
Increased Myocardial MAO-A, Atrogin-1, and IL-1β Expression in Transgenic Mice with Pancreatic Carcinoma—Benefit of MAO-A Inhibition for Cardiac Cachexia

Abstract
1. Introduction
2. Material and Methods
2.1. Transgenic Mouse Model of PDAC
2.2. Western Blotting of MAO-A Expression
2.3. Immunolocalization of Atrogin-1+, IL-1β+, TNF+, COX2+, and CD68+ Cells
2.4. Myocardial Capillary Density
2.5. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
2.6. Statistical Analyses
3. Results
3.1. Group Characteristics
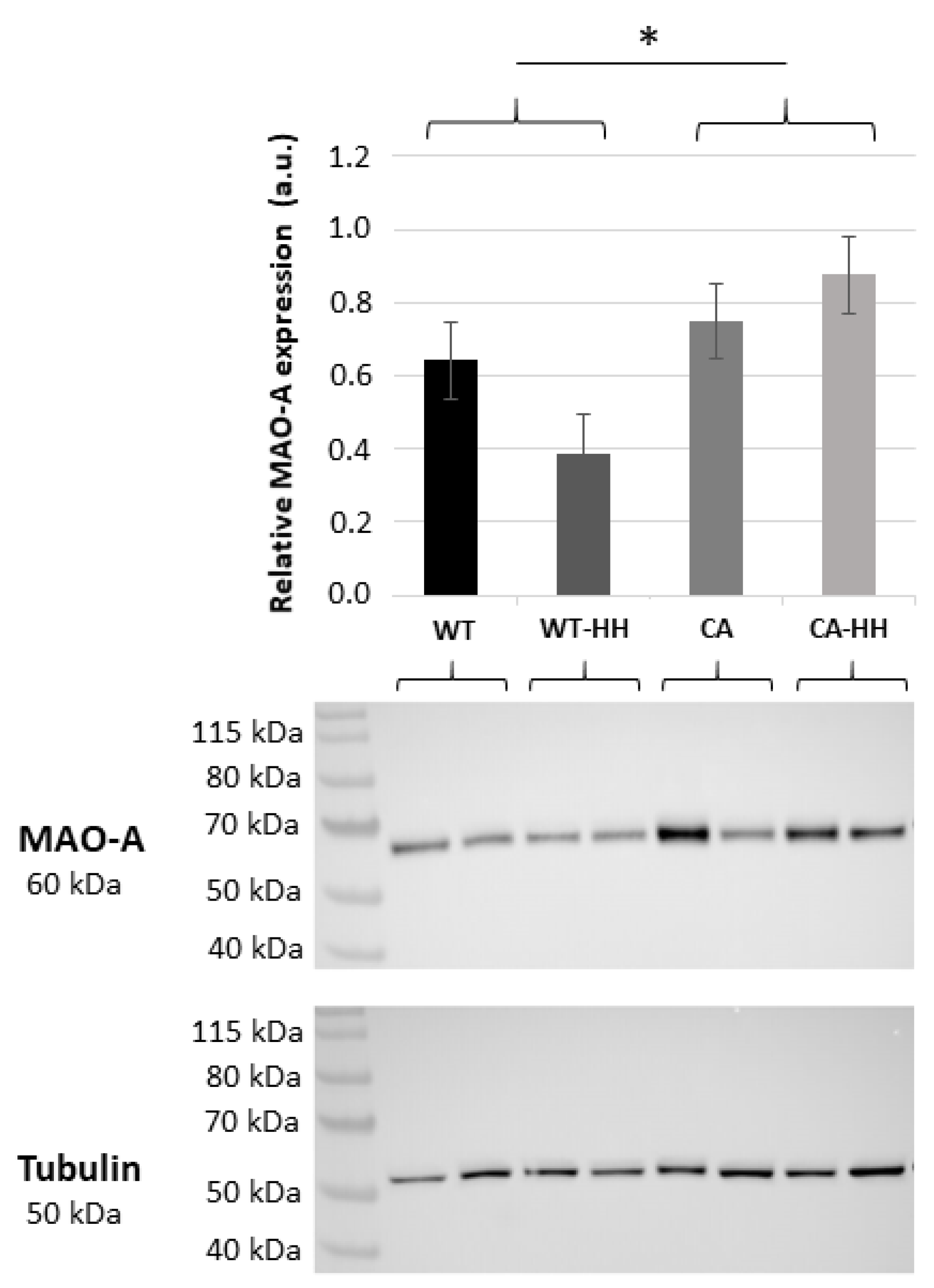
3.2. Increased MAO-A Protein Content in PDAC Compared to WT Mice
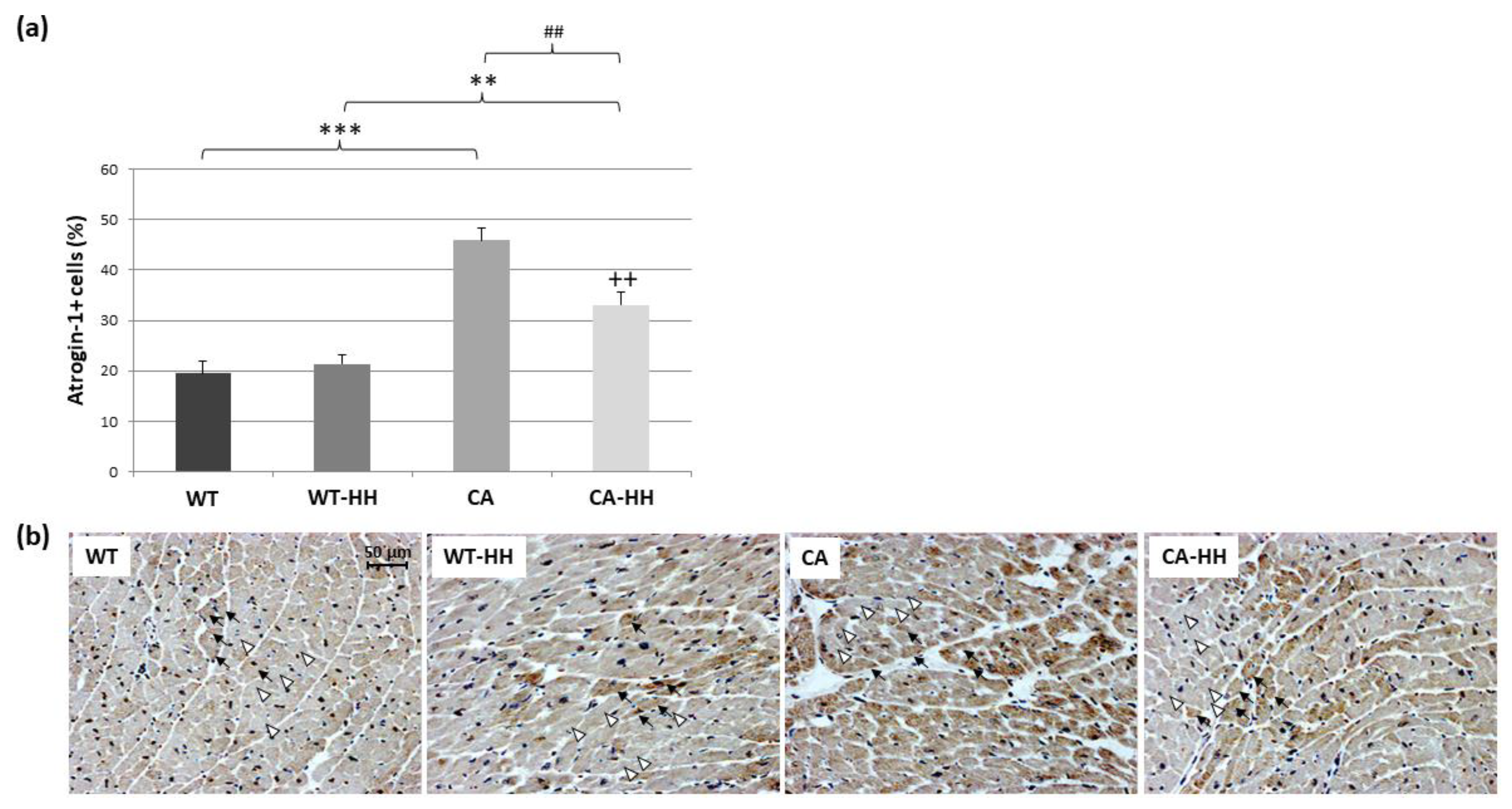
3.3. Percentage of Atrogin-1+ Cells Is Increased in PDAC and Reversed by HH-Treatment
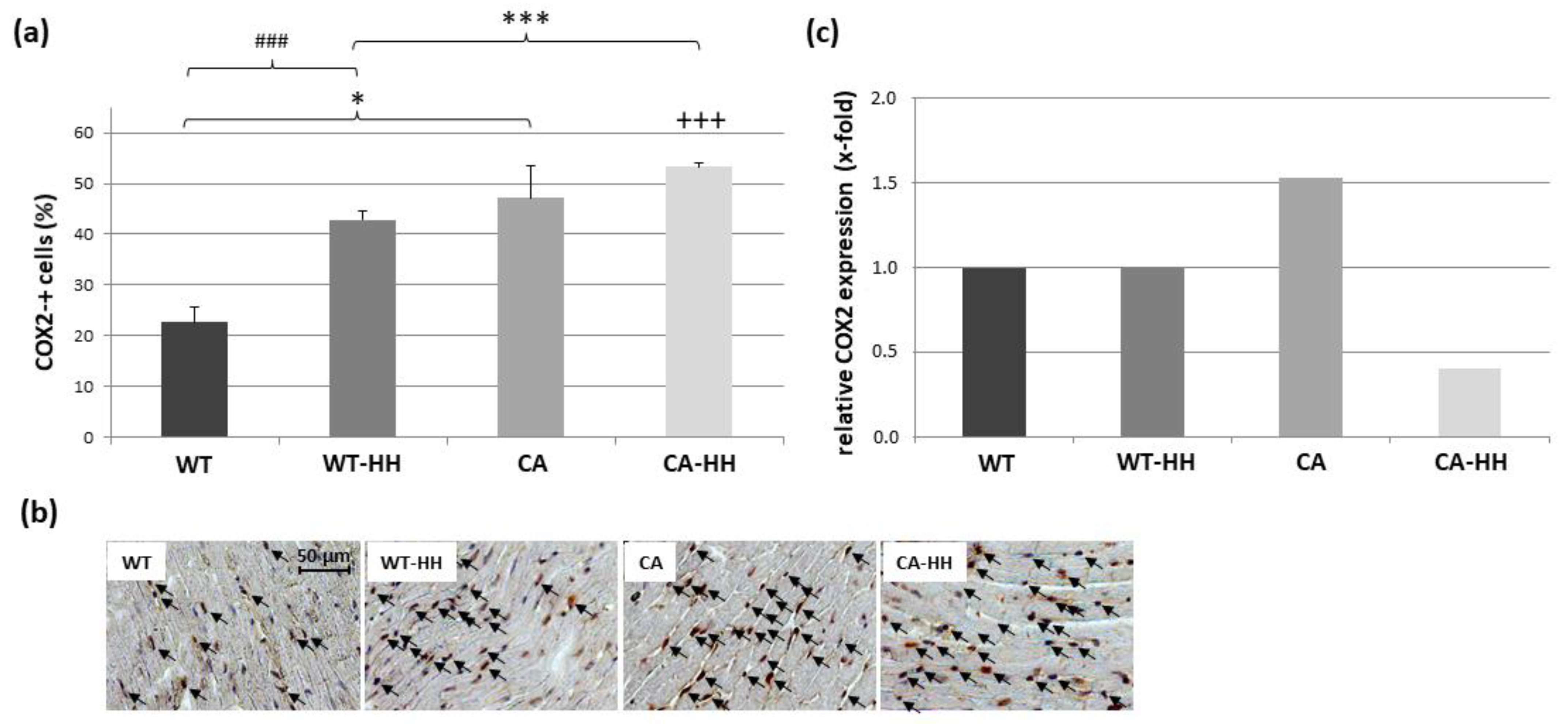
3.4. Upregulation of IL-1β, TNF, COX2, CD68, and SOCS with PDAC and Differential Anti-Inflammatory Effect of HH Treatment
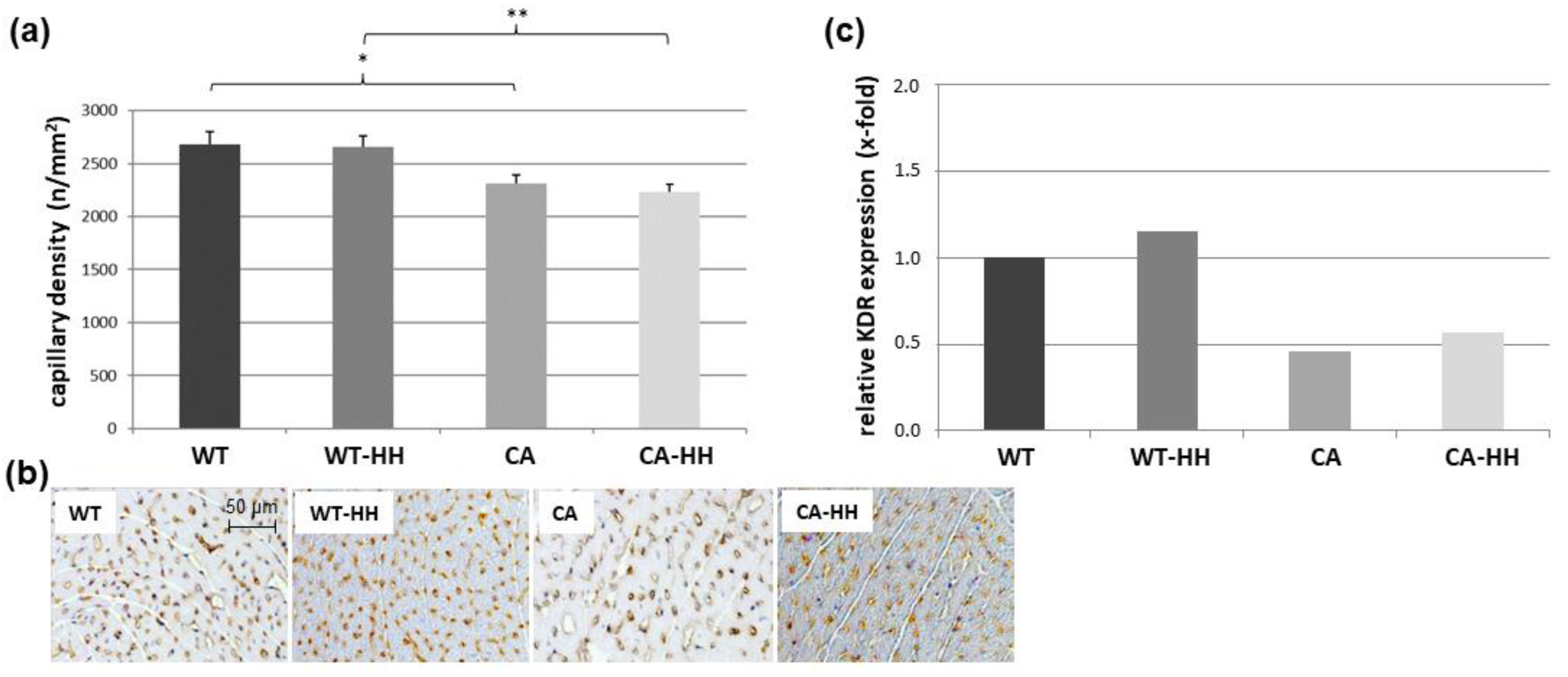
3.5. Effect of PDAC and HH on Myocardial Capillary Density and Angiogenic Signals
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Primer 2018, 4, 17105. [Google Scholar] [CrossRef] [PubMed]
- Johns, N.; Hatakeyama, S.; Stephens, N.A.; Degen, M.; Degen, S.; Frieauff, W.; Lambert, C.; Ross, J.A.; Roubenoff, R.; Glass, D.J.; et al. Clinical Classification of cancer cachexia: Phenotypic correlates in human skeletal muscle. PLoS ONE 2014, 9, e83618. [Google Scholar] [CrossRef]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Argilés, J.M.; Anker, S.D.; Evans, W.J.; Morley, J.E.; Fearon, K.C.; Strasser, F.; Muscaritoli, M.; Baracos, V.E. Consensus on cachexia definition. J. Am. Med. Dir. Assoc. 2010, 11, 229–230. [Google Scholar] [CrossRef]
