
Infliximab in Inflammatory Bowel Disease: Leveraging Physiologically Based Pharmacokinetic Modeling in the Clinical Context


Abstract
1. Introduction
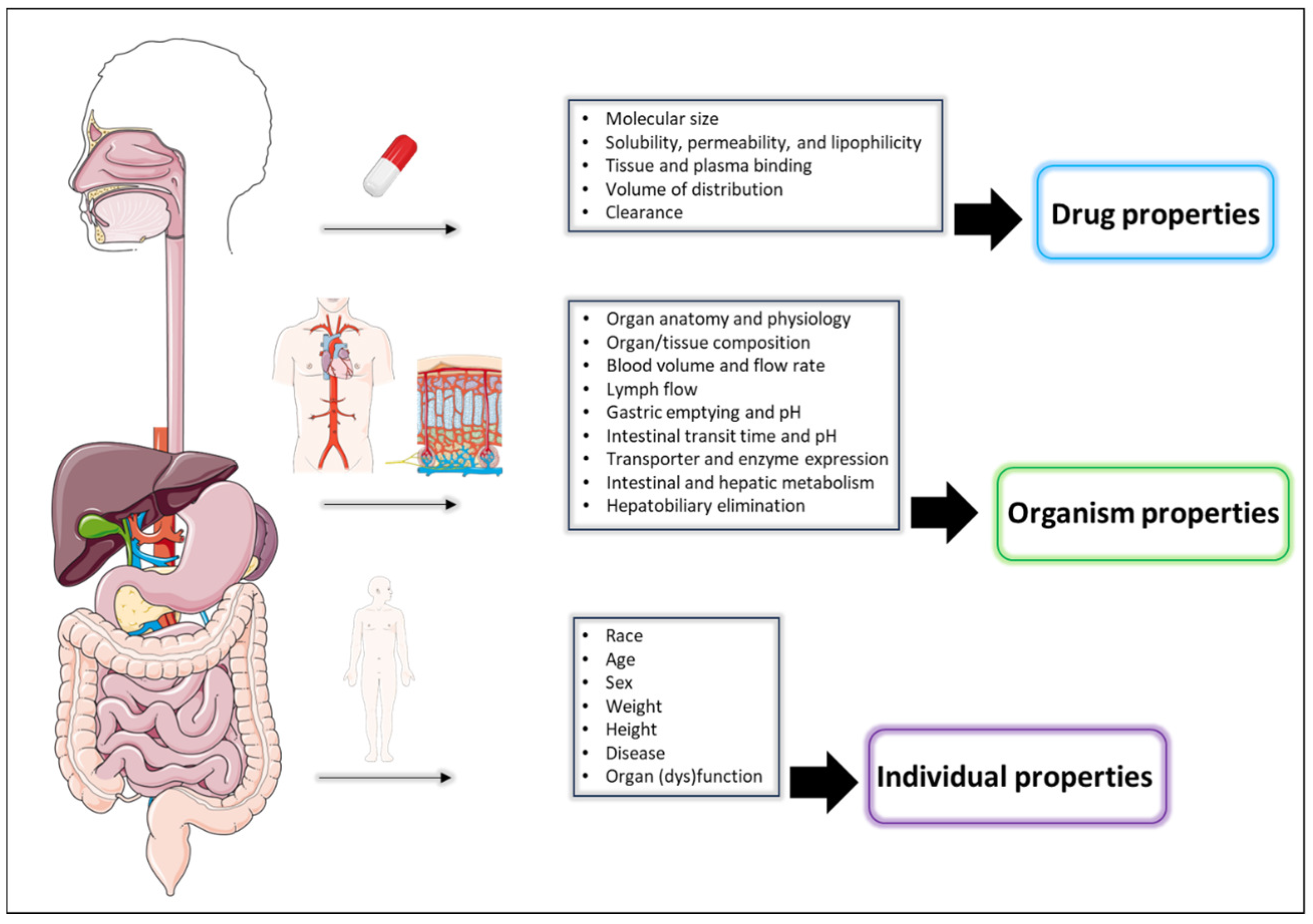
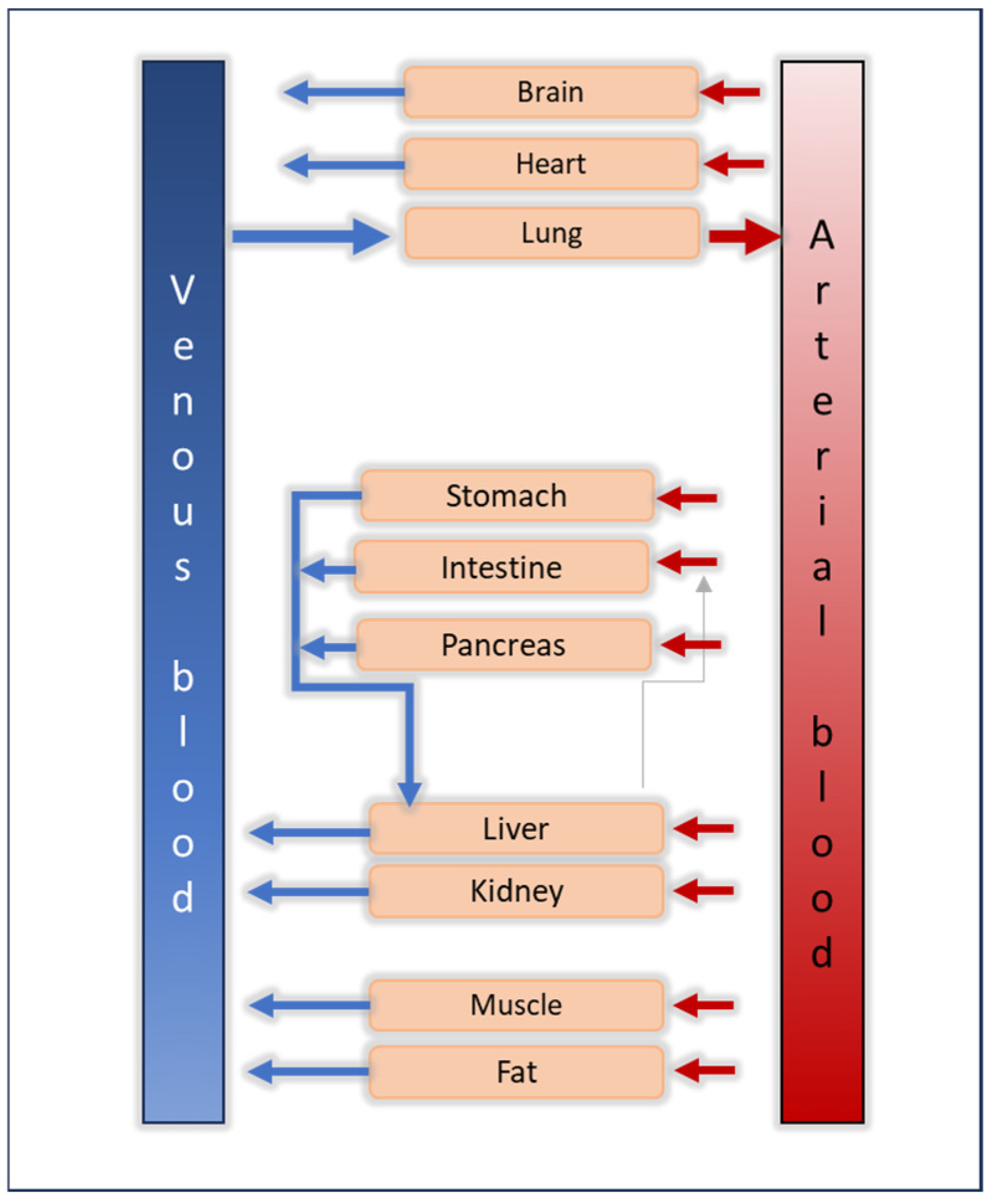
Quantitative Clinical Pharmacology Tools
2. Materials and Methods
3. Results and Discussion
3.1. PBPK Model Development for a Virtual Healthy Adult after Single Intravenous Administration of Infliximab (5 mg/kg)
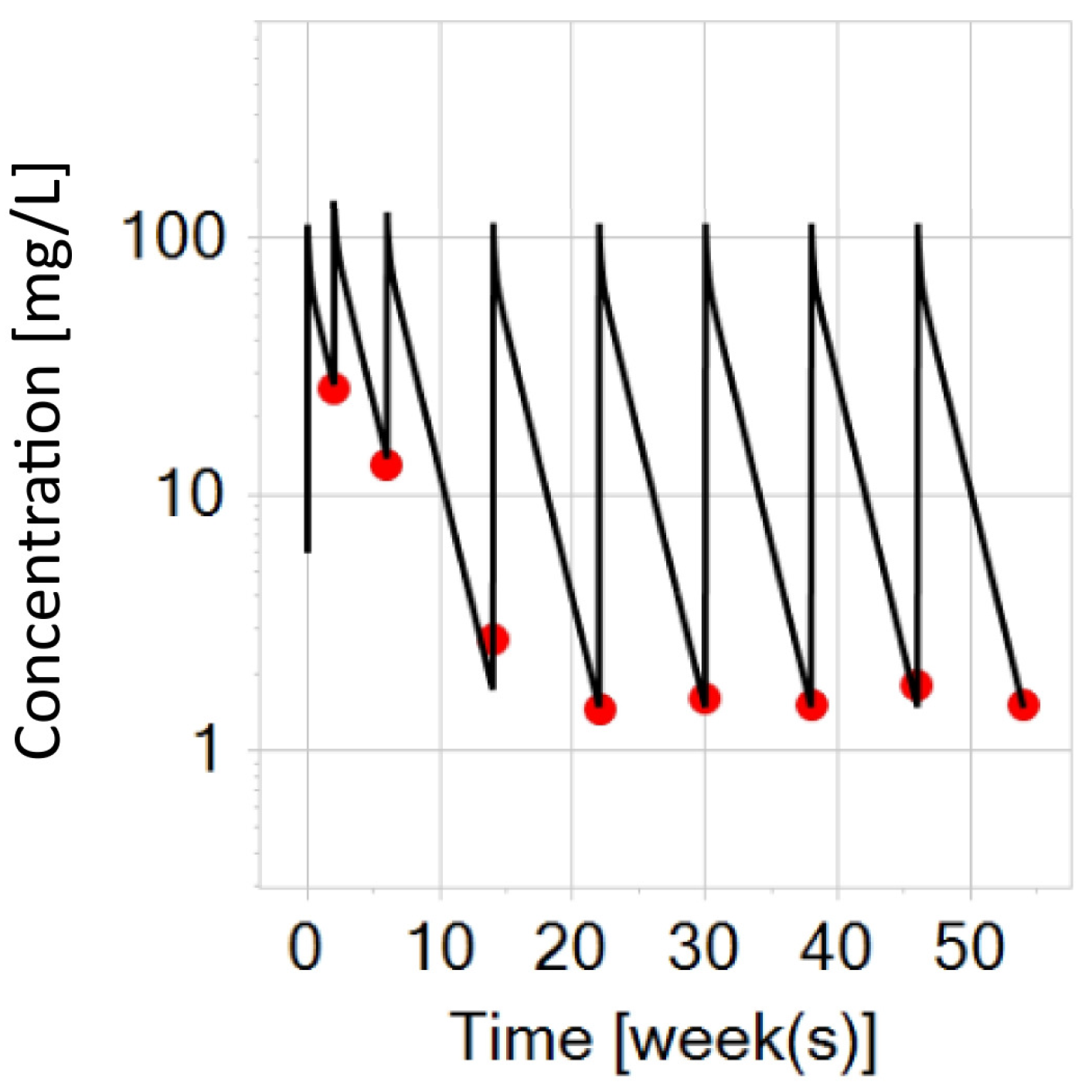
3.2. PBPK Model Development for a Virtual Adult with IBD after Intravenous Administrations of Infliximab (5 mg/kg)
3.3. PBPK Modeling Exploration for Virtual Adults with IBD (Typical and Heavy) after Switching to Subcutaneous Infliximab
