
Understanding the Complex Dynamics of Immunosenescence in Multiple Sclerosis: From Pathogenesis to Treatment
 and
and
Abstract
:1. Introduction
2. Immune System Aging
2.1. Immunosenescence: Adaptive Immunity
2.1.1. Immunosenescence: T Cells
2.1.2. Immunosenescence: B Cells
2.2. Immunosenescence: Innate Immunity
2.2.1. Immunosenescence: Neutrophils
2.2.2. Immunosenescence: Monocytes and Macrophages
2.2.3. Immunosenescence: Natural Killer Cells
2.2.4. Immunosenescence: Dendritic Cells
3. Immunosenescence and Inflammaging
- Senescent cells accumulated within various tissues release pro-inflammatory mediators that have the potential to spread the senescent phenotype to neighboring cells, thereby contributing to age-related inflammation;
- There is an increase in cell debris levels from cell death or damage. Examples of these components include nucleic acids, mitochondrial DNA (mtDNA), cardiolipin, mitochondria, and heat-shock proteins. “Garb-aging” refers to the build-up of DAMPs with aging, which can lead to innate immunity and the release of pro-inflammatory cytokines;
- Age-related declines in autophagy and proteasome activity play a role in the accumulation of misfolded protein aggregates. Consequently, these aggregates serve as triggers for inflammatory pathways.
- The presence of pro-inflammatory circulating microRNA (inflammaMIR) and the age-related accumulation of agalactosylated N-glycans in the blood (one of the most potent indicators of human biological age);
- Nuclear DNA damage and telomere shortening caused by ROS and other agents set off a DNA repair response and stimulate the synthesis of pro-inflammatory molecules;
- Impaired regulation of the complement pathway can cause a local inflammatory response;
- The excessive availability of energy and nutrients can fuel an inflammatory process mediated by metabolic cells, a phenomenon known as “metaflammation”;
4. Immunosenescence in Multiple Sclerosis
4.1. Immunosenescence in Experimental Autoimmune Encephalomyelitis Models
4.2. Particularities of Immunosenescence in Multiple Sclerosis
5. Immunosenescence and Treatment Strategies
5.1. Current Treatments
5.2. Future Prospectives
6. Discussion
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple Sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Confavreux, C.; Vukusic, S. The clinical course of multiple sclerosis. Handb. Clin. Neurol. 2014, 122, 343–369. [Google Scholar] [CrossRef] [PubMed]
- Vaughn, C.B.; Jakimovski, D.; Kavak, K.S.; Ramanathan, M.; Benedict, R.H.B.; Zivadinov, R.; Weinstock-Guttman, B. Epidemiology and treatment of multiple sclerosis in elderly populations. Nat. Rev. Neurol. 2019, 15, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Confavreux, C.; Vukusic, S. Natural history of multiple sclerosis: A unifying concept. Brain 2006, 129 Pt 3, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Sanai, S.A.; Saini, V.; Benedict, R.H.; Zivadinov, R.; Teter, B.E.; Ramanathan, M.; Weinstock-Guttman, B. Aging and multiple sclerosis. Mult. Scler. 2016, 22, 717–725. [Google Scholar] [CrossRef]
- Sospedra, M.; Martin, R. Immunology of Multiple Sclerosis. Semin. Neurol. 2016, 36, 115–127. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Tylutka, A.; Zembroń-Łacny, A. Immunological aging and clinical consequences. Postepy Hig. Med. Dosw. 2020, 74, 260–271. [Google Scholar] [CrossRef]
- Rossi, D.J.; Bryder, D.; Zahn, J.M.; Ahlenius, H.; Sonu, R.; Wagers, A.J.; Weissman, I.L. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc. Natl. Acad. Sci. USA 2005, 102, 9194–9199. [Google Scholar] [CrossRef] [PubMed]
- Bleve, A.; Motta, F.; Durante, B.; Pandolfo, C.; Selmi, C.; Sica, A. Immunosenescence, Inflammaging, and Frailty: Role of Myeloid Cells in Age-Related Diseases. Clin. Rev. Allerg. Immunol. 2022, 64, 123–144. [Google Scholar] [CrossRef] [PubMed]
- Dema, M.; Eixarch, H.; Villar, L.M.; Montalban, X.; Espejo, C. Immunosenescence in multiple sclerosis: The identification of new therapeutic targets. Autoimmun. Rev. 2021, 20, 102893. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.A.; Flores, R.R.; Jang, I.H.; Saathoff, A.; Robbins, P.D. Immune Senescence, Immunosenescence and Aging. Front. Aging 2022, 3, 900028. [Google Scholar] [CrossRef] [PubMed]
