner. Cell Mol. Immunol. 2017, 14, 895–908. [Google Scholar] [CrossRef]
- Marahleh, A.; Kitaura, H.; Ohori, F.; Noguchi, T.; Nara, Y.; Pramusita, A.; Kinjo, R.; Ma, J.; Kanou, K.; Mizoguchi, I. Effect of TNF-α on Osteocyte RANKL Expression during Orthodontic Tooth Movement. J. Dent. Sci. 2021, 16, 1191–1197. [Google Scholar] [CrossRef]
- Ohori, F.; Kitaura, H.; Marahleh, A.; Kishikawa, A.; Ogawa, S.; Qi, J.; Shen, W.-R.; Noguchi, T.; Nara, Y.; Mizoguchi, I. Effect of TNF- α -Induced Sclerostin on Osteocytes during Orthodontic Tooth Movement. J. Immunol. Res. 2019, 2019, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Tang, Y.; Wu, Q.; Ji, Y.; Feng, Z.; Kang, F. HIF-1α Facilitates Osteocyte-mediated Osteoclastogenesis by Activating JAK2/STAT3 Pathway In Vitro. J. Cell Physiol. 2019, 234, 21182–21192. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zhao, J.; Qiu, M.; Zhang, L.; Yang, K.; Chang, L.; Jia, P.; Qi, J.; Deng, L.; Li, C. Osteocytic HIF-1α Pathway Manipulates Bone Micro-Structure and Remodeling via Regulating Osteocyte Terminal Differentiation. Front. Cell Dev. Biol. 2022, 9, 721561. [Google Scholar] [CrossRef]
- Stegen, S.; Stockmans, I.; Moermans, K.; Thienpont, B.; Maxwell, P.H.; Carmeliet, P.; Carmeliet, G. Osteocytic Oxygen Sensing Controls Bone Mass through Epigenetic Regulation of Sclerostin. Nat. Commun. 2018, 9, 2557. [Google Scholar] [CrossRef]
- Bakker, A.D.; da Silva, V.C.; Krishnan, R.; Bacabac, R.G.; Blaauboer, M.E.; Lin, Y.C.; Marcantonio, R.A.C.; Cirelli, J.A.; Klein-Nulend, J. Tumor Necrosis Factor α and Interleukin-1β Modulate Calcium and Nitric Oxide Signaling in Mechanically Stimulated Osteocytes. Arthritis Rheum. 2009, 60, 3336–3345. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Jin, L.; Jiang, C.; Yan, Z.; Cao, Y. IL-1β Contributes to the Secretion of Sclerostin by Osteocytes and Targeting Sclerostin Promotes Spinal Fusion at Early Stages. J. Orthop. Surg. Res. 2023, 18, 162. [Google Scholar] [CrossRef]
- Pathak, J.L.; Bravenboer, N.; Luyten, F.P.; Verschueren, P.; Lems, W.F.; Klein-Nulend, J.; Bakker, A.D. Mechanical Loading Reduces Inflammation-Induced Human Osteocyte-to-Osteoclast Communication. Calcif. Tissue Int. 2015, 97, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Zhang, C.; Jin, L.; Yang, Y. IL-17 Alters the Mesenchymal Stem Cell Niche towards Osteogenesis in Cooperation with Osteocytes. J. Cell Physiol. 2020, 235, 4466–4480. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, Z.; Pan, S.; Feng, Y.; He, H.; Cheng, S.; Wang, L.; Wang, L.; Pathak, J.L. Resveratrol Alleviates Diabetic Periodontitis-Induced Alveolar Osteocyte Ferroptosis Possibly via Regulation of SLC7A11/GPX4. Nutrients 2023, 15, 2115. [Google Scholar] [CrossRef]
- Al Rifai, O.; Susan-Resiga, D.; Essalmani, R.; Creemers, J.W.M.; Seidah, N.G.; Ferron, M. In Vivo Analysis of the Contribution of Proprotein Convertases to the Processing of FGF23. Front. Endocrinol. 2021, 12, 690681. [Google Scholar] [CrossRef]
- Yin, J.; Hao, Z.; Ma, Y.; Liao, S.; Li, X.; Fu, J.; Wu, Y.; Shen, J.; Zhang, P.; Li, X.; et al. Concomitant Activation of the PI3K/Akt and ERK1/2 Signalling Is Involved in Cyclic Compressive Force-induced IL-6 Secretion in MLO-Y4 Cells. Cell Biol. Int. 2014, 38, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Bakker, A.D.; Kulkarni, R.N.; Klein-Nulend, J.; Lems, W.F. IL-6 Alters Osteocyte Signaling toward Osteoblasts but Not Osteoclasts. J. Dent. Res. 2014, 93, 394–399. [Google Scholar] [CrossRef]
- Takagi, R.; Sakamoto, E.; Kido, J.; Inagaki, Y.; Hiroshima, Y.; Naruishi, K.; Yumoto, H. S100A9 Increases IL-6 and RANKL Expressions through MAPKs and STAT3 Signaling Pathways in Osteocyte-Like Cells. Biomed Res. Int. 2020, 2020, 7149408. [Google Scholar] [CrossRef] [PubMed]
- Gardinier, J.D.; Chougule, A.; Zhang, C. The Mechanotransduction of MLO-Y4 Cells Is Disrupted by the Senescence-associated Secretory Phenotype of Neighboring Cells. J. Cell Physiol. 2022, 237, 2249–2257. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Ma, Y.; Li, X.; Wu, X.; Liu, W.; Li, X.; Shen, J.; Wang, H. Lipopolysaccharide Increases IL-6 Secretion via Activation of the ERK1/2 Signaling Pathway to Up-regulate RANKL Gene Expression in MLO-Y4 Cells. Cell Biol. Int. 2017, 41, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.X.; Kukita, T.; Kukita, A.; Otsuka, T.; Niho, Y.; Iijima, T. Interleukin-10 Selectively Inhibits Osteoclastogenesis by Inhibiting Differentiation of Osteoclast Progenitors into Preosteoclast-like Cells in Rat Bone Marrow Culture System. J. Cell Physiol. 1995, 165, 624–629. [Google Scholar] [CrossRef]