- Tisdale, M.J. Mechanisms of cancer cachexia. Physiol. Rev. 2009, 89, 381–410. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, J.; Ketterer, K.; Marsch, C.; Fechtner, K.; Krakowski-Roosen, H.; Büchler, M.W.; Friess, H.; Martignoni, M.E. Pancreatic cancer related cachexia: Influence on metabolism and correlation to weight loss and pulmonary function. BMC Cancer 2009, 9, 255. [Google Scholar] [CrossRef]
- Bachmann, J.; Heiligensetzer, M.; Krakowski-Roosen, H.; Büchler, M.W.; Friess, H.; Martignoni, M.E. Cachexia worsens prognosis in patients with resectable pancreatic cancer. J. Gastrointest. Surg. 2008, 1287, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Neshan, M.; Tsilimigras, D.I.; Han, X.; Zhu, H.; Pawlik, T.M. Molecular mechanism of cachexia: A review. Cells 2024, 13, 252. [Google Scholar] [CrossRef]
- Hildebrandt, W.; Keck, J.; Schmich, S.; Bonaterra, G.A.; Wilhelm, B.; Schwarzbach, H.; Eva, A.; Bertoune, M.; Slater, E.P.; Fendrich, V.; et al. Inflammation and wasting of skeletal muscles in Kras-p53-mutant mice with intraepithelial neoplasia and pancreatic cancer—When does cachexia start? Cells 2022, 11, 1607. [Google Scholar] [CrossRef]
- Martin, A.; Freyssenet, D. Phenotypic features of cancer cachexia-related loss of skeletal muscle mass and function. J. Cachexia Sarcopenia Muscle 2021, 12, 252–273. [Google Scholar] [CrossRef]
- Argiles, J.M.; Busquets, S.; Stemmler, B.; Lopez-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Johns, N.; Stephens, N.A.; Fearon, K.C.H. Muscle wasting in cancer. Int. J. Biochem. Cell Biol. 2013, 45, 2215–2229. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S.; et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef]
- Weber, M.A.; Krakowski-Roosen, H.; Schröder, L.; Kinscherf, R.; Krix, M.; Kopp-Schneider, A.; Essig, M.; Bachert, P.; Kauczor, H.U.; Hildebrandt, W.; et al. Morphology, metabolism, microcirculation, and strength of skeletal muscles in cancer-related cachexia. Acta Oncol. 