- Ostolaza Ibáñez, A.; Corroza Laviñeta, J.; Ayuso Blanco, T. Inmunosenescencia: El rol de la edad en la esclerosis múltiple. Neurología 2023, 38, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.; Yamada, T.; Miller, R.A. Pgp-1 hi T lymphocytes accumulate with age in mice and respond poorly to concanavalin A. Eur. J. Immunol. 1989, 19, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, J.S. Aging of the CD4 T Cell Compartment. Open Longev. Sci. 2012, 6, 83–91. [Google Scholar] [CrossRef]
- Parish, S.T.; Wu, J.E.; Effros, R.B. Sustained CD28 Expression Delays Multiple Features of Replicative Senescence in Human CD8 T Lymphocytes. J. Clin. Immunol. 2010, 30, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Elgueta, R.; Benson, M.J.; De Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef] [PubMed]
- Lages, C.S.; Suffia, I.; Velilla, P.A.; Huang, B.; Warshaw, G.; Hildeman, D.A.; Belkaid, Y.; Chougnet, C. Functional Regulatory T Cells Accumulate in Aged Hosts and Promote Chronic Infectious Disease Reactivation. J. Immunol. 2008, 181, 1835–1848. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.; Liu, Y.; Cheng, Y.; Glanville, J.; Zhang, D.; Lee, J.Y.; Olshen, R.A.; Weyand, C.M.; Boyd, S.D.; Goronzy, J.J. Diversity and clonal selection in the human T-cell repertoire. Proc. Natl. Acad. Sci. USA 2014, 111, 13139–13144. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.O.; Govind, S.; Aspinall, R. Reversing T cell immunosenescence: Why, who, and how. AGE 2013, 35, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Derhovanessian, E.; Maier, A.B.; Hähnel, K.; Beck, R.; de Craen, A.J.M.; Slagboom, E.P.; Westendorp, R.G.J.; Pawelec, G. Infection with cytomegalovirus but not herpes simplex virus induces the accumulation of late-differentiated CD4+ and CD8+ T-cells in humans. J. Gen. Virol. 2011, 92, 2746–2756. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, B.; Song, R.; Hao, Y.; Wang, D.; Li, Y.; Jiang, Y.; Xu, L.; Ma, Y.; Zheng, H.; et al. T-cell Immunoglobulin and ITIM Domain Contributes to CD8+ T-cell Immunosenescence. Aging Cell 2018, 17, e12716. [Google Scholar] [CrossRef] [PubMed]
- Callender, L.A.; Carroll, E.C.; Beal, R.W.J.; Chambers, E.S.; Nourshargh, S.; Akbar, A.N.; Henson, S.M. Human CD8+ EMRA T cells display a senescence-associated secretory phenotype regulated by p38 MAPK. Aging Cell 2018, 17, e12675. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.I.; De Maeyer, R.P.H.; Covre, L.P.; Nehar-Belaid, D.; Lanna, A.; Ward, S.; Marches, R.; Chambers, E.S.; Gomes, D.C.O.; Riddell, N.E.; et al. Sestrins induce natural killer function in senescent-like CD8+ T cells. Nat. Immunol. 2020, 21, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, G. T-cell immunity in the aging human. Haematologica 2014, 99, 795–797. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, G. Age and immunity: What is “immunosenescence”? Exp. Gerontol. 2018, 105, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, G. The human immunosenescence phenotype: Does it exist? Semin. Immunopathol. 2020, 42, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Caruso, C.; Buffa, S.; Candore, G.; Colonna-Romano, G.; Dunn-Walters, D.; Kipling, D.; Pawelec, G. Mechanisms of immunosenescence. Immun. Ageing 2009, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Frasca, D.; Diaz, A.; Romero, M.; Blomberg, B.B. The generation of memory B cells is maintained, but the antibody response is not, in the elderly after repeated influenza immunizations. Vaccine 2016, 34, 2834–2840. [Google Scholar] [CrossRef] [PubMed]
- Tabibian-Keissar, H.; Hazanov, L.; Schiby, G.; Rosenthal, N.; Rakovsky, A.; Michaeli, M.; Shahaf, G.L.; Pickman, Y.; Rosenblatt, K.; Melamed, D.; et al. Aging affects B-cell antigen receptor repertoire diversity in primary and secondary lymphoid tissues. Eur. J. Immunol. 2016, 46, 480–492. [Google Scholar] [CrossRef] [PubMed]
- Frasca, D.; Diaz, A.; Romero, M.; Blomberg, B.B. Human peripheral late/exhausted memory B cells express a senescent-associated secretory phenotype and preferentially utilize metabolic signaling pathways. Exp. Gerontol. 2017, 87, 113–120. [Google Scholar] [CrossRef]