- Liu, D.; Yao, S.; Wise, G.E. Effect of Interleukin-10 on Gene Expression of Osteoclastogenic Regulatory Molecules in the Rat Dental Follicle. Eur. J. Oral. Sci. 2006, 114, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Pacifici, R. The Role of IL-17 and TH17 Cells in the Bone Catabolic Activity of PTH. Front. Immunol. 2016, 7, 57. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Yu, M.; Tyagi, A.M.; Vaccaro, C.; Hsu, E.; Adams, J.; Bellido, T.; Weitzmann, M.N.; Pacifici, R. IL-17 Receptor Signaling in Osteoblasts/Osteocytes Mediates PTH-Induced Bone Loss and Enhances Osteocytic RANKL Production. J. Bone Miner. Res. 2019, 34, 349–360. [Google Scholar] [CrossRef]
- Liao, C.; Cheng, T.; Wang, S.; Zhang, C.; Jin, L.; Yang, Y. Shear Stress Inhibits IL-17A-Mediated Induction of Osteoclastogenesis via Osteocyte Pathways. Bone 2017, 101, 10–20. [Google Scholar] [CrossRef]
- Noguchi, S.; Yamasaki, R.; Nagai-Yoshioka, Y.; Sato, T.; Kuroishi, K.; Gunjigake, K.; Ariyoshi, W.; Kawamoto, T. The Mechanism of Interleukin 33-Induced Stimulation of Interleukin 6 in MLO-Y4 Cells. Int. J. Mol. Sci. 2023, 24, 14842. [Google Scholar] [CrossRef] [PubMed]
- Mun, S.H.; Ko, N.Y.; Kim, H.S.; Kim, J.W.; Kim, D.K.; Kim, A.-R.; Lee, S.H.; Kim, Y.G.; Lee, C.K.; Lee, S.H.; et al. Interleukin-33 Stimulates Formation of Functional Osteoclasts from Human CD14+ Monocytes. Cell. Mol. Life Sci. 2010, 67, 3883–3892. [Google Scholar] [CrossRef]
- Dr. Gonda, K.; Nakaoka, T.; Yoshimura, K.; Otawara-Hamamoto, Y.; Harrii, K. Heterotopic Ossification of Degenerating Rat Skeletal Muscle Induced by Adenovirus-Mediated Transfer of Bone Morphogenetic Protein-2 Gene. J. Bone Miner. Res. 2000, 15, 1056–1065. [Google Scholar] [CrossRef] [PubMed]
- Tshamala, M.; van Bree, H. Osteoinductive Properties of the Bone Marrow Myth or Reality. Vet. Comp. Orthop. Traumatol. 2006, 19, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Devescovi, V.; Leonardi, E.; Ciapetti, G.; Cenni, E. Growth Factors in Bone Repair. Chir. Organi Mov. 2008, 92, 161–168. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, T.L.; Centrella, M. Regulation of IGF Activity in Bone. In Current Directions in Insulin-Like Growth Factor Research; Springer: Berlin/Heidelberg, Germany, 1994; pp. 407–414. [Google Scholar]
- Bichell, D.P.; Rotwein, P.; McCarthy, T.L. Prostaglandin E2 Rapidly Stimulates Insulin-like Growth Factor-I Gene Expression in Primary Rat Osteoblast Cultures: Evidence for Transcriptional Control. Endocrinology 1993, 133, 1020–1028. [Google Scholar] [CrossRef] [PubMed]
- Locklin, R.M.; Khosla, S.; Turner, R.T.; Riggs, B.L. Mediators of the Biphasic Responses of Bone to Intermittent and Continuously Administered Parathyroid Hormone. J. Cell Biochem. 2003, 89, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Canalis, E.; Deregowski, V.; Pereira, R.C.; Gazzerro, E. Signals That Determine the Fate of Osteoblastic Cells. J. Endocrinol. Investig. 2005, 28, 3–7. [Google Scholar]
- Niu, T.; Rosen, C.J. The Insulin-like Growth Factor-I Gene and Osteoporosis: A Critical Appraisal. Gene 2005, 361, 38–56. [Google Scholar] [CrossRef]
- Clevers, H. Wnt/β-Catenin Signaling in Development and Disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef]
- Kassem, M.; Blum, W.; Ristelli, J.; Mosekilde, L.; Eriksen, E.F. Growth Hormone Stimulates Proliferation and Differentiation of Normal Human Osteoblast-like Cells In Vitro. Calcif. Tissue Int. 1993, 52, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.; Riggs, B.L.; Melton, L.J.; Achenbach, S.J.; Atkinson, E.J.; Khosla, S. High Serum IGFBP-2 Is Predictive of Increased Bone Turnover in Aging Men and Women. J. Bone Miner. Res. 2007, 22, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Choi, M.; Kim, L. The Relationship among Circulating Insulin-like Growth Factor Components, Biochemical Markers of Bone Turnover and Bone Mineral Density in Postmenopausal Women under the Age of 60. Clin. Endocrinol. 