2009, 48, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Marcondes, M.C.; Tisdale, M.J. Induction of protein catabolism and the ubiquitin-proteasome pathway by mild oxidative stress. Cancer Lett. 2002, 180, 69–74. [Google Scholar] [CrossRef]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Li, B.; Xu, Y.; Meng, S.; Wang, Y.; Jiang, Y. Luteolin reduces cancer-induced skeletal and cardiac muscle atrophy in a Lewis lung cancer mouse model. Oncol. Rep. 2018, 40, 1129–1137. [Google Scholar] [CrossRef]
- Saha, S.; Kumar Singh, P.; Roy, P.; Kakar, S.S. Cardiac cachexia: Unaddressed aspect in cancer patients. Cells 2022, 11, 990. [Google Scholar] [CrossRef]
- Lena, A.; Wilkenshoff, U.; Hadzibegovic, S.; Porthun, J.; Rösnick, L.; Fröhlich, A.K.; Zeller, T.; Karakas, M.; Keller, U.; Ahn, J.; et al. Clinical and prognostic relevance of cardiac wasting in patients with advanced cancer. J. Am. Coll. Cardiol. 2023, 81, 1569–1586. [Google Scholar] [CrossRef]
- Diba, P.; Sattler, A.L.; Korzun, T.; Habecker, B.A.; Marks, D.L. Unraveling the lost balance: Adrenergic dysfunction in cancer cachexia. Auton. Neurosci. 2024, 251, 103136. [Google Scholar] [CrossRef]
- Knapp, F.; Niemann, B.; Li, L.; Molenda, N.; Kracht, M.; Schulz, R.; Rohrbach, S. Differential effects of right and left heart failure on skeletal muscle in rats. J. Cachexia Sarcopenia Muscle 2020, 11, 1830–1849. [Google Scholar] [CrossRef]
- Wiggs, M.P.; Beaudry, A.G.; Law, M.L. Cardiac Remodeling in Cancer-Induced Cachexia: Functional, Structural, and Metabolic Contributors. Cells 2022, 11, 1931. [Google Scholar] [CrossRef]
- Mühlfeld, C.; Kumar Das, S.K.; Heinzel, F.R.; Schmidt, A.; Post, H.; Schauer, S.; Papadakis, T.; Kummer, W.; Hoefler, G. Cancer induces cardiomyocyte remodeling and hypoinnervation in the left ventricle of the mouse heart. PLoS ONE 2011, 6, e20424. [Google Scholar] [CrossRef]
- Shih, J.C.; Chen, K.; Ridd, M.J. Monoamine oxidase: From genes to behavior. Annu. Rev. Neurosci. 1999, 22, 197–217. [Google Scholar] [CrossRef]
