
Variability of Mitochondrial DNA Heteroplasmy: Association with Asymptomatic Carotid Atherosclerosis

 ,
,  , , , ,
, , , ,  and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Study Participants
2.2. Ultrasonography of Carotid Arteries
2.3. MtDNA Heteroplasmy Measurements
- for mutations m.5178C>A, m.652delG, and m.652insG—60 °C;
- for mutations m.12315G>A, m.14459G>A, and m.1555A>G—50 °C;
- for mutations m.14846G>A, m.13513G>A, m.15059G>A, and m.3256C>T—55 °C.
2.4. Statistics
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics-2017 Update: A Report from the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zeng, Y.; Yang, H.; Hu, Y.; Hu, Y.; Chen, W.; Ying, Z.; Sun, Y.; Qu, Y.; Li, Q.; et al. Familial factors, diet, and risk of cardiovascular disease: A cohort analysis of the UK Biobank. Am. J. Clin. Nutr. 2021, 114, 1837–1846. [Google Scholar] [CrossRef] [PubMed]
- Pieters, M.; Ferreira, M.; de Maat, M.P.M.; Ricci, C. Biomarker association with cardiovascular disease and mortality—The role of fibrinogen. A report from the NHANES study. Thromb. Res. 2021, 198, 182–189. [Google Scholar] [CrossRef]
- Bansal, M.; Ranjan, S.; Kasliwal, R.R. Cardiovascular Risk Calculators and their Applicability to South Asians. Curr. Diabetes Rev. 2021, 17, e100120186497. [Google Scholar] [CrossRef]
- Speed, S.; Sun, Z.; Liu, Z. A focus group study of older Chinese people with CVD patients in the North West of the UK. Prim. Heal. Care Res. Dev. 2021, 22, e24. [Google Scholar] [CrossRef]
- Jebari-Benslaiman, S.; Galicia-García, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martín, C. Pathophysiology of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3346. [Google Scholar] [CrossRef]
- Sobenin, I.A.; Zhelankin, A.V.; Mitrofanov, K.Y.; Sinyov, V.V.; Sazonova, M.A.; Postnov, A.Y.; Orekhov, A.N. Mutations of Mitochondrial DNA in Atherosclerosis and Atherosclerosis-Related Diseases. Curr. Pharm. Des. 2015, 21, 1158–1163. [Google Scholar] [CrossRef]
- Markina, Y.V.; Kirichenko, T.V.; Tolstik, T.V.; Bogatyreva, A.I.; Zotova, U.S.; Cherednichenko, V.R.; Postnov, A.Y.; Markin, A.M. Target and Cell Therapy for Atherosclerosis and CVD. Int. J. Mol. Sci. 2023, 24, 10308. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Kawakami, R.; Sakamoto, A.; Cornelissen, A.; Mori, M.; Kawai, K.; Ghosh, S.; Romero, M.E.; Kolodgie, F.D.; Finn, A.V.; et al. Sex Differences in Coronary Atherosclerosis. Curr. Atheroscler. Rep. 2022, 24, 23–32. [Google Scholar] [CrossRef]
- Gupta, K.; Balachandran, I.; Foy, J.; Hermel, M.; Latif, A.; Krittanawong, C.; Slipczuk, L.; Baloch, F.; Samad, Z.; Virani, S.S. Highlights of Cardiovascular Disease Prevention Studies Presented at the 2023 American College of Cardiology Conference. Curr. Atheroscler. Rep. 2023, 25, 309–321. [Google Scholar] [CrossRef]
- Jabczyk, M.; Nowak, J.; Hudzik, B.; Zubelewicz-Szkodzińska, B. Curcumin in Metabolic Health and Disease. Nutrients 2021, 13, 4440. [Google Scholar] [CrossRef] [PubMed]
- Akodad, M.; Sicard, P.; Fauconnier, J.; Roubille, F. Colchicine and myocardial infarction: A review. Arch. Cardiovasc. Dis. 2020, 113, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ma, X.; Ilyas, I.; Zheng, X.; Luo, S.; Little, P.J.; Kamato, D.; Sahebkar, A.; Wu, W.; Weng, J.; et al. Impact of sodium glucose cotransporter 2 (SGLT2) inhibitors on atherosclerosis: From pharmacology to pre-clinical and clinical therapeutics. Theranostics 2021, 11, 4502–4515. [Google Scholar] [CrossRef] [PubMed]
- Gluba-Brzózka, A.; Franczyk, B.; Rysz-Górzyńska, M.; Ławiński, J.; Rysz, J. Emerging Anti-Atherosclerotic Therapies. Int. J. Mol. Sci. 2021, 22, 12109. [Google Scholar] [CrossRef] [PubMed]