3.4. PBPK Modeling Exploration for an Older Virtual Child (14-Year-Old, 60 kg) with IBD after Intravenous Administrations of Infliximab (5 mg/kg)
3.5. PBPK Modeling Exploration for an Older Virtual Child (14-Year-Old, 60 kg) with IBD after Switching to a Flat-Fixed Dose of Subcutaneous Infliximab (120 mg)
3.6. PBPK Modeling Exploration for a Virtual Pediatric Population Stratified by Weight Bands after Switching to Subcutaneous Infliximab: A PBPK-Model-Informed Dose Suggestion
3.7. Limitations and Future Perspectives of the Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Petric, Z.; Goncalves, J.; Paixao, P. Under the Umbrella of Clinical Pharmacology: Inflammatory Bowel Disease, Infliximab and Adalimumab, and a Bridge to an Era of Biosimilars. Pharmaceutics 2022, 14, 1766. [Google Scholar] [CrossRef]
- Ungar, B.; Mazor, Y.; Weisshof, R.; Yanai, H.; Ron, Y.; Goren, I.; Waizbard, A.; Yavzori, M.; Fudim, E.; Picard, O.; et al. Induction infliximab levels among patients with acute severe ulcerative colitis compared with patients with moderately severe ulcerative colitis. Aliment. Pharmacol. Ther. 2016, 43, 1293–1299. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency (EMA). Remicade (Infliximab) Assessment Report. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/remicade (accessed on 26 June 2024).