1999, 51, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Ormarsdóttir, S.; Ljunggren, Ö.; Mallmin, H.; Olofsson, H.; Blum, W.F.; Lööf, L. Circulating Levels of Insulin-like Growth Factors and Their Binding Proteins in Patients with Chronic Liver Disease: Lack of Correlation with Bone Mineral Density. Liver 2001, 21, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.; Riggs, B.L.; Atkinson, E.J.; Oberg, A.L.; Melton, L.J.; Khosla, S. A Potentially Deleterious Role of IGFBP-2 on Bone Density in Aging Men and Women. J. Bone Miner. Res. 2004, 19, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Riggs, B.L.; Melton, L.J.; Robb, R.A.; Camp, J.J.; Atkinson, E.J.; McDaniel, L.; Amin, S.; Rouleau, P.A.; Khosla, S. A Population-Based Assessment of Rates of Bone Loss at Multiple Skeletal Sites: Evidence for Substantial Trabecular Bone Loss in Young Adult Women and Men. J. Bone Miner. Res. 2008, 23, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, H.; Hakeda, Y.; Wakatsuki, N.; Usui, N.; Akashi, S.; Sato, T.; Tanaka, K.; Kumegawa, M. Insulin-like Growth Factor-I Supports Formation and Activation of Osteoclasts. Endocrinology 1992, 131, 1075–1080. [Google Scholar] [CrossRef] [PubMed]
- Rubin, J.; Ackert-Bicknell, C.L.; Zhu, L.; Fan, X.; Murphy, T.C.; Nanes, M.S.; Marcus, R.; Holloway, L.; Beamer, W.G.; Rosen, C.J. IGF-I Regulates Osteoprotegerin (OPG) and Receptor Activator of Nuclear Factor-κB Ligand In Vitro and OPG In Vivo. J. Clin. Endocrinol. Metab. 2002, 87, 4273–4279. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.A.; Coker, R. Molecules in Focus Transforming Growth Factor-Beta (TGF-β). Int. J. Biochem. Cell Biol. 1998, 30, 293–298. [Google Scholar] [CrossRef]
- Gaba, S.; Jain, U. Advanced Biosensors for Nanomaterial-Based Detection of Transforming Growth Factor Alpha and Beta, a Class of Major Polypeptide Regulators. Int. J. Biol. Macromol. 2024, 257, 128622. [Google Scholar] [CrossRef]
- Ichioka, N.; Inaba, M.; Kushida, T.; Esumi, T.; Takahara, K.; Inaba, K.; Ogawa, R.; Iida, H.; Ikehara, S. Prevention of Senile Osteoporosis in SAMP6 Mice by Intrabone Marrow Injection of Allogeneic Bone Marrow Cells. Stem Cells 2002, 20, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Poniatowski, Ł.A.; Wojdasiewicz, P.; Gasik, R.; Szukiewicz, D. Transforming Growth Factor Beta Family: Insight into the Role of Growth Factors in Regulation of Fracture Healing Biology and Potential Clinical Applications. Mediat. Inflamm. 2015, 2015, 137823. [Google Scholar] [CrossRef] [PubMed]
- Thompson, N.L.; Flanders, K.C.; Smith, J.M.; Ellingsworth, L.R.; Roberts, A.B.; Sporn, M.B. Expression of Transforming Growth Factor-Beta 1 in Specific Cells and Tissues of Adult and Neonatal Mice. J. Cell Biol. 1989, 108, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Bonewald, L.F.; Dallas, S.L. Role of Active and Latent Transforming Growth Factor β in Bone Formation. J. Cell Biochem. 1994, 55, 350–357. [Google Scholar] [CrossRef]
- Maeda, S.; Hayashi, M.; Komiya, S.; Imamura, T.; Miyazono, K. Endogenous TGF-β Signaling Suppresses Maturation of Osteoblastic Mesenchymal Cells. EMBO J. 2004, 23, 552–563. [Google Scholar] [CrossRef]
- Zhen, G.; Wen, C.; Jia, X.; Li, Y.; Crane, J.L.; Mears, S.C.; Askin, F.B.; Frassica, F.J.; Chang, W.; Yao, J.; et al. Inhibition of TGF-β Signaling in Mesenchymal Stem Cells of Subchondral Bone Attenuates Osteoarthritis. Nat. Med. 2013, 19, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Myint, O.; Sakunrangsit, N.; Pholtaisong, J.; Toejing, P.; Pho-on, P.; Leelahavanichkul, A.; Sridurongrit, S.; Aporntewan, C.; Greenblatt, M.B.; Lotinun, S. Differential Gene Expression Involved in Bone Turnover of Mice Expressing Constitutively Active TGFβ Receptor Type I. Int. J. Mol. Sci. 2024, 25, 5829. [Google Scholar] [CrossRef] [PubMed]
- Geiser, A.; Zeng, Q.; Sato, M.; Helvering, L.; Hirano, T.; Turner, C. Decreased Bone Mass and Bone Elasticity in Mice Lacking the Transforming Growth Factor-Β1 Gene. Bone 1998, 23, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, N.; Kobayashi, T.; Mochida, Y.; Yu, P.B.; Yamauchi, M.; Kronenberg, H.M.; Mishina, Y. Wnt Inhibitors Dkk1 and Sost Are Downstream Targets of BMP Signaling through the Type IA Receptor (BMPRIA) in Osteoblasts. J. Bone Miner. Res. 2010, 25, 200–210. [Google Scholar] [CrossRef]
- Kamiya, N.; Ye, L.; Kobayashi, T.; Lucas, D.J.; Mochida, Y.; Yamauchi, M.; Kronenberg, H.M.; Feng, J.Q.; Mishina, Y. Disruption of BMP Signaling in Osteoblasts through Type IA Receptor (BMPRIA) Increases Bone Mass. J. Bone Miner. Res. 2008, 23, 2007–2017. [Google Scholar] [CrossRef]
- Kamiya, N.; Shuxian, L.; Yamaguchi, R.; Phipps, M.; Aruwajoye, O.; Adapala, N.S.; Yuan, H.; Kim, H.K.W.; Feng, J.Q. Targeted Disruption of BMP Signaling through Type IA Receptor (BMPR1A) in Osteocyte Suppresses SOST and RANKL, Leading to Dramatic Increase in Bone Mass, Bone Mineral Density and Mechanical Strength. Bone 2016, 91, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Novais, A.; Chatzopoulou, E.; Chaussain, C.; Gorin, C. The Potential of FGF-2 in Craniofacial Bone Tissue Engineering: A Review. Cells 2021, 10, 932. [Google Scholar] [CrossRef] [PubMed]