- Huang, P. Proanthocyanidins may be potential therapeutic agents for the treatment of carotid atherosclerosis: A review. J. Int. Med Res. 2023, 51, 3000605231167314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Chen, J.; Tang, X.; Luo, Q.; Xu, D.; Yu, B. Interaction between adipocytes and high-density lipoprotein: New insights into the mechanism of obesity-induced dyslipidemia and atherosclerosis. Lipids Heal. Dis. 2019, 18, 223. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.S.; Ray, K.K.; Raal, F.J.; Kallend, D.G.; Jaros, M.; Koenig, W.; Leiter, L.A.; Landmesser, U.; Schwartz, G.G.; Friedman, A.; et al. ORION Phase III Investigators. Pooled Patient-Level Analysis of Inclisiran Trials in Patients With Familial Hypercholesterolemia or Atherosclerosis. J. Am. Coll. Cardiol. 2021, 77, 1182–1193. [Google Scholar] [CrossRef] [PubMed]
- Doroschuk, N.A.; Postnov, A.Y.; Doroschuk, A.D.; Ryzhkova, A.I.; Sinyov, V.V.; Sazonova, M.D.; Khotina, V.A.; Orekhov, A.N.; Sobenin, I.A.; Sazonova, M.A. An original biomarker for the risk of developing cardiovascular diseases and their complications: Telomere length. Toxicol. Rep. 2021, 8, 499–504. [Google Scholar] [CrossRef]
- Hu, H.; Garcia-Barrio, M.; Jiang, Z.S.; Chen, Y.E.; Chang, L. Roles of Perivascular Adipose Tissue in Hypertension and Atherosclerosis. Antioxid. Redox Signal. 2021, 34, 736–749. [Google Scholar] [CrossRef]
- den Hartigh, L.J. Conjugated Linoleic Acid Effects on Cancer, Obesity, and Atherosclerosis: A Review of Pre-Clinical and Human Trials with Current Perspectives. Nutrients 2019, 11, 370. [Google Scholar] [CrossRef] [PubMed]
- Dabravolski, S.A.; Khotina, V.A.; Sukhorukov, V.N.; Kalmykov, V.A.; Mikhaleva, L.M.; Orekhov, A.N. The Role of Mitochondrial DNA Mutations in Cardiovascular Diseases. Int. J. Mol. Sci. 2022, 23, 952. [Google Scholar] [CrossRef] [PubMed]
- Sazonova, M.A.; Ryzhkova, A.I.; Sinyov, V.V.; Galitsyna, E.V.; Melnichenko, A.A.; Demakova, N.A.; Sobenin, I.A.; Shkurat, T.P.; Orekhov, A.N. Mitochondrial Genome Mutations Associated with Myocardial Infarction. Dis. Markers 2018, 2018, 9749457. [Google Scholar] [CrossRef]
- Sinyov, V.V.; Sazonova, M.A.; Ryzhkova, A.I.; Galitsyna, E.V.; Melnichenko, A.A.; Postnov, A.Y.; Orekhov, A.N.; Grechko, A.V.; Sobenin, I.A. Potential use of buccal epithelium for genetic diagnosis of atherosclerosis using mtDNA mutations. Vessel Plus 2017, 1, 145–150. [Google Scholar] [CrossRef]
- Ivanova, M.M.; Borodachev, E.N.; Sazonova, M.A. Human pathologies associated with mutations of mitochondrial genome. In Patologicheskaia Fiziologiia i Eksperimental’naia Terapiia; Izdatelstvo Meditsina: Moscow, Russia, 2012; Volume 3, pp. 115–122. (In Russian) [Google Scholar]
- Sazonova, M.A.; Ryzhkova, A.I.; Sinyov, V.V.; Sazonova, M.D.; Khasanova, Z.B.; Nikitina, N.A.; Karagodin, V.P.; Orekhov, A.N.; Sobenin, I.A. Creation of Cultures Containing Mutations Linked with Cardiovascular Diseases using Transfection and Genome Editing. Curr. Pharm. Des. 2019, 25, 693–699. [Google Scholar] [CrossRef]
- Cai, H.; Liu, Y.; Men, H.; Zheng, Y. Protective Mechanism of Humanin Against Oxidative Stress in Aging-Related Cardiovascular Diseases. Front. Endocrinol. 2021, 12, 683151. [Google Scholar] [CrossRef]
- Daiber, A.; Hahad, O.; Andreadou, I.; Steven, S.; Daub, S.; Münzel, T. Redox-related biomarkers in human cardiovascular disease—Classical footprints and beyond. Redox Biol. 2021, 42, 101875. [Google Scholar] [CrossRef]
- Dominic, E.A.; Ramezani, A.; Anker, S.D.; Verma, M.; Mehta, N.; Rao, M. Mitochondrial cytopathies and cardiovascular disease. Heart 2014, 100, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Lin, Y.; Xu, X.; Lin, S.; Chen, X.; Wang, S. The alterations of mitochondrial DNA in coronary heart disease. Exp. Mol. Pathol. 2020, 114, 104412. [Google Scholar] [CrossRef]
- Dominic, A.; Banerjee, P.; Hamilton, D.J.; Le, N.T.; Abe, J.I. Time-dependent replicative senescence vs. disturbed flow-induced pre-mature aging in atherosclerosis. Redox Biol. 2020, 37, 101614. [Google Scholar] [CrossRef] [PubMed]