- Smith, P.J.; Fumery, M.; Leong, R.W.; Novak, K.; Dignass, A. Real-world experience with subcutaneous infliximab: Broadening treatment strategies for inflammatory bowel disease. Expert. Rev. Clin. Immunol. 2023, 19, 1143–1156. [Google Scholar] [CrossRef] [PubMed]
- Vandenplas, Y.; Broekaert, I.; Domellöf, M.; Indrio, F.; Lapillonne, A.; Pienar, C.; Ribes-Koninckx, C.; Shamir, R.; Szajewska, H.; Thapar, N.; et al. An ESPGHAN position paper on the diagnosis, management and prevention of cow’s milk allergy. J. Pediatr. Gastroenterol. Nutr. 2024, 78, 386–413. [Google Scholar] [CrossRef] [PubMed]
- Mould, D.R.; Upton, R.N. Basic concepts in population modeling, simulation, and model-based drug development. CPT Pharmacomet. Syst. Pharmacol. 2012, 1, 1–14. [Google Scholar] [CrossRef]
- Jones, H.; Rowland-Yeo, K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.A. Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulations: Principles, Methods, and Applications in the Pharmaceutical Industry; John Wiley & Sons: Hoboken, NJ, USA, 2022. [Google Scholar]
- Open Systems Pharmacology Suite, Manual. Available online: https://docs.open-systems-pharmacology.org/ (accessed on 26 June 2024).
- Servier—Servier Medical Art licensed under CC BY 4.0. Available online: https://smart.servier.com/ (accessed on 26 June 2024).
- Perry, C.; Davis, G.; Conner, T.M.; Zhang, T. Utilization of Physiologically Based Pharmacokinetic Modeling in Clinical Pharmacology and Therapeutics: An Overview. Curr. Pharmacol. Rep. 2020, 6, 71–84. [Google Scholar] [CrossRef]
- Luzon, E.; Blake, K.; Cole, S.; Nordmark, A.; Versantvoort, C.; Berglund, E.G. Physiologically based pharmacokinetic modeling in regulatory decision-making at the European Medicines Agency. Clin. Pharmacol. Ther. 2017, 102, 98–105. [Google Scholar] [CrossRef]
- Rowland Yeo, K.; Gil Bergland, E.; Chen, Y. Dose Optimization Informed by PBPK Modeling: State-of-the Art and Future. Clin. Pharmacol. Ther. 2024, 116, 563–576. [Google Scholar] [CrossRef]
- Malik, P.R.V.; Edginton, A.N. Physiologically-Based Pharmacokinetic Modeling vs. Allometric Scaling for the Prediction of Infliximab Pharmacokinetics in Pediatric Patients. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 835–844. [Google Scholar] [CrossRef]
- Rippe, B.; Haraldsson, B. Transport of macromolecules across microvascular walls: The two-pore theory. Physiol. Rev. 1994, 74, 163–219. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.E.; Haegebaert, R.M.S.; Østergaard, J.; Jensen, H. Size-based characterization of adalimumab and TNF-α interactions using flow induced dispersion analysis: Assessment of avidity-stabilized multiple bound species. Sci. Rep. 2021, 11, 4754. [Google Scholar] [CrossRef] [PubMed]
- Hanzel, J.; Bukkems, L.H.; Gecse, K.B.; D’Haens, G.R.; Mathôt, R.A.A. Population pharmacokinetics of subcutaneous infliximab CT-P13 in Crohn’s disease and ulcerative colitis. Aliment. Pharmacol. Ther. 2021, 54, 1309–1319. [Google Scholar] [CrossRef]
- EMA, Clinical Trial Register. Available online: https://clinicaldata.ema.europa.eu/web/cdp (accessed on 25 June 2024).