- Shih, J.C.; Wu, J.B.; Chen, K. Transcriptional regulation and multiple functions of MAO genes. J. Neural Transm. 2011, 118, 979–986. [Google Scholar] [CrossRef]
- Bortolato, M.; Chen, K.; Shih, J.C. Monoamine oxidase inactivation: From pathophysiology to therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1527–1533. [Google Scholar] [CrossRef]
- Manni, M.E.; Rigacci, S.; Borchi, E.; Bargelli, V.; Miceli, C.; Giordano, C.; Raimondi, L.; Nediani, C. Monoamine oxidase is overactivated in left and right ventricles from ischemic hearts: An intriguing therapeutic target. Oxidative Med. Cell. Longev. 2016, 2016, 4375418. [Google Scholar] [CrossRef]
- Umbarkar, P.; Singh, S.; Arkat, S.; Bodhankar, S.L.; Lohidasan, S.; Sitasawad, S.L. Monoamine oxidase-A is an important source of oxidative stress and promotes cardiac dysfunction, apoptosis, and fibrosis in diabetic cardiomyopathy. Free Radic. Biol. Med. 2015, 87, 263–273. [Google Scholar] [CrossRef]
- Kaludercic, N.; Takimoto, E.; Nagayama, T.; Feng, N.; Lai, E.W.; Bedja, D.; Chen, K.; Gabrielson, K.L.; Blakely, R.D.; Shih, J.C.; et al. Monoamine oxidase A-mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ. Res. 2010, 106, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Sivasubramaniam, S.D.; Finch, C.C.; Rodriguez, M.J.; Mahy, N.; Billett, E.E. A comparative study of the expression of monoamine oxidase-A and -B mRNA and protein in non-CNS human tissues. Cell Tissue Res. 2003, 313, 291–300. [Google Scholar] [CrossRef]
- Mialet-Perez, J.; Santin, Y.; Parini, A. Monoamine oxidase-A, serotonin and norepinephrine: Synergistic players in cardiac physiology and pathology. J. Neural Transm. 2018, 125, 1627–1634. [Google Scholar] [CrossRef] [PubMed]