- Filograna, R.; Mennuni, M.; Alsina, D.; Larsson, N.G. Mitochondrial DNA copy number in human disease: The more the better? FEBS Lett. 2021, 595, 976–1002. [Google Scholar] [CrossRef]
- O’Hara, R.; Tedone, E.; Ludlow, A.; Huang, E.; Arosio, B.; Mari, D.; Shay, J.W. Quantitative mitochondrial DNA copy number determination using droplet digital PCR with single-cell resolution. Genome Res. 2019, 29, 1878–1888. [Google Scholar] [CrossRef] [PubMed]
- Zhelankin, A.V.; Sazonova, M.A. Association of the mutations in the human mitochondrial genome with chronic non-inflammatory diseases: Type 2 diabetes, hypertension and different types of cardiomyopathy. Patol. Fiziol. Eksp. Ter. 2012, 3, 123–128. (In Russian) [Google Scholar] [PubMed]
- Picard, M. Blood mitochondrial DNA copy number: What are we counting? Mitochondrion 2021, 60, 1–11. [Google Scholar] [CrossRef]
- Pillalamarri, V.; Shi, W.; Say, C.; Yang, S.; Lane, J.; Guallar, E.; Pankratz, N.; Arking, D.E. Whole-exome sequencing in 415,422 individuals identifies rare variants associated with mitochondrial DNA copy number. HGG Adv. 2022, 4, 100147. [Google Scholar] [CrossRef] [PubMed]
- McNally, E.M.; Golbus, J.R.; Puckelwartz, M.J. Genetic mutations and mechanisms in dilated cardiomyopathy. J. Clin. Investig. 2013, 123, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Sazonova, M.A.; Shkurat, T.P.; Demakova, N.A.; Zhelankin, A.V.; Barinova, V.A.; Sobenin, I.A.; Orekhov, A.N. Mitochondrial genome sequencing in atherosclerosis: What’s next? Curr. Pharm. Des. 2016, 22, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.C.; Tseng, L.M.; Lee, H.C. Role of mitochondrial dysfunction in cancer progression. Exp. Biol. Med. 2016, 241, 1281–1295. [Google Scholar] [CrossRef]
- Novoselov, V.V.; Sazonova, M.A.; Ivanova, E.A.; Orekhov, A.N. Study of the activated macrophage transcriptome. Exp. Mol. Pathol. 2015, 99, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.A.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E268–E285. [Google Scholar] [CrossRef]
- Sazonova, M.A.; Sinyov, V.V.; Ryzhkova, A.I.; Galitsyna, E.V.; Khasanova, Z.B.; Postnov, A.Y.; Yarygina, E.I.; Orekhov, A.N.; Sobenin, I.A. Role of Mitochondrial Genome Mutations in Pathogenesis of Carotid Atherosclerosis. Oxid. Med. Cell. Longev. 2017, 2017, 6934394. [Google Scholar] [CrossRef]
- Sazonova, M.A.; Chicheva, M.M.; Zhelankin, A.V.; Sobenin, I.A.; Bobryshev, Y.V.; Orekhov, A.N. Association of mutations in the mitochondrial genome with the subclinical carotid atherosclerosis in women. Exp. Mol. Pathol. 2015, 99, 25–32. [Google Scholar] [CrossRef]
- Touboul, P.J.; Hennerici, M.G.; Meairs, S.; Adams, H.; Amarenco, P.; Bornstein, N.; Csiba, L.; Desvarieux, M.; Ebrahim, S.; Hernandez Hernandez, R.; et al. Mannheim carotid intima-media thickness and plaque consensus (2004-2006-2011). An update on behalf of the advisory board of the 3rd, 4th and 5th watching the risk symposia, at the 13th, 15th and 20th European Stroke Conferences, Mannheim, Germany, 2004, Brussels, Belgium, 2006, and Hamburg, Germany, 2011. Cerebrovasc Dis. 2012, 34, 290–296. [Google Scholar] [CrossRef]
- Çetin, N.; Güneş Tatar, İ.; Yüceege, M.; Ergun, O.; Hekimoğlu, B. Ultrasonographic evaluation of abdominal wall fat index, carotid intima-media thickness and plaque score in obstructive sleep apnea syndrome. Med. Ultrason. 2019, 21, 422–426. [Google Scholar] [CrossRef]
- Sazonova, M.; Budnikov, E.; Khasanova, Z.; Sobenin, I.; Postnov, A.; Orekhov, A. Studies of the human aortic intima by a direct quantitative assay of mutant alleles in the mitochondrial genome. Atherosclerosis 2009, 204, 184–190. [Google Scholar] [CrossRef]