- Zunino, C.; Perrier, J.; Gualano, V.; Zhou, H.; Lukacova, V.; Le Merdy, M. Validation of PBPK-Based Translation to Predict Monoclonal Antibody Pharmacokinetics in Pediatric Populations. In Proceedings of the 2023 AAPS National Biotechnology Conference, Philadelphia, PA, USA, 23–26 April 2023. [Google Scholar]
- Reinisch, W.; Jang, B.I.; Borzan, V.; Lahat, A.; Pukitis, A.; Osipenko, M.; Mostovoy, Y.; Schreiber, S.; Ben-Horin, S.; Lee, S.J.; et al. DOP62 A novel formulation of CT-P13 (infliximab biosimilar) for subcutaneous administration: 1-year result from a Phase I open-label randomised controlled trial in patients with active Crohn’s disease. J. Crohns Colitis 2019, 13, S066–S067. [Google Scholar] [CrossRef]
- Adedokun, O.J.; Sandborn, W.J.; Feagan, B.G.; Rutgeerts, P.; Xu, Z.; Marano, C.W.; Johanns, J.; Zhou, H.; Davis, H.M.; Cornillie, F.; et al. Association Between Serum Concentration of Infliximab and Efficacy in Adult Patients with Ulcerative Colitis. Gastroenterology 2014, 147, 1296–1307.e5. [Google Scholar] [CrossRef]
- Gianolio, L.; Armstrong, K.; Swann, E.; Shepherd, R.; Henderson, P.; Wilson, D.C.; Russell, R.K. Effectiveness of Switching to Subcutaneous Infliximab in Pediatric Inflammatory Bowel Disease Patients on Intravenous Maintenance Therapy. J. Pediatr. Gastroenterol. Nutr. 2023, 77, 235–239. [Google Scholar] [CrossRef]
- McInnes, S.J.P.; Turner, C.T.; Al-Bataineh, S.A.; Airaghi Leccardi, M.J.I.; Irani, Y.; Williams, K.A.; Cowin, A.J.; Voelcker, N.H. Surface engineering of porous silicon to optimise therapeutic antibody loading and release. J. Mater. Chem. B 2015, 3, 4123–4133. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Ishii-Watabe, A.; Tada, M.; Kobayashi, T.; Kanayasu-Toyoda, T.; Kawanishi, T.; Yamaguchi, T. Importance of neonatal FcR in regulating the serum half-life of therapeutic proteins containing the Fc domain of human IgG1: A comparative study of the affinity of monoclonal antibodies and Fc-fusion proteins to human neonatal FcR. J. Immunol. 2010, 184, 1968–1976. [Google Scholar] [CrossRef]
- Grevys, A.; Frick, R.; Mester, S.; Flem-Karlsen, K.; Nilsen, J.; Foss, S.; Sand, K.M.K.; Emrich, T.; Fischer, J.A.A.; Greiff, V.; et al. Antibody variable sequences have a pronounced effect on cellular transport and plasma half-life. iScience 2022, 25, 103746. [Google Scholar] [CrossRef]
- Scallon, B.J.; Moore, M.A.; Trinh, H.; Knight, D.M.; Ghrayeb, J. Chimeric anti-TNF-α monoclonal antibody cA2 binds recombinant transmembrane TNF-α and activates immune effector functions. Cytokine 1995, 7, 251–259. [Google Scholar] [CrossRef]
- Li, X.; Jusko, W.; Cao, Y. Role of interstitial fluid turnover on target suppression by therapeutic biologics using a minimal physiologically-based pharmacokinetic (mPBPK) model. J. Pharmacol. Exp. Ther. 2018, 367, 1–8. [Google Scholar] [CrossRef]
- Niederalt, C.; Kuepfer, L.; Solodenko, J.; Eissing, T.; Siegmund, H.U.; Block, M.; Willmann, S.; Lippert, J. A generic whole body physiologically based pharmacokinetic model for therapeutic proteins in PK-Sim. J. Pharmacokinet. Pharmacodyn. 2018, 45, 235–257. [Google Scholar] [CrossRef]
- Children’s Health Queensland: Estimated Weight Based on Age. Available online: https://www.childrens.health.qld.gov.au/ (accessed on 26 June 2024).
- Lambert, J.; Wyand, M.; Lassen, C.; Shneyer, L.; Thomson, E.; Knight, A.; Willers, J.; Kay, J. Bioavailability, safety and immunogenicity of biosimilar infliximab (BOW015) compared to reference infliximab. Int. J. Clin. Pharmacol. Ther. 2016, 54, 315–322. [Google Scholar] [CrossRef]
- Marônek, M.; Marafini, I.; Gardlík, R.; Link, R.; Troncone, E.; Monteleone, G. Metalloproteinases in Inflammatory Bowel Diseases. J. Inflamm. Res. 2021, 14, 1029–1041. [Google Scholar] [CrossRef]
- Klotz, U.; Teml, A.; Schwab, M. Clinical Pharmacokinetics and Use of Infliximab. Clin. Pharmacokinet. 2007, 46, 645–660. [Google Scholar] [CrossRef]
- Schreiber, S.; Ben-Horin, S.; Leszczyszyn, J.; Dudkowiak, R.; Lahat, A.; Gawdis-Wojnarska, B.; Pukitis, A.; Horynski, M.; Farkas, K.; Kierkus, J.; et al. Randomized Controlled Trial: Subcutaneous vs Intravenous Infliximab CT-P13 Maintenance in Inflammatory Bowel Disease. Gastroenterology 2021, 160, 2340–2353. [Google Scholar] [CrossRef]
- Schreiber, S.; Jang, B.; Borzan, V.; Lahat, A.; Pukitis, A.; Osipenko, M.; Mostovoy, Y.; Ben-Horin, S.; Ye, B.D.; Lee, S.J.; et al. Tu2018—Novel Formulation of CT-P13 (Infliximab Biosimilar) for Subcutaneous Administration: Initial Results from a Phase I Open-Label Randomized Controlled Trial in Patients with Active Crohn’s Disease. Gastroenterology 2018, 6, S-1371. [Google Scholar] [CrossRef]
- European Medicines Agency (EMA). Assessment Report on Remsima (Infliximab). Available online: https://www.ema.europa.eu/en/documents/assessment-report/remsima-epar-public-assessment-report_en.pdf (accessed on 26 June 2024).