- Frasca, D.; Diaz, A.; Romero, M.; D’Eramo, F.; Blomberg, B.B. Aging effects on T-bet expression in human B cell subsets. Cell. Immunol. 2017, 321, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, E.C.; Bartley, J.M.; Haynes, L. The impact of aging on CD4+ T cell responses to influenza infection. Biogerontology 2018, 19, 437–446. [Google Scholar] [CrossRef]
- Busse, P.J.; Mathur, S.K. Age-related changes in immune function: Effect on airway inflammation. J. Allergy Clin. Immunol. 2010, 126, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging as Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2018, 8, 1960. [Google Scholar] [CrossRef] [PubMed]
- Vidya, M.K.; Kumar, V.G.; Sejian, V.; Bagath, M.; Krishnan, G.; Bhatta, R. Toll-like receptors: Significance, ligands, signaling pathways, and functions in mammals. Int. Rev. Immunol. 2018, 37, 20–36. [Google Scholar] [CrossRef] [PubMed]
- Kufer, T.A.; Nigro, G.; Sansonetti, P.J. Multifaceted Functions of NOD-Like Receptor Proteins in Myeloid Cells at the Intersection of Innate and Adaptive Immunity. Microbiol. Spectr. 2016, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Bientinesi, E.; Monti, D. Immunosenescence and inflammaging in the aging process: Age-related diseases or longevity? Ageing Res. Rev. 2021, 71, 101422. [Google Scholar] [CrossRef] [PubMed]
- Hazeldine, J.; Harris, P.; Chapple, I.L.; Grant, M.; Greenwood, H.; Livesey, A.; Sapey, E.; Lord, J.M. Impaired neutrophil extracellular trap formation: A novel defect in the innate immune system of aged individuals. Aging Cell 2014, 13, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.B.; Lopes, L.B.; Da Silveira Antunes, R.N.; Fukasawa, J.T.; Cavaretto, D.d.A.; Calamita, Z. Effects of Immunosenescence on the Lower Expression of Surface Molecules in Neutrophils and Lymphocytes. Curr. Aging Sci. 2019, 11, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Sendama, W. The effect of ageing on the resolution of inflammation. Ageing Res. Rev. 2020, 57, 101000. [Google Scholar] [CrossRef] [PubMed]
- Barkaway, A.; Rolas, L.; Joulia, R.; Bodkin, J.; Lenn, T.; Owen-Woods, C.; Reglero-Real, N.; Stein, M.; Vázquez-Martínez, L.; Girbl, T.; et al. Age-related changes in the local milieu of inflamed tissues cause aberrant neutrophil trafficking and subsequent remote organ damage. Immunity 2021, 54, 1494–1510.e7. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, L.P.; Teixeira, V.R.; Alencar-Silva, T.; Simonassi-Paiva, B.; Pereira, R.W.; Pogue, R.; Carvalho, J.L. Hallmarks of aging and immunosenescence: Connecting the dots. Cytokine Growth Factor Rev. 2021, 59, 9–21. [Google Scholar] [CrossRef]
- Kohno, K.; Koya-Miyata, S.; Harashima, A.; Tsukuda, T.; Katakami, M.; Ariyasu, T.; Ushio, S.; Iwaki, K. Inflammatory M1-like macrophages polarized by NK-4 undergo enhanced phenotypic switching to an anti-inflammatory M2-like phenotype upon co-culture with apoptotic cells. J. Inflamm. 2021, 18, 2. [Google Scholar] [CrossRef]
- Moss, C.E.; Phipps, H.; Wilson, H.L.; Kiss-Toth, E. Markers of the ageing macrophage: A systematic review and meta-analysis. Front. Immunol. 2023, 14, 1222308. [Google Scholar] [CrossRef]
- Moss, C.E.; Johnston, S.A.; Kimble, J.V.; Clements, M.; Codd, V.; Hamby, S.; Goodall, A.H.; Deshmukh, S.; Sudbery, I.; Coca, D.; et al. Aging-related defects in macrophage function are driven by MYC and USF1 transcriptional programs. Cell Rep. 2024, 43, 114073. [Google Scholar] [CrossRef]
- Van Beek, A.A.; Van Den Bossche, J.; Mastroberardino, P.G.; De Winther, M.P.J.; Leenen, P.J.M. Metabolic Alterations in Aging Macrophages: Ingredients for Inflammaging? Trends Immunol. 2019, 40, 113–127. [Google Scholar] [CrossRef]
- Müller, L.; Fülöp, T.; Pawelec, G. Immunosenescence in vertebrates and invertebrates. Immun. Ageing 2013, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Brauning, A.; Rae, M.; Zhu, G.; Fulton, E.; Admasu, T.D.; Stolzing, A.; Sharma, A. Aging of the Immune System: Focus on Natural Killer Cells Phenotype and Functions. Cells 2022, 11, 1017. [Google Scholar] [CrossRef]
- Crooke, S.N.; Ovsyannikova, I.G.; Poland, G.A.; Kennedy, R.B. Immunosenescence: A systems-level overview of immune cell biology and strategies for improving vaccine responses. Exp. Gerontol. 2019, 124, 110632. [Google Scholar] [CrossRef]