- Choksi, P.; Jepsen, K.J.; Clines, G.A. The Challenges of Diagnosing Osteoporosis and the Limitations of Currently Available Tools. Clin. Diabetes Endocrinol. 2018, 4, 12. [Google Scholar] [CrossRef]
- Ramli, F.F.; Chin, K.Y. A Review of the Potential Application of Osteocyte-Related Biomarkers, Fibroblast Growth Factor-23, Sclerostin, and Dickkopf-1 in Predicting Osteoporosis and Fractures. Diagnostics 2020, 10, 145. [Google Scholar] [CrossRef] [PubMed]
- Montero, A.; Okada, Y.; Tomita, M.; Ito, M.; Tsurukami, H.; Nakamura, T.; Doetschman, T.; Coffin, J.D.; Hurley, M.M. Disruption of the Fibroblast Growth Factor-2 Gene Results in Decreased Bone Mass and Bone Formation. J. Clin. Investig. 2000, 105, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Z.; Zhang, J.; Yuan, D.L.; Xie, W.Q.; Ladel, C.H.; Mobasheri, A.; Li, Y.S. Role of Signaling Pathways in Age-Related Orthopedic Diseases: Focus on the Fibroblast Growth Factor Family. Mil. Med. Res. 2024, 11, 40. [Google Scholar] [CrossRef]
- Coffin, J.D.; Homer-Bouthiette, C.; Hurley, M.M. Fibroblast Growth Factor 2 and Its Receptors in Bone Biology and Disease. J. Endocr. Soc. 2018, 2, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Jilka, R.L. Molecular and Cellular Mechanisms of the Anabolic Effect of Intermittent PTH. Bone 2007, 40, 1434–1446. [Google Scholar] [CrossRef]
- Jelenik, T.; Dille, M.; Müller-Lühlhoff, S.; Kabra, D.G.; Zhou, Z.; Binsch, C.; Hartwig, S.; Lehr, S.; Chadt, A.; Peters, E.M.J.; et al. FGF21 Regulates Insulin Sensitivity Following Long-Term Chronic Stress. Mol. Metab. 2018, 16, 126–138. [Google Scholar] [CrossRef]
- Zhao, Y.X.; Song, Y.W.; Zhang, L.; Zheng, F.J.; Wang, X.M.; Zhuang, X.H.; Wu, F.; Liu, J. Association between Bile Acid Metabolism and Bone Mineral Density in Postmenopausal Women. Clinics 2020, 75, e1486. [Google Scholar] [CrossRef]
- Kir, S.; Beddow, S.A.; Samuel, V.T.; Miller, P.; Previs, S.F.; Suino-Powell, K.; Xu, H.E.; Shulman, G.I.; Kliewer, S.A.; Mangelsdorf, D.J. FGF19 as a Postprandial, Insulin-Independent Activator of Hepatic Protein and Glycogen Synthesis. Science 2011, 331, 1621–1624. [Google Scholar] [CrossRef]
- Guridi, M.; Tintignac, L.A.; Lin, S.; Kupr, B.; Castets, P.; Rüegg, M.A. Activation of MTORC1 in Skeletal Muscle Regulates Whole-Body Metabolism through FGF21. Sci. Signal. 2015, 8, ra113. [Google Scholar] [CrossRef]
- Kim, A.M.; Somayaji, V.R.; Dong, J.Q.; Rolph, T.P.; Weng, Y.; Chabot, J.R.; Gropp, K.E.; Talukdar, S.; Calle, R.A. Once-weekly Administration of a Long-acting Fibroblast Growth Factor 21 Analogue Modulates Lipids, Bone Turnover Markers, Blood Pressure and Body Weight Differently in Obese People with Hypertriglyceridaemia and in Non-human Primates. Diabetes Obes. Metab. 2017, 19, 1762–1772. [Google Scholar] [CrossRef]
- Charoenphandhu, N.; Suntornsaratoon, P.; Krishnamra, N.; Sa-nguanmoo, P.; Tanajak, P.; Wang, X.; Liang, G.; Li, X.; Jiang, C.; Chattipakorn, N.; et al. Fibroblast Growth Factor-21 Restores Insulin Sensitivity but Induces Aberrant Bone Microstructure in Obese Insulin-Resistant Rats. J. Bone Miner. Metab. 2017, 35, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.T.; Hsu, B.G.; Wang, C.H.; Lin, Y.L.; Lai, Y.H.; Kuo, C.H. Lower Serum Fibroblast Growth Factor 21 Levels Are Associated with Normal Lumbar Spine Bone Mineral Density in Hemodialysis Patients. Int. J. Environ. Res. Public. Health 2020, 17, 1938. [Google Scholar] [CrossRef]
- Hao, R.H.; Gao, J.L.; Li, M.; Huang, W.; Zhu, D.L.; Thynn, H.N.; Dong, S.S.; Guo, Y. Association between Fibroblast Growth Factor 21 and Bone Mineral Density in Adults. Endocrine 2018, 59, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; He, J.; Fu, W.; Wang, C.; Yue, H.; Gu, J.; Zhang, H.; Zhang, Z. Fibroblast Growth Factor 21 Is Associated with Bone Mineral Density, but Not with Bone Turnover Markers and Fractures in Chinese Postmenopausal Women. J. Clin. Densitom. 2019, 22, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Lui, D.T.W.; Lee, C.H.; Chau, V.W.K.; Fong, C.H.Y.; Yeung, K.M.Y.; Lam, J.K.Y.; Lee, A.C.H.; Chow, W.S.; Tan, K.C.B.; Woo, Y.C.; et al. Potential Role of Fibroblast Growth Factor 21 in the Deterioration of Bone Quality in Impaired Glucose Tolerance. J. Endocrinol. Investig. 2021, 44, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Freitas, I.; Owen, B.M. Metabolic Roles of Endocrine Fibroblast Growth Factors. Curr. Opin. Pharmacol. 2015, 25, 30–35. [Google Scholar] [CrossRef]