- Pan, X.; Stader, F.; Abduljalil, K.; Gill, K.L.; Johnson, T.N.; Gardner, I.; Jamei, M. Development and Application of a Physiologically-Based Pharmacokinetic Model to Predict the Pharmacokinetics of Therapeutic Proteins from Full-term Neonates to Adolescents. AAPS J. 2020, 22, 76. [Google Scholar] [CrossRef]
- Roblin, X.; Veyrard, P.; Bastide, L.; Berger, A.E.; Barrau, M.; Paucelle, A.-S.; Waeckel, L.; Kwiatek, S.; Flourie, B.; Nancey, S.; et al. Subcutaneous injection of infliximab CT-P13 results in stable drug levels within 14-day treatment cycle in Crohn’s disease. Aliment. Pharmacol. Ther. 2022, 56, 77–83. [Google Scholar] [CrossRef]
- Little, R.D.; Ward, M.G.; Wright, E.; Jois, A.J.; Boussioutas, A.; Hold, G.L.; Gibson, P.R.; Sparrow, M.P. Therapeutic Drug Monitoring of Subcutaneous Infliximab in Inflammatory Bowel Disease—Understanding Pharmacokinetics and Exposure Response Relationships in a New Era of Subcutaneous Biologics. J. Clin. Med. 2022, 11, 6173. [Google Scholar] [CrossRef]
- Falquina, I.C.M.; Chumillas, R.M.S.; Garcia, L.A.; González, B.V.; Cuesta, N.R.; Hernández, L.A.; Pascual, L.A.; Aladrén, B.S. P617 Switching from an intensified regimen of infliximab to a subcutaneous standard dose in adults with Inflammatory Bowel Disease: Our experience in a tertiary hospital. J. Crohns. Colitis. 2022, 16, i544–i545. [Google Scholar] [CrossRef]
- Malik, P.; Edginton, A. Pediatric physiology in relation to the pharmacokinetics of monoclonal antibodies. Expert. Opin. Drug. Metab. Toxicol. 2018, 14, 585–599. [Google Scholar] [CrossRef]
- Cornillie, F.; Shealy, D.; D’Haens, G.; Geboes, K.; Van Assche, G.; Ceuppens, J.; Wagner, C.; Schaible, T.; Plevy, S.E.; Targan, S.R.; et al. Infliximab induces potent anti-inflammatory and local immunomodulatory activity but no systemic immune suppression in patients with Crohn’s disease. Aliment. Pharmacol. Ther. 2001, 15, 463–473. [Google Scholar] [CrossRef]
- Ternant, D.; Mulleman, D.; Degenne, D.; Willot, S.; Guillaumin, J.M.; Watier, H.; Goupille, P.; Paintaud, G. An enzyme-linked immunosorbent assay for therapeutic drug monitoring of infliximab. Ther. Drug. Monit. 2006, 28, 169–174. [Google Scholar] [CrossRef]
- Adedokun, O.J.; Xu, Z.; Padgett, L.; Blank, M.; Johanns, J.; Griffiths, A.; Ford, J.; Zhou, H.; Guzzo, C.; Davis, H.M.; et al. Pharmacokinetics of infliximab in children with moderate-to-severe ulcerative colitis: Results from a randomized, multicenter, open-label, phase 3 study. Inflamm. Bowel Dis. 2013, 19, 2753–2762. [Google Scholar] [CrossRef]