- IBM SPSS Statistics 27 Documentation. Available online: https://www.ibm.com/support/pages/ibm-spss-statistics-27-documentation (accessed on 27 October 2023).
- Watanabe, T.; Fan, J. Atherosclerosis and inflammation mononuclear cell recruitment and adhesion molecules with reference to the implication of ICAM-1/LFA-1 pathway in atherogenesis. Int. J. Cardiol. 1998, 66 (Suppl. S1), S45–S55. [Google Scholar] [CrossRef]
- Chavan, V.; Willis, J.; Walker, S.K.; Clark, H.R.; Liu, X.; Fox, M.A.; Srivastava, S.; Mukherjee, K. Central presynaptic terminals are enriched in ATP but the majority lack mitochondria. PLoS ONE 2015, 10, e0125185. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Zorita, S.; Romero-Carramiñana, I.; Santacatterina, F.; Esparza-Moltó, P.B.; Simó, C.; Del-Arco, A.; Núñez de Arenas, C.; Saiz, J.; Barbas, C.; Cuezva, J.M. IF1 ablation prevents ATP synthase oligomerization, enhances mitochondrial ATP turnover and promotes an adenosine-mediated pro-inflammatory phenotype. Cell Death Dis. 2023, 14, 413. [Google Scholar] [CrossRef]
- Esparza-Moltó, P.B.; Romero-Carramiñana, I.; Núñez de Arenas, C.; Pereira, M.P.; Blanco, N.; Pardo, B.; Bates, G.R.; Sánchez-Castillo, C.; Artuch, R.; Murphy, M.P.; et al. Generation of mitochondrial reactive oxygen species is controlled by ATPase inhibitory factor 1 and regulates cognition. PLoS Biol. 2021, 19, e3001252. [Google Scholar] [CrossRef]
- Maltzman, J.S. Mitochondria-More than just ATP for CTLs. Sci. Immunol. 2021, 6, eabn0249. [Google Scholar] [CrossRef]
- Annesley, S.J.; Fisher, P.R. Mitochondria in Health and Disease. Cells 2019, 8, 680. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S. Organelle zones in mitochondria. J. Biochem. 2019, 165, 101–107. [Google Scholar] [CrossRef]
- Wong, Y.C.; Kim, S.; Peng, W.; Krainc, D. Regulation and Function of Mitochondria-Lysosome Membrane Contact Sites in Cellular Homeostasis. Trends Cell Biol. 2019, 29, 500–513. [Google Scholar] [CrossRef] [PubMed]
- Elbaz-Alon, Y. Mitochondria-organelle contact sites: The plot thickens. Biochem. Soc. Trans. 2017, 45, 477–488. [Google Scholar] [CrossRef]
- Carlton, J.G.; Jones, H.; Eggert, U.S. Membrane and organelle dynamics during cell division. Nat. Rev. Mol. Cell Biol. 2020, 21, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Sampath, H. Mitochondrial DNA Integrity: Role in Health and Disease. Cells 2019, 8, 100. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, V.F. Mitochondrial Genetics. Adv. Exp. Med. Biol. 2019, 1158, 247–255. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial genetic medicine. Nat. Genet. 2018, 50, 1642–1649. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.C.; Bwiza, C.P.; Lee, C. Mitonuclear genomics and aging. Hum. Genet. 2020, 139, 381–399. [Google Scholar] [CrossRef]
- Lee, S.R.; Han, J. Mitochondrial Mutations in Cardiac Disorders. Adv. Exp. Med. Biol. 2017, 982, 81–111. [Google Scholar] [CrossRef]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA heteroplasmy in disease and targeted nuclease-based therapeutic approaches. EMBO Rep. 2020, 21, e49612. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Kanai, M.; Durham, T.J.; Tsuo, K.; McCoy, J.G.; Kotrys, A.V.; Zhou, W.; Chinnery, P.F.; Karczewski, K.J.; Calvo, S.E.; et al. Nuclear genetic control of mtDNA copy number and heteroplasmy in humans. Nature 2023, 620, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.V.; Gitschlag, B.L.; Patel, M.R. Cellular mechanisms of mtDNA heteroplasmy dynamics. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 510–525. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.V.; Moraes, C.T. Current strategies towards therapeutic manipulation of mtDNA heteroplasmy. Front. Biosci. 2017, 22, 991–1010. [Google Scholar] [CrossRef]