- Ewendt, F.; Feger, M.; Föller, M. Role of Fibroblast Growth Factor 23 (FGF23) and AKlotho in Cancer. Front. Cell Dev. Biol. 2021, 8, 601006. [Google Scholar] [CrossRef]
- Sirikul, W.; Siri-Angkul, N.; Chattipakorn, N.; Chattipakorn, S.C. Fibroblast Growth Factor 23 and Osteoporosis: Evidence from Bench to Bedside. Int. J. Mol. Sci. 2022, 23, 2500. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; He, X.; Olauson, H.; Larsson, T.E.; Lindgren, U. FGF23 Affects the Lineage Fate Determination of Mesenchymal Stem Cells. Calcif. Tissue Int. 2013, 93, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Coulson, J.; Bagley, L.; Barnouin, Y.; Bradburn, S.; Butler-Browne, G.; Gapeyeva, H.; Hogrel, J.Y.; Maden-Wilkinson, T.; Maier, A.B.; Meskers, C.; et al. Circulating Levels of Dickkopf-1, Osteoprotegerin and Sclerostin Are Higher in Old Compared with Young Men and Women and Positively Associated with Whole-Body Bone Mineral Density in Older Adults. Osteoporos. Int. 2017, 28, 2683–2689. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Cai, X.; Lee, J.; Katz, R.; Cauley, J.A.; Fried, L.F.; Hoofnagle, A.N.; Satterfield, S.; Harris, T.B.; Shlipak, M.G.; et al. Associations of FGF23 with Change in Bone Mineral Density and Fracture Risk in Older Individuals. J. Bone Miner. Res. 2016, 31, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Kalem, M.N.; Kalem, Z.; Akgun, N.; Bakırarar, B. The Relationship between Postmenopausal Women’s Sclerostin Levels and Their Bone Density, Age, Body Mass Index, Hormonal Status, and Smoking and Consumption of Coffee and Dairy Products. Arch. Gynecol. Obstet. 2017, 295, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Imel, E.A.; Liu, Z.; McQueen, A.K.; Acton, D.; Acton, A.; Padgett, L.R.; Peacock, M.; Econs, M.J. Serum Fibroblast Growth Factor 23, Serum Iron and Bone Mineral Density in Premenopausal Women. Bone 2016, 86, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Bai, X.; Han, L.; Sun, X.; Chen, X. The Relationship between Serum Fibroblast Growth Factor 23, Klotho, and Lumbar Spine Bone Mineral Density in Northern Chinese Postmenopausal Women. Menopause 2019, 26, 546–553. [Google Scholar] [CrossRef]
- Celik, E.; Guzel, S.; Abali, R.; Guzelant, A.Y.; Celik Guzel, E.; Kuçukyalcin, V. The Relationship between Fibroblast Growth Factor 23 and Osteoporosis in Postmenopausal Women. Minerva Med. 2013, 104, 497–504. [Google Scholar] [PubMed]
- Lane, N.E.; Parimi, N.; Corr, M.; Yao, W.; Cauley, J.A.; Nielson, C.M.; Ix, J.H.; Kado, D.; Orwoll, E. Association of Serum Fibroblast Growth Factor 23 (FGF23) and Incident Fractures in Older Men: The Osteoporotic Fractures in Men (MrOS) Study. J. Bone Miner. Res. 2013, 28, 2325–2332. [Google Scholar] [CrossRef]
- Mirza, M.A.; Karlsson, M.K.; Mellström, D.; Orwoll, E.; Ohlsson, C.; Ljunggren, Ö.; Larsson, T.E. Serum Fibroblast Growth Factor-23 (FGF-23) and Fracture Risk in Elderly Men. J. Bone Miner. Res. 2011, 26, 857–864. [Google Scholar] [CrossRef]
- Cruz-Lopez, O.; Conejo-Garcia, A.; C Nunez, M.; Kimatrai, M.; E. Garcia-Rubino, M.; Morales, F.; Gomez-Perez, V.M.; Campos, J. Novel Substituted Quinazolines for Potent EGFR Tyrosine Kinase Inhibitors. Curr. Med. Chem. 2011, 18, 943–963. [Google Scholar] [CrossRef]
- De Luca, A.; Gallo, M.; Aldinucci, D.; Ribatti, D.; Lamura, L.; D’Alessio, A.; De Filippi, R.; Pinto, A.; Normanno, N. Role of the EGFR Ligand/Receptor System in the Secretion of Angiogenic Factors in Mesenchymal Stem Cells. J. Cell Physiol. 2011, 226, 2131–2138. [Google Scholar] [CrossRef] [PubMed]
- Ebert, R.; Benisch, P.; Krug, M.; Zeck, S.; Meißner-Weigl, J.; Steinert, A.; Rauner, M.; Hofbauer, L.; Jakob, F. Acute Phase Serum Amyloid A Induces Proinflammatory Cytokines and Mineralization via Toll-like Receptor 4 in Mesenchymal Stem Cells. Stem Cell Res. 2015, 15, 231–239. [Google Scholar] [CrossRef]