- Deshwal, S.; Di Sante, M.; Di Lisa, F.; Kaludercic, N. Emerging role of monoamine oxidase as a therapeutic target for cardiovascular disease. Curr. Opin. Pharmacol. Actions 2017, 33, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, P.; Kunduzova, O.; Masini, E.; Cambon, C.; Bani, D.; Raimondi, L.; Seguelas, M.H.; Nistri, S.; Colucci, W.; Leducq, N.; et al. Oxidative stress by monoamine oxidase mediates receptor-independent cardiomyocyte apoptosis by serotonin and postischemic myocardial injury. Circulation 2005, 112, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Huuskonen, C.; Hämäläinen, M.; Paavonen, T.; Moilanen, E.; Mennander, A. Monoamine oxidase A inhibition protects the myocardium after experimental acute volume overload. Anatol. J. Cardiol. 2019, 21, 39–45. [Google Scholar] [CrossRef]
- Vuohelainen, V.; Hämäläinen, M.; Paavonen, T.; Karlsson, S.; Moilanen, E.; Mennander, A. Inhibition of monoamine oxidase A increases recovery after experimental cardiac arrest. Interact. Cardiovasc. Thorac. Surg. 2015, 21, 441–449. [Google Scholar] [CrossRef]
- Shi, Q.; Malik, H.; Crawford, R.M.; Streeter, J.; Wang, J.; Huo, R.; Shih, J.C.; Chen, B.; Hall, D.; Abel, E.D.; et al. Cardiac MAO-A inhibition protects against catecholamine-induced ventricular arrhythmias via enhanced diastolic calcium control. Cardiovasc. Res. 2024, 10, 596–611. [Google Scholar] [CrossRef]
- Manoli, I.; Le, H.; Alesci, S.; McFann, K.K.; Su, Y.A.; Kino, T.; Chrousos, G.P.; Blackman, M.R. Monoamine oxidase-A is a major target gene for glucocorticoids in human skeletal muscle cells. FASEB J. 2005, 19, 1359–1361. [Google Scholar] [CrossRef]
- Schmich, S.K.P.; Keck, J.; Bonaterra, G.A.; Bertoune, M.; Adam, A.; Wilhelm, B.; Slater, E.P.; Schwarzbach, H.; Fendrich, V.; Kinscherf, R.; et al. Effects of monoamino-oxidase-A (MAO-A) inhibition on skeletal muscle inflammation and wasting through pancreatic ductal adenocarcinoma in triple transgenic mice. Biomedicines 2023, 11, 912. [Google Scholar] [CrossRef]