- Grady, J.P.; Pickett, S.J.; Ng, Y.S.; Alston, C.L.; Blakely, E.L.; Hardy, S.A.; Feeney, C.L.; Bright, A.A.; Schaefer, A.M.; Gorman, G.S.; et al. mtDNA heteroplasmy level and copy number indicate disease burden in m.3243A>G mitochondrial disease. EMBO Mol. Med. 2018, 10, e8262. [Google Scholar] [CrossRef] [PubMed]
- Lorca, R.; Aparicio, A.; Gómez, J.; Álvarez-Velasco, R.; Pascual, I.; Avanzas, P.; González-Urbistondo, F.; Alen, A.; Vázquez-Coto, D.; González-Fernández, M.; et al. Mitochondrial Heteroplasmy as a Marker for Premature Coronary Artery Disease: Analysis of the Poly-C Tract of the Control Region Sequence. J. Clin. Med. 2023, 12, 2133. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial cytopathies. Cell Calcium 2016, 60, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Mullin, N.K.; Voigt, A.P.; Flamme-Wiese, M.J.; Liu, X.; Riker, M.J.; Varzavand, K.; Stone, E.M.; Tucker, B.A.; Mullins, R.F. Multimodal single-cell analysis of nonrandom heteroplasmy distribution in human retinal mitochondrial disease. JCI Insight 2023, 8, e165937. [Google Scholar] [CrossRef] [PubMed]
- Klein, H.U.; Trumpff, C.; Yang, H.S.; Lee, A.J.; Picard, M.; Bennett, D.A.; De Jager, P.L. Characterization of mitochondrial DNA quantity and quality in the human aged and Alzheimer’s disease brain. Mol. Neurodegener. 2021, 16, 75. [Google Scholar] [CrossRef]
- Almarzooqi, F.; Vallance, H.; Mezei, M.; Lehman, A.; Horvath, G.; Rakic, B.; Zypchen, L.; Mattman, A. Macrocytosis in Mitochondrial DNA Deletion Syndromes. Acta Haematol. 2023, 146, 220–225. [Google Scholar] [CrossRef]
- Snyder, R.J.; Kleeberger, S.R. Role of Mitochondrial DNA in Inflammatory Airway Diseases. Compr. Physiol. 2021, 11, 1485–1499. [Google Scholar] [CrossRef]
- Masingue, M.; Rucheton, B.; Bris, C.; Romero, N.B.; Procaccio, V.; Eymard, B. Highly asymmetrical distribution of muscle wasting correlates to the heteroplasmy in a patient carrying a large-scale mitochondrial DNA deletion: A novel pathophysiological mechanism for explaining asymmetry in mitochondrial myopathies. Neuromuscul. Disord. NMD 2022, 32, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Heidari, M.M.; Keshmirshekan, A.; Bidakhavidi, M.; Khosravi, A.; Bandari, Z.; Khatami, M.; Nafissi, S. A novel heteroplasmic mutation in mitochondrial tRNAArg gene associated with non-dystrophic myotonias. Acta Neurol. Belg. 2020, 120, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, M.A.; McCormick, E.M.; Perera, L.; Longley, M.J.; Bai, R.; Kong, J.; Dulik, M.; Shen, L.; Goldstein, A.C.; McCormack, S.E.; et al. Mitochondrial single-stranded DNA binding protein novel de novo SSBP1 mutation in a child with single large-scale mtDNA deletion (SLSMD) clinically manifesting as Pearson, Kearns-Sayre, and Leigh syndromes. PLoS ONE 2019, 14, e0221829. [Google Scholar] [CrossRef]
- Yan, C.; Duanmu, X.; Zeng, L.; Liu, B.; Song, Z. Mitochondrial DNA: Distribution, Mutations, and Elimination. Cells 2019, 8, 379. [Google Scholar] [CrossRef]
- Protasoni, M.; Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 2021, 22, 586. [Google Scholar] [CrossRef] [PubMed]
- Ryzhkova, A.I.; Sazonova, M.A.; Sinyov, V.V.; Galitsyna, E.V.; Chicheva, M.M.; Melnichenko, A.A.; Grechko, A.V.; Postnov, A.Y.; Orekhov, A.N.; Shkurat, T.P. Mitochondrial diseases caused by mtDNA mutations: A mini-review. Ther. Clin. Risk Manag. 2018, 14, 1933–1942. [Google Scholar] [CrossRef]
- Alves, C.A.P.F.; Gonçalves, F.G.; Grieb, D.; Lucato, L.T.; Goldstein, A.C.; Zuccoli, G. Neuroimaging of Mitochondrial Cytopathies. Top. Magn. Reson. Imaging TMRI 2018, 27, 219–240. [Google Scholar] [CrossRef]
- Viering, D.H.H.M.; Vermeltfoort, L.; Bindels, R.J.M.; Deinum, J.; de Baaij, J.H.F. Electrolyte Disorders in Mitochondrial Cytopathies: A Systematic Review. J. Am. Soc. Nephrol. JASN 2023, 34, 1875–1888. [Google Scholar] [CrossRef]
- Turnbull, D.M.; Rustin, P. Genetic and biochemical intricacy shapes mitochondrial cytopathies. Neurobiol. Dis. 2016, 92 Pt A, 55–63. [Google Scholar] [CrossRef]