- Jongsma, M.M.E.; Winter, D.A.; Huynh, H.Q.; Norsa, L.; Hussey, S.; Kolho, K.L.; Bronsky, J.; Assa, A.; Cohen, S.; Lev-Tzion, R.; et al. Infliximab in young paediatric IBD patients: It is all about the dosing. Eur. J. Pediatr. 2020, 179, 1935–1944. [Google Scholar] [CrossRef]
- Argüelles-Arias, F.; Fernández Álvarez, P.; Castro Laria, L.; Maldonado Pérez, B.; Belvis Jiménez, M.; Merino-Bohórquez, V.; Caunedo Álvarez, Á.; Calleja Hernández, M. Switch to subcutaneous infliximab during the SARS-CoV-2 pandemic: Preliminary results. Rev. Esp. Enferm. Dig. 2022, 114, 118–119. [Google Scholar] [CrossRef]
- Derraik, J.G.; Rademaker, M.; Cutfield, W.S.; Pinto, T.E.; Tregurtha, S.; Faherty, A.; Peart, J.M.; Drury, P.L.; Hofman, P.L. Effects of age, gender, BMI, and anatomical site on skin thickness in children and adults with diabetes. PLoS ONE 2014, 9, e86637. [Google Scholar] [CrossRef]
- Davis, J.D.; Bravo Padros, M.; Conrado, D.J.; Ganguly, S.; Guan, X.; Hassan, H.E.; Hazra, A.; Irvin, S.C.; Jayachandran, P.; Kosloski, M.P.; et al. Subcutaneous Administration of Monoclonal Antibodies: Pharmacology, Delivery, Immunogenicity, and Learnings from Applications to Clinical Development. Clin. Pharmacol. Ther. 2024, 115, 422–439. [Google Scholar] [CrossRef]
- Shebley, M.; Sandhu, P.; Emami Riedmaier, A.; Jamei, M.; Narayanan, R.; Patel, A.; Peters, S.A.; Reddy, V.P.; Zheng, M.; de Zwart, L.; et al. Physiologically Based Pharmacokinetic Model Qualification and Reporting Procedures for Regulatory Submissions: A Consortium Perspective. Clin. Pharmacol. Ther. 2018, 104, 88–110. [Google Scholar] [CrossRef]
- Basu, S.; Lien, Y.T.K.; Vozmediano, V.; Schlender, J.F.; Eissing, T.; Schmidt, S.; Niederalt, C. Physiologically Based Pharmacokinetic Modeling of Monoclonal Antibodies in Pediatric Populations Using PK-Sim. Front. Pharmacol. 2020, 11, 868. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
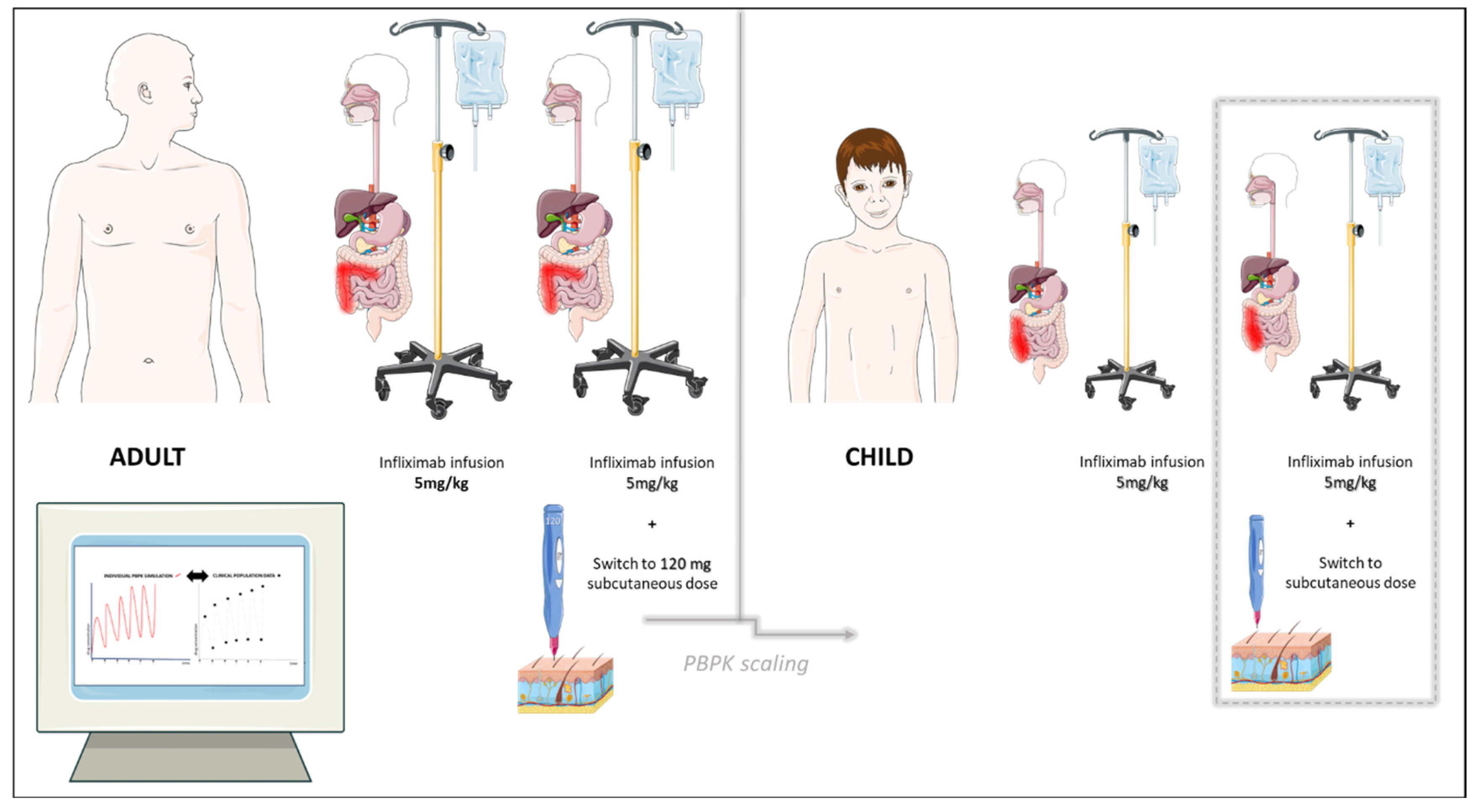
| 1. Clinical trial in healthy adults, single intravenous dose 5 mg/kg [18] |
| 2. Clinical trial in adult patients with IBD [20] intravenous dose 5 mg/kg induction + maintenance up to week 54 subcutaneous switch, dose: 120 mg (skin of abdomen or upper thigh) subcutaneous switch, dose: 240 mg (skin of abdomen or upper thigh) |