- Metcalf, T.U.; Cubas, R.A.; Ghneim, K.; Cartwright, M.J.; Grevenynghe, J.V.; Richner, J.M.; Olagnier, D.P.; Wilkinson, P.A.; Cameron, M.J.; Park, B.S.; et al. Global analyses revealed age-related alterations in innate immune responses after stimulation of pathogen recognition receptors. Aging Cell 2015, 14, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 2007, 128, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Vitale, G.; Capri, M.; Salvioli, S. Inflammaging and ‘Garb-aging’. Trends Endocrinol. Metab. 2017, 28, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Perdaens, O.; Van Pesch, V. Molecular Mechanisms of Immunosenescene and Inflammaging: Relevance to the Immunopathogenesis and Treatment of Multiple Sclerosis. Front. Neurol. 2022, 12, 811518. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, C.E.; Cavanagh, M.M.; Jin, J.; Weyand, C.M.; Goronzy, J.J. Functional pathways regulated by microRNA networks in CD8 T-cell aging. Aging Cell 2019, 18, e12879. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Lu, K.; Ye, T.; Zhang, Z. MicroRNA 223 attenuates LPS induced inflammation in an acute lung injury model via the NLRP3 inflammasome and TLR4/NF κB signaling pathway via RHOB. Int. J. Mol. Med. 2019, 43, 1467–1477. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Hu, B.; Jadhav, R.R.; Jin, J.; Zhang, H.; Cavanagh, M.M.; Akondy, R.S.; Ahmed, R.; Weyand, C.M.; Goronzy, J.J. Activation of miR-21-Regulated Pathways in Immune Aging Selects against Signatures Characteristic of Memory T Cells. Cell Rep. 2018, 25, 2148–2162.e5. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Jadhav, R.R.; Gustafson, C.E.; Smithey, M.J.; Hirsch, A.J.; Uhrlaub, J.L.; Hildebrand, W.H.; Nikolich-Žugich, J.; Weyand, C.M.; Goronzy, J.J. Defects in Antiviral T Cell Responses Inflicted by Aging-Associated miR-181a Deficiency. Cell Rep. 2019, 29, 2202–2216.e5. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liang, Q.; Ren, Y.; Guo, C.; Ge, X.; Wang, L.; Cheng, Q.; Luo, P.; Zhang, Y.; Han, X. Immunosenescence: Molecular mechanisms and diseases. Sig. Transduct. Target Ther. 2023, 8, 200. [Google Scholar] [CrossRef]
- Mogilenko, D.A.; Shpynov, O.; Andhey, P.S.; Arthur, L.; Swain, A.; Esaulova, E.; Brioschi, S.; Shchukina, I.; Kerndl, M.; Bambouskova, M.; et al. Comprehensive Profiling of an Aging Immune System Reveals Clonal GZMK+ CD8+ T Cells as Conserved Hallmark of Inflammaging. Immunity 2021, 54, 99–115.e12. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef] [PubMed]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wang, H.; Ren, S.; Wang, B.; Yang, W.; Lv, L.; Sha, X.; Li, W.; Wang, Y. Identification of crucial inflammaging related risk factors in multiple sclerosis. Front. Mol. Neurosci. 2024, 17, 1398665. [Google Scholar] [CrossRef] [PubMed]
- Cantuti-Castelvetri, L.; Gokce, O.; Simons, M. Reparative inflammation in multiple sclerosis. Semin. Immunol. 2022, 59, 101630. [Google Scholar] [CrossRef] [PubMed]
- Touil, H.; Li, R.; Zuroff, L.; Moore, C.S.; Healy, L.; Cignarella, F.; Piccio, L.; Ludwin, S.; Prat, A.; Gommerman, J.; et al. Cross-talk between B cells, microglia and macrophages, and implications to central nervous system compartmentalized inflammation and progressive multiple sclerosis. eBioMedicine 2023, 96, 104789. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Xu, H.; Guo, S.; Mertens-Talcott, S.U.; Sun, Y. Ghrelin Signaling in Immunometabolism and Inflamm-Aging. Adv. Exp. Med. Biol. 2018, 1090, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Correale, J. Immunosuppressive Amino-Acid Catabolizing Enzymes in Multiple Sclerosis. Front. Immunol. 2021, 11, 600428. [Google Scholar] [CrossRef] [PubMed]
- Bolton, C.; Smith, P.A. The influence and impact of ageing and immunosenescence (ISC) on adaptive immunity during multiple sclerosis (MS) and the animal counterpart experimental autoimmune encephalomyelitis (EAE). Ageing Res. Rev. 2018, 41, 64–81. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.J.; Andrews, N.; Ball, D.; Gray, J.; Hachoumi, L.; Holmes, A.; Latcham, J.; Petrie, A.; Potter, P.; Rice, A.; et al. Does age matter? The impact of rodent age on study outcomes. Lab. Anim. 2017, 51, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Bjelobaba, I.; Begovic-Kupresanin, V.; Pekovic, S.; Lavrnja, I. Animal models of multiple sclerosis: Focus on experimental autoimmune encephalomyelitis. J. Neurosci. Res. 2018, 96, 1021–1042. [Google Scholar] [CrossRef] [PubMed]