- Sazonova, M.A. Association of mitochondrial genome mutations with lipofibrous plaques in human aortic intima. Patol. Fiziol. Eksp. Ter. 2015, 59, 17–28. (In Russian) [Google Scholar] [CrossRef] [PubMed]
- Nissanka, N.; Minczuk, M.; Moraes, C.T. Mechanisms of Mitochondrial DNA Deletion Formation. Trends Genet. TIG 2019, 35, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Puertas, M.J.; González-Sánchez, M. Insertions of mitochondrial DNA into the nucleus-effects and role in cell evolution. Genome 2020, 63, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Frazier, A.E.; Compton, A.G.; Kishita, Y.; Hock, D.H.; Welch, A.E.; Amarasekera, S.S.C.; Rius, R.; Formosa, L.E.; Imai-Okazaki, A.; Francis, D.; et al. Fatal perinatal mitochondrial cardiac failure caused by recurrent de novo duplications in the ATAD3 locus. Med. N. Y. 2021, 2, 49–73. [Google Scholar] [CrossRef] [PubMed]
- Güney, A.I.; Javadova, D.; Kırac, D.; Ulucan, K.; Koc, G.; Ergec, D.; Tavukcu, H.; Tarcan, T. Detection of Y chromosome microdeletions and mitochondrial DNA mutations in male infertility patients. Genet. Mol. Res. GMR 2012, 11, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Sazonova, M.A.; Postnov, A.I.; Orekhov, A.N.; Sobenin, I.A. A new method of quantitative estimation of mutant allele in mitochondrial genome. Patol. Fiziol. Eksp. Ter. 2011, 4, 81–84. (In Russian) [Google Scholar] [PubMed]
- Sobenin, I.A.; Mitrofanov, K.Y.; Zhelankin, A.V.; Sazonova, M.A.; Postnov, A.Y.; Revin, V.V.; Bobryshev, Y.V.; Orekhov, A.N. Quantitative assessment of heteroplasmy of mitochondrial genome: Perspectives in diagnostics and methodological pitfalls. Biomed. Res. Int. 2014, 2014, 292017. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Venegas, V.; Li, F.; Wong, L.J. Analysis of mitochondrial DNA point mutation heteroplasmy by ARMS quantitative PCR. Curr. Protoc. Hum. Genet. 2011, 68, 19.6. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, P.; Hamidkhani, A.; Asgarani, E. Comparative Analysis of Denaturing Gradient Gel Electrophoresis and Temporal Temperature Gradient Gel Electrophoresis Profiles as a Tool for the Differentiation of Candida Species. Jundishapur J. Microbiol. 2015, 8, e22249. [Google Scholar] [CrossRef]
- Kristinsson, R.; Lewis, S.E.; Danielson, P.B. Comparative analysis of the HV1 and HV2 regions of human mitochondrial DNA by denaturing high-performance liquid chromatography. J. Forensic Sci. 2009, 54, 28–36. [Google Scholar] [CrossRef]
- Ryan, S.E.; Ryan, F.; O’Dwyer, V.; Neylan, D. A real-time ARMS PCR/high-resolution melt curve assay for the detection of the three primary mitochondrial mutations in Leber’s hereditary optic neuropathy. Mol. Vis. 2016, 22, 1169–1175. [Google Scholar]
- Mukae, S.; Aoki, S.; Itoh, S.; Sato, R.; Nishio, K.; Iwata, T.; Katagiri, T. Mitochondrial 5178A/C genotype is associated with acute myocardial infarction. Circ. J. Off. J. Jpn. Circ. Soc. 2003, 67, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Hefti, E.; Blanco, J.G. Mitochondrial DNA heteroplasmy in cardiac tissue from individuals with and without coronary artery disease. Mitochondrial DNA Part A DNA Mapp. Seq. Anal. 2018, 29, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Vecoli, C.; Borghini, A.; Pulignani, S.; Mercuri, A.; Turchi, S.; Carpeggiani, C.; Picano, E.; Andreassi, M.G. Prognostic value of mitochondrial DNA4977 deletion and mitochondrial DNA copy number in patients with stable coronary artery disease. Atherosclerosis 2018, 276, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Battle, S.L.; Puiu, D.; TOPMed mtDNA Working Group; Verlouw, J.; Broer, L.; Boerwinkle, E.; Taylor, K.D.; Rotter, J.I.; Rich, S.S.; Grove, M.L.; et al. A bioinformatics pipeline for estimating mitochondrial DNA copy number and heteroplasmy levels from whole genome sequencing data. NAR Genom. Bioinform. 2022, 4, lqac034. [Google Scholar] [CrossRef] [PubMed]