- Eccles, S.A. The Epidermal Growth Factor Receptor/Erb-B/HER Family in Normal and Malignant Breast Biology. Int. J. Dev. Biol. 2011, 55, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Müller-Deubert, S.; Seefried, L.; Krug, M.; Jakob, F.; Ebert, R. Epidermal Growth Factor as a Mechanosensitizer in Human Bone Marrow Stromal Cells. Stem Cell Res. 2017, 24, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Holfeld, J.; Schaden, W.; Orgill, D.; Ogawa, R. Mechanotherapy: Revisiting Physical Therapy and Recruiting Mechanobiology for a New Era in Medicine. Trends Mol. Med. 2013, 19, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and Extracellular Matrix Homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Iskratsch, T.; Wolfenson, H.; Sheetz, M.P. Appreciating Force and Shape—The Rise of Mechanotransduction in Cell Biology. Nat. Rev. Mol. Cell Biol. 2014, 15, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Kerpedjieva, S.S.; Kim, D.S.; Barbeau, D.J.; Tamama, K. EGFR Ligands Drive Multipotential Stromal Cells to Produce Multiple Growth Factors and Cytokines via Early Growth Response-1. Stem. Cells Dev. 2012, 21, 2541–2551. [Google Scholar] [CrossRef]
- Kara, F.; Yildirim, A.; Gumusdere, M.; Karatay, S.; Yildirim, K.; Bakan, E. Association between Hepatocyte Growth Factor (HGF) Gene Polymorphisms and Serum HGF Levels in Patients with Rheumatoid Arthritis. Eurasian J. Med. 2014, 46, 176–185. [Google Scholar] [CrossRef]
- Ikebuchi, F.; Oka, K.; Mizuno, S.; Fukuta, K.; Hayata, D.; Ohnishi, H.; Nakamura, T. Dissociation of C-Met Phosphotyrosine Sites in Human Cells in Response to Mouse Hepatocyte Growth Factor but Not Human Hepatocyte Growth Factor: The Possible Roles of Different Amino Acids in Different Species. Cell Biochem. Funct. 2013, 31, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zheng, Y.; Bai, J.; Shi, C.; Shi, X.; Shan, H.; Zhou, X. Hepatocyte Growth Factor Overexpression Promotes Osteoclastogenesis and Exacerbates Bone Loss in CIA Mice. J. Orthop. Translat 2021, 27, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Aenlle, K.K.; Curtis, K.M.; Roos, B.A.; Howard, G.A. Hepatocyte Growth Factor and P38 Promote Osteogenic Differentiation of Human Mesenchymal Stem Cells. Mol. Endocrinol. 2014, 28, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Grano, M.; Galimi, F.; Zambonin, G.; Colucci, S.; Cottone, E.; Zallone, A.Z.; Comoglio, P.M. Hepatocyte Growth Factor Is a Coupling Factor for Osteoclasts and Osteoblasts In Vitro. Proc. Natl. Acad. Sci. USA 1996, 93, 7644–7648. [Google Scholar] [CrossRef] [PubMed]
- Sensebé, L.; Bourin, P. Mesenchymal Stem Cells for Therapeutic Purposes. Transplantation 2009, 87, S49–S53. [Google Scholar] [CrossRef] [PubMed]
- Levrero, M.; De Laurenzi, V.; Costanzo, A.; Sabatini, S.; Gong, J.; Wang, J.Y.J.; Melino, G. The P53/P63/P73 Family of Transcription Factors: Overlapping and Distinct Functions. J. Cell Sci. 2000, 113, 1661–1670. [Google Scholar] [CrossRef]
- Bourdon, J.C. P53 Family Isoforms. Curr. Pharm. Biotechnol. 2007, 8, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Aenlle, K.K.; Curtis, K.M.; Roos, B.A.; Howard, G.A. Hepatocyte Growth Factor (HGF) and 1,25-Dihydroxyvitamin D Together Stimulate Human Bone Marrow-Derived Stem Cells toward the Osteogenic Phenotype by HGF-Induced up-Regulation of VDR. Bone 2012, 51, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Cross, M.J.; Dixelius, J.; Matsumoto, T.; Claesson-Welsh, L. VEGF-Receptor Signal Transduction. Trends Biochem. Sci. 2003, 28, 488–494. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The Biology of VEGF and Its Receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Sleeman, J.P. Lymphangiogenesis Reviews the Relationship between Tumors and the Lymphatics: What More Is There to Know? Lymphology 2006, 39, 62. [Google Scholar]
- Yonemura, Y.; Endo, Y.; Tabata, K.; Kawamura, T.; Yun, H.Y.; Bandou, E.; Sasaki, T.; Miura, M. Role of VEGF-C and VEGF-D in Lymphangiogenesis in Gastric Cancer. Int. J. Clin. Oncol. 2005, 10, 318–327. [Google Scholar] [CrossRef]
- Thi, M.M.; Suadicani, S.O.; Spray, D.C. Fluid Flow-Induced Soluble Vascular Endothelial Growth Factor Isoforms Regulate Actin Adaptation in Osteoblasts. J. Biol. Chem. 2010, 285, 30931–30941. [Google Scholar] [CrossRef]
- Nakai, T.; Yoshimura, Y.; Deyama, Y.; Suzuki, K.; Iida, J. Mechanical Stress Up-Regulates RANKL Expression via the VEGF Autocrine Pathway in Osteoblastic MC3T3-E1 Cells. Mol. Med. Rep. 2009, 2, 229–234. [Google Scholar] [CrossRef]
- Jung, Y.D.; Liu, W.; Reinmuth, N.; Ahmad, S.A.; Fan, F.; Gallick, G.E.; Ellis, L.M. Vascular Endothelial Growth Factor Is Upregulated by Interleukin-1β in Human Vascular Smooth Muscle Cells via the P38 Mitogen-Activated Protein Kinase Pathway. Angiogenesis 2001, 4, 155–162. [Google Scholar] [CrossRef]
- Deckers, M.M.L.; Karperien, M.; van der Bent, C.; Yamashita, T.; Papapoulos, S.E.; Löwik, C.W.G.M. Expression of Vascular Endothelial Growth Factors and Their Receptors during Osteoblast Differentiation. Endocrinology 2000, 141, 1667–1674. [Google Scholar] [CrossRef]