- Sierra, H.; Cordova, M.; Chen, C.S.J.; Rajadhyaksha, M. Confocal Imaging–Guided Laser Ablation of Basal Cell Carcinomas: An Ex Vivo Study. J. Investig. Dermatol. 2015, 135, 612–615. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.E.; Hasan, M.; Han, J.S.; Kang, M.J.; Jung, B.H.; Kwok, S.K.; Kim, H.Y.; Kwon, O.S. Experimental autoimmune encephalomyelitis and age-related correlations of NADPH oxidase, MMP-9, and cell adhesion molecules: The increased disease severity and blood–brain barrier permeability in middle-aged mice. J. Neuroimmunol. 2015, 287, 43–53. [Google Scholar] [CrossRef]
- Hampton, D.W.; Innes, N.; Merkler, D.; Zhao, C.; Franklin, R.J.M.; Chandran, S. Focal Immune-Mediated White Matter Demyelination Reveals an Age-Associated Increase in Axonal Vulnerability and Decreased Remyelination Efficiency. Am. J. Pathol. 2012, 180, 1897–1905. [Google Scholar] [CrossRef] [PubMed]
- Goyne, C.E.; Fair, A.E.; Sumowski, P.E.; Graves, J.S. The Impact of Aging on Multiple Sclerosis. Curr. Neurol. Neurosci. Rep. 2024, 24, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Thakolwiboon, S.; Mills, E.A.; Yang, J.; Doty, J.; Belkin, M.I.; Cho, T.; Schultz, C.; Mao-Draayer, Y. Immunosenescence and multiple sclerosis: Inflammaging for prognosis and therapeutic consideration. Front. Aging 2023, 4, 1234572. [Google Scholar] [CrossRef] [PubMed]
- Graves, J.S.; Krysko, K.M.; Hua, L.H.; Absinta, M.; Franklin, R.J.M.; Segal, B.M. Ageing and multiple sclerosis. Lancet Neurol. 2023, 22, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, D.; Magliozzi, R.; Mitsikostas, D.D.; Gorgoulis, V.G.; Nicholas, R.S. Aging, Cellular Senescence, and Progressive Multiple Sclerosis. Front. Cell. Neurosci. 2020, 14, 178. [Google Scholar] [CrossRef] [PubMed]
- Zuroff, L.; Rezk, A.; Shinoda, K.; Espinoza, D.A.; Elyahu, Y.; Zhang, B.; Chen, A.A.; Shinohara, R.T.; Jacobs, D.; Alcalay, R.N.; et al. Immune aging in multiple sclerosis is characterized by abnormal CD4 T cell activation and increased frequencies of cytotoxic CD4 T cells with advancing age. eBioMedicine 2022, 82, 104179. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.T.; Howell, J.C.; Ozturk, T.; Gangishetti, U.; Kollhoff, A.L.; Hatcher-Martin, J.M.; Anderson, A.M.; Tyor, W.R. CSF Cytokines in Aging, Multiple Sclerosis, and Dementia. Front. Immunol. 2019, 10, 480. [Google Scholar] [CrossRef] [PubMed]
- Rawji, K.S.; Young, A.M.H.; Ghosh, T.; Michaels, N.J.; Mirzaei, R.; Kappen, J.; Kolehmainen, K.L.; Alaeiilkhchi, N.; Lozinski, B.; Mishra, M.K.; et al. Niacin-mediated rejuvenation of macrophage/microglia enhances remyelination of the aging central nervous system. Acta Neuropathol. 2020, 139, 893–909. [Google Scholar] [CrossRef]
- Prozorovski, T.; Ingwersen, J.; Lukas, D.; Göttle, P.; Koop, B.; Graf, J.; Schneider, R.; Franke, K.; Schumacher, S.; Britsch, S.; et al. Regulation of sirtuin expression in autoimmune neuroinflammation: Induction of SIRT1 in oligodendrocyte progenitor cells. Neurosci. Lett. 2019, 704, 116–125. [Google Scholar] [CrossRef]
- Willis, C.M.; Nicaise, A.M.; Bongarzone, E.R.; Givogri, M.; Reiter, C.R.; Heintz, O.; Jellison, E.R.; Sutter, P.A.; TeHennepe, G.; Ananda, G.; et al. Astrocyte Support for Oligodendrocyte Differentiation can be Conveyed via Extracellular Vesicles but Diminishes with Age. Sci. Rep. 2020, 10, 828. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Misra Sen, J.; Gorospe, M.; et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef]
- McMahon, J.; McQuaid, S.; Reynolds, R.; FitzGerald, U. Increased expression of ER stress- and hypoxia-associated molecules in grey matter lesions in multiple sclerosis. Mult. Scler. 2012, 18, 1437–1447. [Google Scholar] [CrossRef]
- Vollmer, T.; Signorovitch, J.; Huynh, L.; Galebach, P.; Kelley, C.; DiBernardo, A.; Sasane, R. The natural history of brain volume loss among patients with multiple sclerosis: A systematic literature review and meta-analysis. J. Neurol. Sci. 2015, 357, 8–18. [Google Scholar] [CrossRef]