- Wu, J.B.; Shao, C.; Li, X.; Li, Q.; Hu, P.; Shi, C.; Li, Y.; Chen, Y.T.; Yin, F.; Liao, C.P.; et al. Monoamine oxidase A mediates prostate tumorigenesis and cancer metastasis. J. Clin. Investig. 2014, 124, 2891–2908. [Google Scholar] [CrossRef]
- Bharti, R.; Dey, G.; Das, A.K.; Mandal, M. Differential expression of IL-6/IL-6R and MAO-A regulates invasion/angiogenesis in breast cancer. Br. J. Cancer 2018, 118, 1442–1452. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef]
- Pchejetski, D.; Kunduzova, O.; Dayon, A.; Calise, D.; Seguelas, M.H.; Leducq, N.; Seif, I.; Parini, A.; Cuvillier, O. Oxidative stress-dependent sphingosine kinase-1 inhibition mediates monoamine oxidase A-associated cardiac cell apoptosis. Circ. Res. 2007, 100, 41–49. [Google Scholar] [CrossRef]
- Veltkamp, R.; Uhlmann, S.; Marinescu, M.; Sticht, C.; Finke, D.; Gretz, N.; Gröne, H.J.; Katus, H.A.; Backs, J.; Lehmann, L.H. Experimental ischaemic stroke induces transient cardiac atrophy and dysfunction. J. Cachexia Sarcopenia Muscle 2019, 10, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Li, H.H.; Willis, M.S.; Lockyer, P.; Miller, N.; McDonough, H.; Glass, D.J.; Patterson, C. Atrogin-1 inhibits Akt-dependent cardiac hypertrophy in mice via ubiquitin-dependent coactivation of forkhead proteins. J. Clin. Investig. 2022, 132, e157373. [Google Scholar] [CrossRef]
- Ni, Y.G.; Berenji, K.; Wang, N.; Oh, M.; Sachan, N.; Dey, A.; Patterson, C. Foxo transcription factors blunt cardiac hypertrophy by inhibiting calcineurin signaling. Circulation 2006, 114, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- Gallot, Y.S.; Durieux, A.C.; Castells, J.; Desgeorges, M.M.; Vernus, B.; Plantureux, L.; Rémond, D.; Jahnke, V.E.; Lefai, E.; Dardevet, D.; et al. Myostatin gene inactivation prevents skeletal muscle wasting in cancer. Cancer Res. 2014, 74, 7344–7356. [Google Scholar] [CrossRef]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L.A. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Skurk, C.; Izumiya, Y.; Maatz, H.; Razeghi, P.; Shiojima, I.; Sandri, M.; Sato, K.; Zeng, L.; Schiekofer, S.; Pimentel, D.; et al. The FOXO3a transcription factor regulates cardiac myocyte size downstream of AKT signaling. J. Biol. Chem. 2005, 280, 20814–20823. [Google Scholar] [CrossRef]
- Fang, W.Y.; Tseng, Y.T.; Lee, T.Y.; Fu, Y.C.; Chang, W.H.; Lo, W.W.; Lin, C.L.; Lo, Y.C. Triptolide prevents LPS-induced skeletal muscle atrophy via inhibiting NF-κB/TNF-α and regulating protein synthesis/degradation pathway. Br. J. Pharmacol. 2021, 178, 2998–3016. [Google Scholar] [CrossRef]
- Li, Y.P.; Chen, Y.; Li, A.S.; Reid, M.B. Hydrogen peroxide stimulates ubiquitin-conjugating activity and expression of genes for specific E2 and E3 proteins in skeletal muscle myotubes. Am. J. Physiol.-Cell Physiol. 2003, 285, C806–C812. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, M.; Tan, S.; Wang, C.; Fan, S.; Huang, C. Harmine is an inflammatory inhibitor through the suppression of NF-κB signaling. Biochem. Biophys. Res. Commun. 2017, 489, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Barry, J.L.; McMillan, D.; Skipworth, R.J.E.; Fallon, M.T.; Paval, D.R.; McNeish, I.; Gallagher, I.J. The Emerging Role of Interleukin 1β (IL-1β) in Cancer Cachexia. Inflammation 2021, 44, 1223–1228. [Google Scholar]
- Cheung, W.W.; Hao, S.; Zheng, R.; Wang, Z.; Gonzalez, A.; Zhou, P.; Hoffman, H.M.; Mak, R.H. Targeting interleukin-1 for reversing fat browning and muscle wasting in infantile nephropathic cystinosis. J. Cachexia Sarcopenia Muscle 2021, 12, 1296–1311. [Google Scholar] [CrossRef]