- Yeh, L.C.C.; Lee, J.C. Osteogenic Protein-1 Increases Gene Expression of Vascular Endothelial Growth Factor in Primary Cultures of Fetal Rat Calvaria Cells. Mol. Cell Endocrinol. 1999, 153, 113–124. [Google Scholar] [CrossRef]
- Saadeh, P.B.; Mehrara, B.J.; Steinbrech, D.S.; Spector, J.A.; Greenwald, J.A.; Chin, G.S.; Ueno, H.; Gittes, G.K.; Longaker, M.T. Mechanisms of Fibroblast Growth Factor-2 Modulation of Vascular Endothelial Growth Factor Expression by Osteoblastic Cells. Endocrinology 2000, 141, 2075–2083. [Google Scholar] [CrossRef]
- Harada, S.; Nagy, J.A.; Sullivan, K.A.; Thomas, K.A.; Endo, N.; Rodan, G.A.; Rodan, S.B. Induction of Vascular Endothelial Growth Factor Expression by Prostaglandin E2 and E1 in Osteoblasts. J. Clin. Investig. 1994, 93, 2490–2496. [Google Scholar] [CrossRef]
- Kodama, H.; Yamasaki, A.; Nose, M.; Niida, S.; Ohgame, Y.; Abe, M.; Kumegawa, M.; Suda, T. Congenital Osteoclast Deficiency in Osteopetrotic (Op/Op) Mice Is Cured by Injections of Macrophage Colony-Stimulating Factor. J. Exp. Med. 1991, 173, 269–272. [Google Scholar] [CrossRef]
- Niida, S.; Kaku, M.; Amano, H.; Yoshida, H.; Kataoka, H.; Nishikawa, S.; Tanne, K.; Maeda, N.; Nishikawa, S.-I.; Kodama, H. Vascular Endothelial Growth Factor Can Substitute for Macrophage Colony-Stimulating Factor in the Support of Osteoclastic Bone Resorption. J. Exp. Med. 1999, 190, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Mayr-wohlfart, U.; Waltenberger, J.; Hausser, H.; Kessler, S.; Günther, K.P.; Dehio, C.; Puhl, W.; Brenner, R.E. Vascular Endothelial Growth Factor Stimulates Chemotactic Migration of Primary Human Osteoblasts. Bone 2002, 30, 472–477. [Google Scholar] [CrossRef]
- Tan, Y.; Yang, Y.Q.; Chai, L.; Wong, R.; Rabie, A. Effects of Vascular Endothelial Growth Factor (VEGF) on MC3T3-E1. Orthod. Craniofac. Res. 2010, 13, 223–228. [Google Scholar] [CrossRef]
- Mayer, H.; Bertram, H.; Lindenmaier, W.; Korff, T.; Weber, H.; Weich, H. Vascular Endothelial Growth Factor (VEGF-A) Expression in Human Mesenchymal Stem Cells: Autocrine and Paracrine Role on Osteoblastic and Endothelial Differentiation. J. Cell Biochem. 2005, 95, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Street, J.; Lenehan, B. Vascular Endothelial Growth Factor Regulates Osteoblast Survival—Evidence for an Autocrine Feedback Mechanism. J. Orthop. Surg. Res. 2009, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I.; Correa, D. PDGF in Bone Formation and Regeneration: New Insights into a Novel Mechanism Involving MSCs. J. Orthop. Res. 2011, 29, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- Albanese, A.; Licata, M.E.; Polizzi, B.; Campisi, G. Platelet-Rich Plasma (PRP) in Dental and Oral Surgery: From the Wound Healing to Bone Regeneration. Immun. Ageing 2013, 10, 23. [Google Scholar] [CrossRef]
- Greenberg, J.I.; Shields, D.J.; Barillas, S.G.; Acevedo, L.M.; Murphy, E.; Huang, J.; Scheppke, L.; Stockmann, C.; Johnson, R.S.; Angle, N.; et al. A Role for VEGF as a Negative Regulator of Pericyte Function and Vessel Maturation. Nature 2008, 456, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Olsen, B.R. The Roles of Vascular Endothelial Growth Factor in Bone Repair and Regeneration. Bone 2016, 91, 30–38. [Google Scholar] [CrossRef]
- Claesson-Welsh, L.; Heldin, C.H. Platelet-Derived Growth Factor Three Isoforms That Bind to Two Distinct Cell Surface Receptors. Acta Oncol. 1989, 28, 331–334. [Google Scholar] [CrossRef]
- Heldin, C.H.; Westermark, B. Mechanism of Action and In Vivo Role of Platelet-Derived Growth Factor. Physiol. Rev. 1999, 79, 1283–1316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, J.; Jin, D. Platelet-Derived Growth Factor (PDGF)-BB Stimulates Osteoclastic Bone Resorption Directly: The Role of Receptor β. Biochem. Biophys. Res. Commun. 1998, 251, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Leeman, E.; Carnes, D.C.; Graves, D.T. Human Osteoblasts Synthesize and Respond to Platelet-Derived Growth Factor. Am. J. Physiol. Cell Physiol. 1991, 261, C348–C354. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.; Leonidou, A.; Lester, M.; Heliotis, M.; Mantalaris, A.; Tsiridis, E. Investigating the Role of PDGF as a Potential Drug Therapy in Bone Formation and Fracture Healing. Expert Opin. Investig. Drugs 2009, 18, 1633–1654. [Google Scholar] [CrossRef] [PubMed]