- Eschborn, M.; Pawlitzki, M.; Wirth, T.; Nelke, C.; Pfeuffer, S.; Schulte-Mecklenbeck, A.; Lohmann, L.; Rolfes, L.; Pape, K.; Eveslage, M.; et al. Evaluation of Age-Dependent Immune Signatures in Patients with Multiple Sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e1094. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.; Miller, D.H.; Freedman, M.S.; Cree, B.A.; Wolinsky, J.S.; Weiner, H.; Lubetzki, C.; Hartung, H.P.; Montalban, X.; Uitdehaag, B.M.; et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet 2016, 387, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Hawker, K.; O’Connor, P.; Freedman, M.S.; Calabresi, P.A.; Antel, J.; Simon, J.; Hauser, S.; Waubant, E.; Vollmer, T.; Panitch, H.; et al. Rituximab in patients with primary progressive multiple sclerosis: Results of a randomized double-blind placebo-controlled multicenter trial. Ann. Neurol. 2009, 66, 460–471. [Google Scholar] [CrossRef]
- Kalim, H.; Pratama, M.Z.; Mahardini, E.; Winoto, E.S.; Krisna, P.A.; Handono, K. Accelerated immune aging was correlated with lupus-associated brain fog in reproductive-age systemic lupus erythematosus patients. Int. J. Rheum. Dis. 2020, 23, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.A.; Corboy, J.R. Discontinuing Disease-Modifying Therapies in Multiple Sclerosis. Pract. Neurol. 2022, 49–52. [Google Scholar]
- Bsteh, G.; Hegen, H.; Riedl, K.; Altmann, P.; Auer, M.; Berek, K.; Di Pauli, F.; Ehling, R.; Kornek, B.; Monschein, T.; et al. Quantifying the risk of disease reactivation after interferon and glatiramer acetate discontinuation in multiple sclerosis: The VIAADISC score. Eur. J. Neurol. 2021, 28, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, B.L.; Wolf, A.B.; Sillau, S.; Corboy, J.R.; Alvarez, E. Evolution of Disease Modifying Therapy Benefits and Risks: An Argument for De-escalation as a Treatment Paradigm for Patients with Multiple Sclerosis. Front. Neurol. 2022, 12, 799138. [Google Scholar] [CrossRef] [PubMed]
- Hua, L.H.; Fan, T.H.; Conway, D.; Thompson, N.; Kinzy, T.G. Discontinuation of disease-modifying therapy in patients with multiple sclerosis over age 60. Mult. Scler. 2019, 25, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Hua, L.H.; Harris, H.; Conway, D.; Thompson, N.R. Changes in patient-reported outcomes between continuers and discontinuers of disease modifying therapy in patients with multiple sclerosis over age 60. Mult. Scler. Relat. Disord. 2019, 30, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Corboy, J.R.; Fox, R.J.; Kister, I.; Cutter, G.R.; Morgan, C.J.; Seale, R.; Engebretson, E.; Gustafson, T.; Miller, A.E.; DISCOMS Investigators. Risk of new disease activity in patients with multiple sclerosis who continue or discontinue disease-modifying therapies (DISCOMS): A multicentre, randomised, single-blind, phase 4, non-inferiority trial. Lancet Neurol. 2023, 22, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Macaron, G.; Larochelle, C.; Arbour, N.; Galmard, M.; Girard, J.M.; Prat, A.; Duquette, P. Impact of aging on treatment considerations for multiple sclerosis patients. Front. Neurol. 2023, 14, 1197212. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, F.; Laurent, S.; Fink, G.R.; Barnett, M.H.; Reddel, S.; Hartung, H.P.; Warnke, C. Age and the risks of high-efficacy disease modifying drugs in multiple sclerosis. Curr. Opin. Neurol. 2019, 32, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Jordan, A.L.; Yang, J.; Fisher, C.J.; Racke, M.K.; Mao-Draayer, Y. Progressive multifocal leukoencephalopathy in dimethyl fumarate-treated multiple sclerosis patients. Mult. Scler. 2022, 28, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Bar-Or, A.; Cree, B.A.C.; Fox, R.J.; Giovannoni, G.; Gold, R.; Vermersch, P.; Arnold, D.L.; Arnould, S.; Scherz, T.; et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): A double-blind, randomised, phase 3 study. Lancet 2018, 391, 1263–1273. [Google Scholar] [CrossRef]
- D’Amico, E.; Chisari, C.G.; Arena, S.; Zanghì, A.; Toscano, S.; Lo Fermo, S.; Maimone, D.; Castaing, M.; Sciacca, S.; Zappia, M.; et al. Cancer Risk and Multiple Sclerosis: Evidence from a Large Italian Cohort. Front. Neurol. 2019, 10, 337. [Google Scholar] [CrossRef] [PubMed]