- Sazonova, M.A.; Sinyov, V.V.; Barinova, V.A.; Ryzhkova, A.I.; Bobryshev, Y.V.; Orekhov, A.N.; Sobenin, I.A. Association of mitochondrial mutations with the age of patients having atherosclerotic lesions. Exp. Mol. Pathol. 2015, 99, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Fetterman, J.L.; Qian, Y.; Sun, X.; Blackwell, T.W.; Pitsillides, A.; Cade, B.E.; Wang, H.; Raffield, L.M.; Lange, L.A.; et al. NHLBI Trans-Omics for Precision Medicine TOPMed Consortium. Presence and transmission of mitochondrial heteroplasmic mutations in human populations of European and African ancestry. Mitochondrion 2021, 60, 33–42. [Google Scholar] [CrossRef]
- Longchamps, R.J.; Castellani, C.A.; Yang, S.Y.; Newcomb, C.E.; Sumpter, J.A.; Lane, J.; Grove, M.L.; Guallar, E.; Pankratz, N.; Taylor, K.D.; et al. Evaluation of mitochondrial DNA copy number estimation techniques. PLoS ONE 2020, 15, e0228166. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Mutation | Primers | Size of DNA Amplicons |
|---|---|---|
| m.12315G>A | F: bio-CTCATGCCCCCATGTCTAA (12230–12249) R: TTACTTTTATTTGGAGTTGCAC (12337–12317) | 108 bp |
| m.652delG | F: TAGACGGGCTCACATCAC (621–638) R: bio-GGGGTATCTAATCCCAGTTTGGGT (1087–1064) | 467 bp |
| m.14459G>A | F: CAGCTTCCTACACTATTAAAGT (14303–14334) R: bio-GTTTTTTTAATTTATTTAGGGGG (14511–14489) | 209 bp |
| m.5178C>A | F: bio-GCAGTTGAGGTGGATTAAAC (4963–4982) R: GGAGTAGATTAGGCGTAGGTAG (5366–5345) | 383 bp |
| m.13513G>A | F: CCTCACAGGTTTCTACTCCAAA (13491–13512) R: bio-AAGTCCTAGGAAAGTGACAGCGAGG (13825–13806) | 335 bp |
| m.652insG | F: TAGACGGGCTCACATCAC (621–638) R: bio-GGGGTATCTAATCCCAGTTTGGGT (1087–1064) | 467 bp |
| m.3256C>T | F: bio-AGGACAAGAGAAATAAGGCC (3129–3149) R: ACGTTGGGGCCTTTGCGTAG (3422–3403) | 294 bp |
| m.15059G>A | F: bio-CATTATTCTCGCACGGACT (14671–14689) R: GCTATAGTTGCAAGCAGGAG (15120–15100) | 450 bp |
| m.1555A>G | F:TAGGTCAAGGTGTAGCCCATGAGGTGGCAA (1326–1355) R: bio-GTAAGGTGGAGTGGGTTTGGG (1704–1684) | 379 bp |
| m.14846G>A | F: bio-CATTATTCTCGCACGGACT (14671–14689) R: GCTATAGTTGCAAGCAGGAG (15120–15100) | 450 bp |
| Mutation | Primer |
|---|---|
| m.12315G>A | TTTGGAGTTGCAC (12328–12316) |
| m.652delG | CCCATAAACAAATA (639–651) |
| m.14459G>A | GATACTCCTCAATAGCCA (14439–14456) |
| m.5178C>A | ATTAAGGGTGTTAGTCATGT (5200–5181) |
| m.13513G>A | AGGTTTCTACTCCAA (13497–13511) |
| m.652insG | CCCATAAACAAATA (639–651) |
| m.3256C>T | AAGAAGAGGAATTGA (3300–3286) |
| m.15059G>A | TTTCTGAGTAGAGAAATGAT (15080–15061) |
| m.1555A>G | ACGCATTTATATAGAGGA (1537–1554) |
| m.14846G>A | GCGCCAAGGAGTGA (14861–14848) |
| Characteristics | First Sample a | Second Sample b | Difference, p |
|---|---|---|---|
| Sex, male/female | 61/170 | 70/151 | 0.182 |
| Age, years | 63.0 (5.9) | 61.6 (5.0) | 0.058 |
| BMI, kg/m2 | 28.9 (4.9) | 28.7 (4.8) | 0.597 |
| Diabetes, % | 5 | 6 | 0.782 |
| Systolic BP, mm Hg | 130 (16) | 130 (15) | 0.900 |
| Diastolic BP, mm Hg | 80 (9) | 81 (9) | 0.817 |
| Smoking, % | 8 | 10 | 0.436 |
| Carotid IMT, mm | 0.828 (0.115) | 0.694 (0.100) | <0.001 c |
| Total cholesterol, mg/dL | 229.5 (40.1) | 234.2 (45.6) | 0.248 |
| HDL, mg/dL | 49.1 (12.6) | 50.3 (16.4) | 0.387 |
| LDL, mg/dL | 157.6 (34.6) | 160.6 (42.8) | 0.414 |
| TG, mg/dL | 115.0 (60.7) | 119.3 (80.6) | 0.519 |
| mtDNA Mutation | First Sample a | Second Sample b | Difference, p |
|---|---|---|---|
| m.5178C>A | 11.8 (9.6) | 6.8 (7.8) | <0.001 c |
| m.1555A>G | 22.3 (13.1) | 23.0 (10.3) | 0.542 |
| m.13513G>A | 22.7 (11.9) | 32.1 (16.2) | <0.001 c |
| m.652delG | 23.8 (11.0) | 19.9 (10.2) | <0.001 c |
| m.14846G>A | 16.4 (15.7) | 19.7 (16.6) | 0.030 d |
| m.12315G>A | 35.5 (24.2) | 31.1 (21.1) | <0.043 d |