- Hollinger, J.O.; Hart, C.E.; Hirsch, S.N.; Lynch, S.; Friedlaender, G.E. Recombinant Human Platelet-Derived Growth Factor: Biology and Clinical Applications. J. Bone Jt. Surg. 2008, 90, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.C.; Ehrlich, M.G.; McAllister, S.C.; Machan, J.T.; Hart, C.E.; Voigt, C.; Lesieur-Brooks, A.M.; Weber, E.W. Recombinant Human Platelet-Derived Growth Factor-BB Augmentation of New-Bone Formation in a Rat Model of Distraction Osteogenesis. J. Bone Jt. Surg. Am. Vol. 2009, 91, 1973–1984. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Cui, Z.; Wang, L.; Xia, Z.; Hu, Y.; Xian, L.; Li, C.; Xie, L.; Crane, J.; Wan, M.; et al. PDGF-BB Secreted by Preosteoclasts Induces Angiogenesis during Coupling with Osteogenesis. Nat. Med. 2014, 20, 1270–1278. [Google Scholar] [CrossRef]
- Chung, R.; Foster, B.K.; Zannettino, A.C.W.; Xian, C.J. Potential Roles of Growth Factor PDGF-BB in the Bony Repair of Injured Growth Plate. Bone 2009, 44, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Galibert, L.; Tometsko, M.E.; Anderson, D.M.; Cosman, D.; Dougall, W.C. The Involvement of Multiple Tumor Necrosis Factor Receptor (TNFR)-Associated Factors in the Signaling Mechanisms of Receptor Activator of NF-κB, a Member of the TNFR Superfamily. J. Biol. Chem. 1998, 273, 34120–34127. [Google Scholar] [CrossRef]
- Grigoriadis, A.E.; Wang, Z.-Q.; Cecchini, M.G.; Hofstetter, W.; Felix, R.; Fleisch, H.A.; Wagner, E.F. C-Fos: A Key Regulator of Osteoclast-Macrophage Lineage Determination and Bone Remodeling. Science 1994, 266, 443–448. [Google Scholar] [CrossRef]
- Mensah, K.A.; Ritchlin, C.T.; Schwarz, E.M. RANKL Induces Heterogeneous DC-STAMP lo and DC-STAMP hi Osteoclast Precursors of Which the DC-STAMP lo Precursors Are the Master Fusogens. J. Cell Physiol. 2010, 223, 76–83. [Google Scholar] [CrossRef]
- Kim, K.; Lee, J.; Kim, J.H.; Jin, H.M.; Zhou, B.; Lee, S.Y.; Kim, N. Protein Inhibitor of Activated STAT 3 Modulates Osteoclastogenesis by Down-Regulation of NFATc1 and Osteoclast-Associated Receptor. J. Immunol. 2007, 178, 5588–5594. [Google Scholar] [CrossRef] [PubMed]
- Tsubaki, M.; Komai, M.; Itoh, T.; Imano, M.; Sakamoto, K.; Shimaoka, H.; Takeda, T.; Ogawa, N.; Mashimo, K.; Fujiwara, D.; et al. Nitrogen-Containing Bisphosphonates Inhibit RANKL- and M-CSF-Induced Osteoclast Formation through the Inhibition of ERK1/2 and Akt Activation. J. Biomed. Sci. 2014, 21, 10. [Google Scholar] [CrossRef] [PubMed]
- Levi-Montalcini, R.; Angeletti, P.U. Essential Role of the Nerve Growth Factor in the Survival and Maintenance of Dissociated Sensory and Sympathetic Embryonic Nerve Cells In Vitro. Dev. Biol. 1963, 7, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Nepomuceno, T.; De Gregoriis, G.; De Oliveira, F.M.B.; Suarez-Kurtz, G.; Monteiro, A.; Carvalho, M. The Role of PALB2 in the DNA Damage Response and Cancer Predisposition. Int. J. Mol. Sci. 2017, 18, 1886. [Google Scholar] [CrossRef]
- Keselman, A.; Heller, N. Estrogen Signaling Modulates Allergic Inflammation and Contributes to Sex Differences in Asthma. Front. Immunol. 2015, 6, 568. [Google Scholar] [CrossRef] [PubMed]
- Katzer, K.; Hill, J.L.; McIver, K.B.; Foster, M.T. Lipedema and the Potential Role of Estrogen in Excessive Adipose Tissue Accumulation. Int. J. Mol. Sci. 2021, 22, 11720. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, E.; Floras, J.S.; Harvey, P.J. Estrogen Status and the Renin Angiotensin Aldosterone System. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R498–R500. [Google Scholar] [CrossRef]
- Molnar, I. Nerve Growth Factor and Its Role in Immuno-Inflammatory and Endocrine Metabolic Diseases. HOJ Emerg. Intern. Med. 2023, 1, 1–8. [Google Scholar]
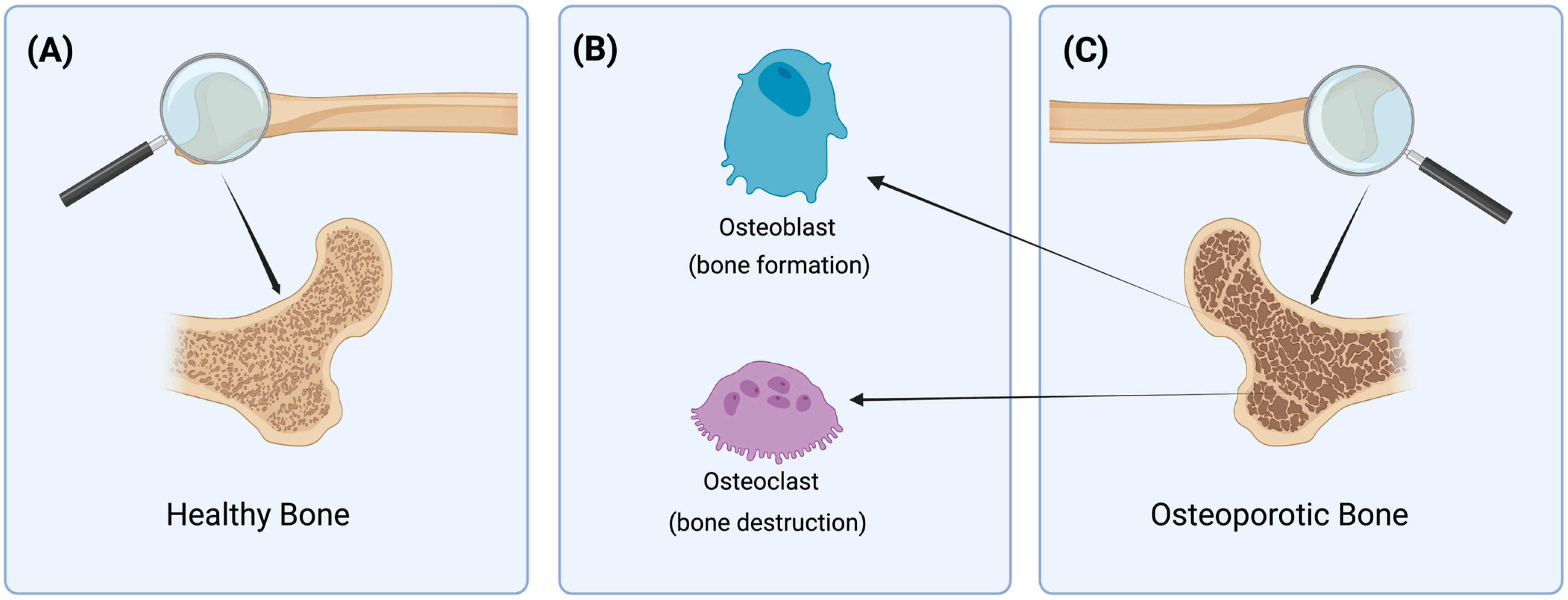


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Umur, E.; Bulut, S.B.; Yiğit, P.; Bayrak, E.; Arkan, Y.; Arslan, F.; Baysoy, E.; Kaleli-Can, G.; Ayan, B. Exploring the Role of Hormones and Cytokines in Osteoporosis Development. Biomedicines 2024, 12, 1830. https://doi.org/10.3390/biomedicines12081830
Umur E, Bulut SB, Yiğit P, Bayrak E, Arkan Y, Arslan F, Baysoy E, Kaleli-Can G, Ayan B. Exploring the Role of Hormones and Cytokines in Osteoporosis Development. Biomedicines. 2024; 12(8):1830. https://doi.org/10.3390/biomedicines12081830
Chicago/Turabian StyleUmur, Egemen, Safiye Betül Bulut, Pelin Yiğit, Emirhan Bayrak, Yaren Arkan, Fahriye Arslan, Engin Baysoy, Gizem Kaleli-Can, and Bugra Ayan. 2024. "Exploring the Role of Hormones and Cytokines in Osteoporosis Development" Biomedicines 12, no. 8: 1830. https://doi.org/10.3390/biomedicines12081830