- Coles, A.J.; Cohen, J.A.; Fox, E.J.; Giovannoni, G.; Hartung, H.P.; Havrdova, E.; Schippling, S.; Selmaj, K.W.; Traboulsee, A.; Compston, D.A.S.; et al. Alemtuzumab CARE-MS II 5-year follow-up: Efficacy and safety findings. Neurology 2017, 89, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Ragonese, P.; Aridon, P.; Vazzoler, G.; Mazzola, M.A.; Lo Re, V.; Lo Re, M.; Realmuto, S.; Alessi, S.; D’Amelio, M.; Savettieri, G.; et al. Association between multiple sclerosis, cancer risk, and immunosuppressant treatment: A cohort study. BMC Neurol. 2017, 17, 155. [Google Scholar] [CrossRef] [PubMed]
- Koutsoudaki, P.N.; Papadopoulos, D.; Passias, P.G.; Koutsoudaki, P.; Gorgoulis, V.G. Cellular senescence and failure of myelin repair in multiple sclerosis. Mech. Ageing Dev. 2020, 192, 111366. [Google Scholar] [CrossRef] [PubMed]
- Oost, W.; Talma, N.; Meilof, J.F.; Laman, J.D. Targeting senescence to delay progression of multiple sclerosis. J. Mol. Med. 2018, 96, 1153–1166. [Google Scholar] [CrossRef] [PubMed]
- Musi, N.; Valentine, J.M.; Sickora, K.R.; Baeuerle, E.; Thompson, C.S.; Shen, Q.; Orr, M.E. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 2018, 17, e12840. [Google Scholar] [CrossRef] [PubMed]
- Krzystyniak, A.; Wesierska, M.; Petrazzo, G.; Gadecka, A.; Dudkowska, M.; Bielak-Zmijewska, A.; Mosieniak, G.; Figiel, I.; Wlodarczyk, J.; Sikora, E. Combination of dasatinib and quercetin improves cognitive abilities in aged male Wistar rats, alleviates inflammation and changes hippocampal synaptic plasticity and histone H3 methylation profile. Aging 2022, 14, 572–595. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, Y.; Liu, Y.; Zheng, P. mTOR Regulation and Therapeutic Rejuvenation of Aging Hematopoietic Stem Cells. Sci. Signal. 2009, 2, ra75. [Google Scholar] [CrossRef] [PubMed]
- Dziedzic, A.; Saluk-Bijak, J.; Miller, E.; Bijak, M. Metformin as a Potential Agent in the Treatment of Multiple Sclerosis. Int. J. Mol. Sci. 2020, 21, 5957. [Google Scholar] [CrossRef] [PubMed]
- De Keersmaecker, A.V.; Van Doninck, E.; Popescu, V.; Willem, L.; Cambron, M.; Laureys, G.; D’Haeseleer, M.; Bjerke, M.; Roelant, E.; Lemmerling, M.; et al. A metformin add-on clinical study in multiple sclerosis to evaluate brain remyelination and neurodegeneration (MACSiMiSE-BRAIN): Study protocol for a multi-center randomized placebo controlled clinical trial. Front. Immunol. 2024, 15, 1362629. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Wang, W.; Su, D.M. Contributions of Age-Related Thymic Involution to Immunosenescence and Inflammaging. Immun. Ageing 2020, 17, 2. [Google Scholar] [CrossRef] [PubMed]
- Dudakov, J.A.; Hanash, A.M.; Jenq, R.R.; Young, L.F.; Ghosh, A.; Singer, N.V.; West, M.L.; Smith, O.M.; Holland, A.M.; Tsai, J.J.; et al. Interleukin-22 Drives Endogenous Thymic Regeneration in Mice. Science 2012, 336, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, M.A.; Zakhary, C.M.; Rushdi, H.; Hamdan, J.A.; Youssef, K.N.; Khan, A.; Khan, S. The Effectiveness of Statins as Potential Therapy for Multiple Sclerosis: A Systematic Review of Randomized Controlled trials. Cureus 2021, 13, e18092. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Thoomukuntla, B.; Bena, J.; Cohen, J.A.; Fox, R.J.; Ontaneda, D. Ibudilast reduces slowly enlarging lesions in progressive multiple sclerosis. Mult. Scler. 2024, 30, 369–380. [Google Scholar] [CrossRef] [PubMed]
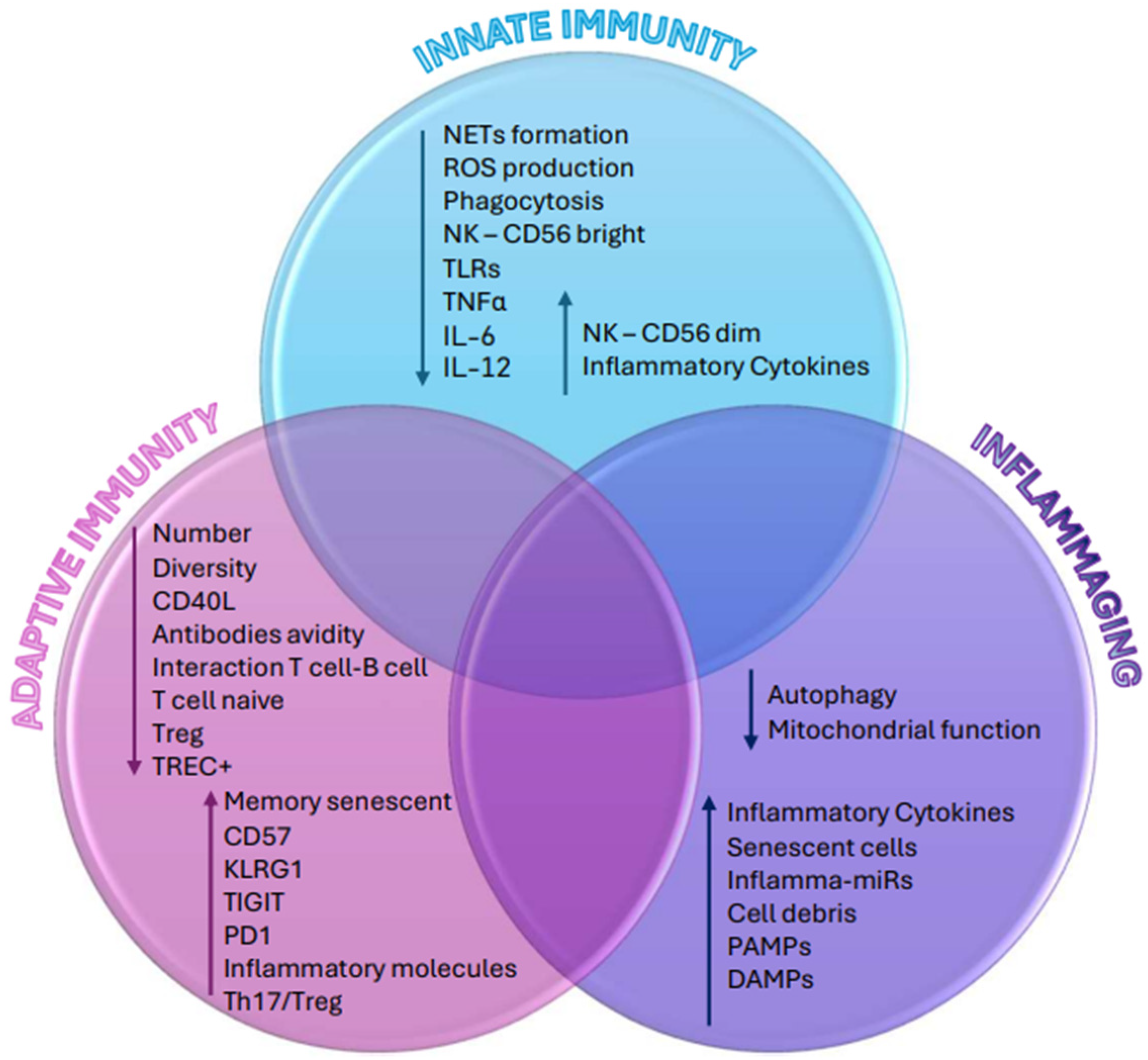

{kind=link}
| Adaptive Immunity Cell | Changes in Immunosenescence |
|---|---|
| T cell | Decreased number of naive T cells |
| Increased memory T cell population | |
| Reduction in TCR repertoire diversity | |
| Diminished presence of the CD28 co-stimulatory molecule | |
| Increased expression of CD57, KLRG1, TIGIT, and PD1 | |
| Distinct cytokine secretion profile (SASP) | |
| Shift toward cytotoxicity mediated by NK receptors | |
| B cell | Decreased number of naive B cells |
| Increased memory B cell population | |
| Reduction in BCR repertoire diversity | |
| Increase in the population of ABCs | |
| Increased autoimmune activity | |
| Increase in the population of age-associated B cells |
| Type of Pattern-Recognition Receptors | Action |
|---|---|
| Toll-like receptors | NF-κB activation, activation of MAP kinases pathway to recruit pro-inflammatory cytokines and co-stimulatory molecules, which promote inflammatory responses |