| 3. Clinical trial in adult patients with IBD, population pharmacokinetic study (subcutaneous switch to 120 mg in adults with IBD) and simulations on smaller, average, and heavier adult patients [17] |
| 4. Clinical trial in pediatric population with IBD (intravenous route 5 mg/kg, ulcerative colitis) [14,21] |
| 5. Clinical trial in older pediatric population with IBD (post-switch to subcutaneous 120 mg, skin of abdomen or upper thigh) [22] |
| TNF-α |
| Reference concentration: 0.2 pm [16] |
| Localization: plasma and interstitial space; relative expression: 1 [14] |
| Infliximab |
| Molecular weight: 149.9 kDa [1] |
| Radius (solute): 5.18 nm [23] |
| Kd (FcRn) in endosomal space: 0.73 µmol/L [24] |
| Kd (FcRn) plasma/interstitial *: 4.54 mmol/L (i.e., negligible binding affinity [25])Kd (TNF-α) *: 120.17 pmol/L (100 pmol/L [26]) |
| Koff (TNF-α) *: 1.331/ms (111/ms [27]) |
| Kass (FcRn)*: 0.72 L/µmol/min (0.80 L/µmol/min [28]) |
| Anatomy and physiology of a virtual healthy adult |
| Fraction endosomal: 0.20 [28] |
| Specific clearance in endosome *: 0.911/min |
| Fraction recycled to plasma: 1 [28] |
| Fraction of endosomal uptake from plasma *: 0.53 |
| Rate constant for endosomal uptake: 0.291/min [28] |
| Rate constant for recycling from endosomal space *: 0.301/min |
| Kd (FcRn, endogenous IgG) in plasma/interstitial space: 10,000 µmol/L [28] |
| Kd (FcRn, endogenous IgG) in endosome: 0.63 µmol/L [28] |
| Kass (FcRn, endogenous IgG): 0.87 L/ µmol/min [28] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petric, Z.; Gonçalves, J.; Paixão, P. Infliximab in Inflammatory Bowel Disease: Leveraging Physiologically Based Pharmacokinetic Modeling in the Clinical Context. Biomedicines 2024, 12, 1974. https://doi.org/10.3390/biomedicines12091974
Petric Z, Gonçalves J, Paixão P. Infliximab in Inflammatory Bowel Disease: Leveraging Physiologically Based Pharmacokinetic Modeling in the Clinical Context. Biomedicines. 2024; 12(9):1974. https://doi.org/10.3390/biomedicines12091974
Chicago/Turabian StylePetric, Zvonimir, João Gonçalves, and Paulo Paixão. 2024. "Infliximab in Inflammatory Bowel Disease: Leveraging Physiologically Based Pharmacokinetic Modeling in the Clinical Context" Biomedicines 12, no. 9: 1974. https://doi.org/10.3390/biomedicines12091974
APA StylePetric, Z., Gonçalves, J., & Paixão, P. (2024). Infliximab in Inflammatory Bowel Disease: Leveraging Physiologically Based Pharmacokinetic Modeling in the Clinical Context. Biomedicines, 12(9), 1974. https://doi.org/10.3390/biomedicines12091974