| m.14459G>A | 5.1 (5.4) | 4.2 (4.7) | 0.055 |
| m.652insG | 1.4 (9.8) | 3.9 (13.5) | 0.027 d |
| m.3256C>T | 16.4 (16.5) | 11.6 (8.6) | 0.001 c |
| m.15059G>A | 11.2 (14.3) | 9.8 (10.1) | 0.226 |
| mtDNA Mutation | r, Pearson’s Correlation Coefficient | Significance, p |
|---|---|---|
| m.5178C>A | 0.274 | <0.001 ** |
| m.1555A>G | −0.029 | 0.542 |
| m.652delG | 0.184 | <0.001 ** |
| m.12315G>A | 0.097 | 0.041 * |
| m.14459G>A | 0.091 | 0.055 |
| m.3256C>T | 0.180 | <0.001 ** |
| m.15059G>A | 0.057 | 0.226 |
| m.13513G>A | −0.316 | <0.001 ** |
| m.652insG | −0.105 | 0.027 * |
| m.14846G>A | −0.102 | 0.030 * |
| Model | Minus Twice the Log Likelihood | Cox and Snell R2 | Nagelkerke R2 |
|---|---|---|---|
| 1 | 147,273 | 0.358 | 0.481 * |
| Model | Detected Cases | Predicted Cases | |||
|---|---|---|---|---|---|
| Association of Atherosclerotic Plaques with Total Burden of 10 Mutation | Percentage of Correct Predictions | ||||
| 0.00 | 1.00 | ||||
| 1 | Association of atherosclerotic plaques with total burden of 10 mutations | 0.00 | 18 | 7 | 24.6 |
| 1.00 | 11 | 37 | 50.6 | ||
| Total percentage value | 75.3 * | ||||
| Analyzed Variables | |||||||
|---|---|---|---|---|---|---|---|
| Mutations | B | S.E. | Wald | df | Sig. | Exp (B) | |
| Model 1 | m.1555A>G | −0.174 | 0.042 | 15.951 | 1 | 0.0001 ** | 0.840 |
| m.3256C>T | 0.022 | 0.051 | 0.557 | 1 | 0.420 | 1.022 | |
| m.14846G>A | −0.021 | 0.029 | 0.795 | 1 | 0.258 | 0.979 | |
| m.5178C>A | 0.034 | 0.045 | 0.760 | 1 | 0.355 | 1.034 | |
| m.652delG | 0.063 | 0.022 | 6.751 | 1 | 0.012 ** | 1.065 | |
| m.12315G>A | 0.133 | 0.017 | 21.961 | 1 | 0.0001 ** | 1.42 | |
| m.13513G>A | −0.051 | 0.019 | 6.871 | 1 | 0.006 ** | 0.950 | |
| m.14459G>A | 0.031 | 0.015 | 2.862 | 1 | 0.043 ** | 1.031 | |
| m.15059G>A | 0.061 | 0.021 | 7.766 | 1 | 0.0001 ** | 1.062 | |
| m.652insG | 0.072 | 0.080 | 0.991 | 1 | 0.453 | 1.074 | |
| Constant | −2.371 | 1.301 | 4.226 | 1 | 0.048 * | 0.093 | |
| Probability of Faultless Prognosis | ||||
|---|---|---|---|---|
| Area under the Curve | Standard Error | Asymptomatic Significance | Asymptomatic Confidence Interval 95% | |
| Lower than 95% | Higher than 95% | |||
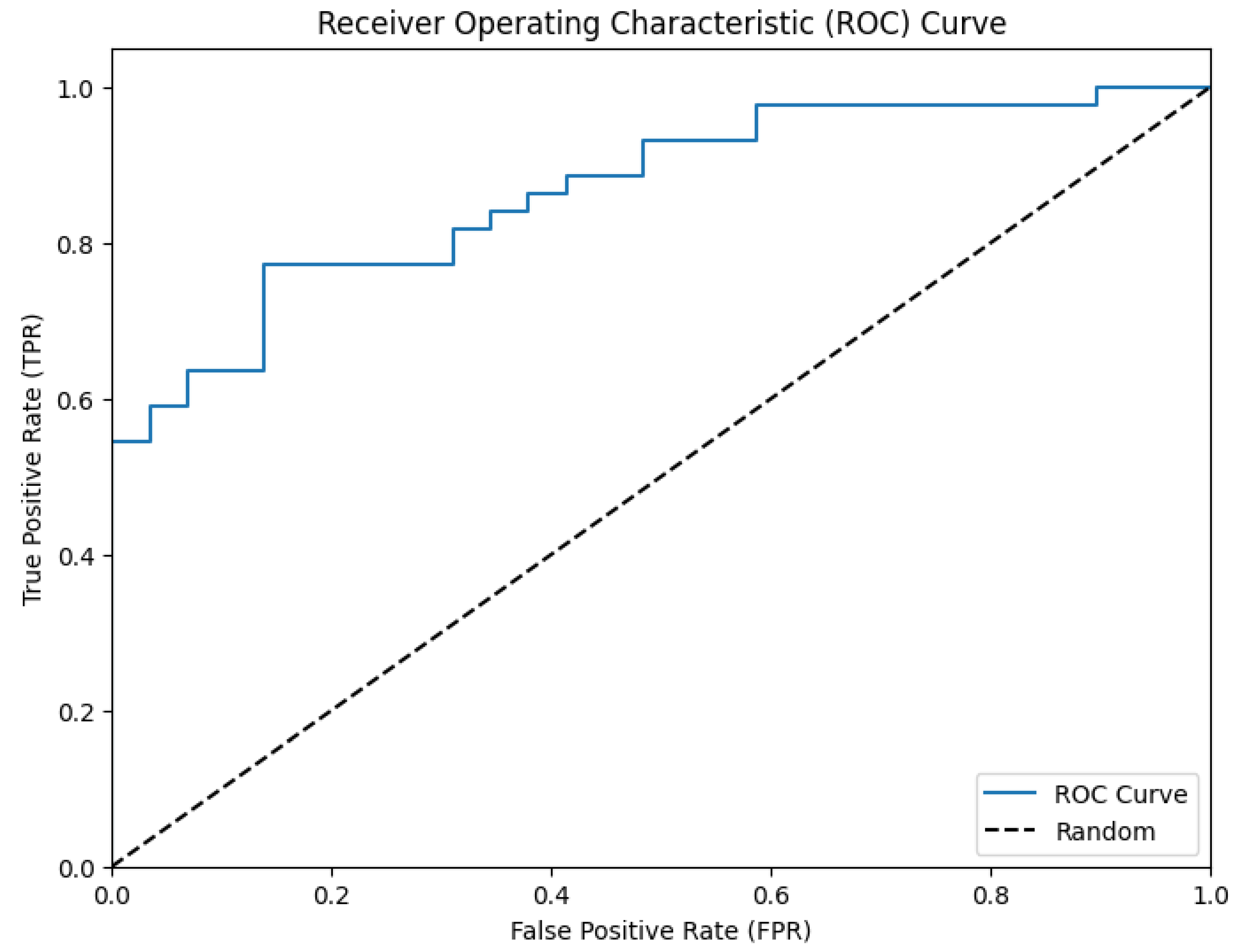
| 0.867 | 0.020 | 0.0015 | 0.771 | 0.910 |
| OR | Risk of ASP in Carotid Arteries | Sensitivity | Specificity |
|---|---|---|---|
| 4.5 (95% CI: 2.4–6.8) | 2.08 (95% CI: 1.51–3.53) | 0.749 | 0.858 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sazonova, M.A.; Kirichenko, T.V.; Ryzhkova, A.I.; Sazonova, M.D.; Doroschuk, N.A.; Omelchenko, A.V.; Nikiforov, N.G.; Ragino, Y.I.; Postnov, A.Y. Variability of Mitochondrial DNA Heteroplasmy: Association with Asymptomatic Carotid Atherosclerosis. Biomedicines 2024, 12, 1868. https://doi.org/10.3390/biomedicines12081868
Sazonova MA, Kirichenko TV, Ryzhkova AI, Sazonova MD, Doroschuk NA, Omelchenko AV, Nikiforov NG, Ragino YI, Postnov AY. Variability of Mitochondrial DNA Heteroplasmy: Association with Asymptomatic Carotid Atherosclerosis. Biomedicines. 2024; 12(8):1868. https://doi.org/10.3390/biomedicines12081868
Chicago/Turabian StyleSazonova, Margarita A., Tatiana V. Kirichenko, Anastasia I. Ryzhkova, Marina D. Sazonova, Natalya A. Doroschuk, Andrey V. Omelchenko, Nikita G. Nikiforov, Yulia I. Ragino, and Anton Yu. Postnov. 2024. "Variability of Mitochondrial DNA Heteroplasmy: Association with Asymptomatic Carotid Atherosclerosis" Biomedicines 12, no. 8: 1868. https://doi.org/10.3390/biomedicines12081868
APA StyleSazonova, M. A., Kirichenko, T. V., Ryzhkova, A. I., Sazonova, M. D., Doroschuk, N. A., Omelchenko, A. V., Nikiforov, N. G., Ragino, Y. I., & Postnov, A. Y. (2024). Variability of Mitochondrial DNA Heteroplasmy: Association with Asymptomatic Carotid Atherosclerosis. Biomedicines, 12(8), 1868. https://doi.org/10.3390/biomedicines12081868