| NOD-like receptors | Stimulation of the inflammasome complex and the production of IL-11, IL-18, and IL-33 |
| Rig-like receptors | Proximal triggering of the IFN pathway |
| Innate Immunity Cell | Changes in Immunosenescence |
|---|---|
| Neutrophils | Reduced ROS production |
| Impaired NET formation | |
| Altered adhesion and phagocytosis | |
| Monocytes/Macrophages | Impaired migration and chemotaxis |
| M2-like over classic M1-like macrophages | |
| Reduced TLR expression | |
| NK Cells | Increased CD56dim and decreased CD56bright subset |
| Reduced cytotoxic activity | |
| Defective degranulation capacity | |
| Dendritic Cells | Increased ROS production |
| Impaired phagocytic capabilities and antigen presentation | |
| Decreased responsiveness to TLR stimulation | |
| Reduced production of type I and II interferons | |
| Enhanced pro-inflammatory cytokine secretion |
| SASP Molecule | Role |
|---|---|
| IL-1 | Pro-inflammatory |
| IL-6 | Pro-inflammatory activity contributing to inflammation and DNA damage |
| IL-7 | Promotes B lymphocyte growth and T lymphocyte activation |
| IL-11 | Hematopoiesis and tissue cell proliferation |
| IL-15 | Necessary for T cell regulation; high levels are pro-inflammatory |
| IL-8 | Chemotaxis and enhancement of pro-inflammatory activity |
| CXCL1 | Neutrophil activation |
| MCP2 | Monocyte chemotaxis |
| TNFα | Induces apoptosis |
| VEGF | Blood vessel formation |
| IGF-1 | Proliferation and apoptosis |
| Molecule | Cut-Off Age | Impact on Annual Relapse Rate Vs. Placebo | Impact on Disability Progression Vs. Placebo |
|---|---|---|---|
| Teriflunomide | <38 years | ↓ | ↓ |
| ≥38 years | ↓ | no impact | |
| Dimethyl fumarate | <40 years | ↓ | ↓ |
| ≥40 years | ↓ | no impact | |
| Fingolimod | <40 years | ↓ | no impact |
| ≥40 years | no impact | no impact | |
| Siponimod | <50 years ≥50 years | n/a | ↓ ↓ |
| Cladribine | <40 years ≥40 years | ↓ ↓ | ↓ ↓ |
| Ocrelizumab | <45 years ≥45 years | ↓ ↓ | ↓ ↓ |
| Value | Score | |
|---|---|---|
| Age | <45 years | 2 points |
| 45–55 years | 1 point | |
| >55 years | 0 points | |
| MRI activity | ≥3 new or enlarged T2 lesions or 1 gadolinium-enhancing lesion | 2 points |
| <3 new or enlarged T2 lesions and no gadolinium-enhancing lesion | 1 point | |
| Stable disease | <4 years | 2 points |
| 4–8 years | 1 point | |
| >8 years | 0 points |
| Treatment | Adverse Events Increased with Age |
|---|---|
| Fingolimod | Reduction of heart rate Hypertension HSV1/VVZ reactivation PML Malignancies (frequently affecting the skin) |
| Natalizumab | PML HSV1/VVZ reactivation |
| Cladribine | HSV1/VVZ reactivation Malignancies (frequently solid tumors) |
| Ocrelizumab | HSV1/VVZ reactivation Hypogammaglobulinemia Malignancies (frequently breast cancer) PML |
| Siponimod | Hypertension Diabetes Macular edema |
| Dimethyl fumarate | Lymphopenia PML (frequently related to grade 3 lymphopenia) |
| SCAP | Senolytic Drug |
|---|---|
| Bcl-2/Bcl-XL | Navitoclax A1331852 A1155463 Fisetin |
| PI3K/Akt/ROS | Quercetin Piperlongumine Fisetin |
| p53/p21/serpine | Quercetin Fisetin FOXO related molecule |
| Ephrins/dependence receptors/tyrosine kinases | Dasatinib Piperlongumine |
| HIF-1α | Quercetin Fisetin |
| HSP-90 | Tanespimycin Geldanamycin Alvespimycin |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neațu, M.; Hera-Drăguț, A.; Ioniță, I.; Jugurt, A.; Davidescu, E.I.; Popescu, B.O. Understanding the Complex Dynamics of Immunosenescence in Multiple Sclerosis: From Pathogenesis to Treatment. Biomedicines 2024, 12, 1890. https://doi.org/10.3390/biomedicines12081890
Neațu M, Hera-Drăguț A, Ioniță I, Jugurt A, Davidescu EI, Popescu BO. Understanding the Complex Dynamics of Immunosenescence in Multiple Sclerosis: From Pathogenesis to Treatment. Biomedicines. 2024; 12(8):1890. https://doi.org/10.3390/biomedicines12081890
Chicago/Turabian StyleNeațu, Monica, Ana Hera-Drăguț, Iulia Ioniță, Ana Jugurt, Eugenia Irene Davidescu, and Bogdan Ovidiu Popescu. 2024. "Understanding the Complex Dynamics of Immunosenescence in Multiple Sclerosis: From Pathogenesis to Treatment" Biomedicines 12, no. 8: 1890. https://doi.org/10.3390/biomedicines12081890