- Li, W.; Moylan, J.S.; Chambers, M.A.; Smith, J.; Reid, M.B. Interleukin-1 stimulates catabolism in C2C12 myotubes. Am. J. Physiol.-Cell Physiol. 2009, 297, C706–C714. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.P.; Chen, Y.; John, J.; Moylan, J.; Jin, B.; Mann, D.L.; Reid, M.B. TNF-alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J. 2005, 19, 362–370. [Google Scholar] [CrossRef]
- Kaur, N.; Gupta, P.; Dutt, V.; Sharma, O.; Gupta, S.; Dua, A.; Injeti, E.; Mittal, A. Cinnamaldehyde attenuates TNF-α induced skeletal muscle loss in C2C12 myotubes via regulation of protein synthesis, proteolysis, oxidative stress and inflammation. Arch. Biochem. Biophys. 2024, 753, 109922. [Google Scholar] [CrossRef] [PubMed]
- Sishi, B.J.; Engelbrecht, A.M. Tumor necrosis factor alpha (TNF-alpha) inactivates the PI3-kinase/PKB pathway and induces atrophy and apoptosis in L6 myotubes. Cytokine 2011, 54, 173–184. [Google Scholar] [CrossRef]
- Silva, K.A.; Dong, J.; Dong, Y.; Dong, Y.; Schor, N.; Tweardy, D.J.; Zhang, L.; Mitch, W.E. Inhibition of Stat3 activation suppresses caspase-3 and the ubiquitin-proteasome system, leading to preservation of muscle mass in cancer cachexia. J. Biol. Chem. 2015, 290, 11177–11187. [Google Scholar] [CrossRef]
- Ng, I.H.; Yeap, Y.Y.; Ong, L.S.; Jans, D.A.; Bogoyevitch, M.A. Oxidative stress impairs multiple regulatory events to drive persistent cytokine-stimulated STAT3 phosphorylation. Biochim. Biophys. Acta 2014, 1843, 483–494. [Google Scholar] [CrossRef]
- Bonetto, A.; Aydogdu, T.; Kunzevitzky, N.; Guttridge, D.C.; Khuri, S.; Koniaris, L.G.; Zimmers, T.A. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS ONE 2011, 6, e22538. [Google Scholar] [CrossRef] [PubMed]
- Oba, T.; Yasukawa, H.; Hoshijima, M.; Sasaki, K.; Futamata, N.; Fukui, D.; Mawatari, K.; Nagata, T.; Kyogoku, S.; Ohshima, H.; et al. Cardiac-specific deletion of SOCS-3 prevents development of left ventricular remodeling after acute myocardial infarction. J. Am. Coll. Cardiol. 2012, 59, 838–852. [Google Scholar] [CrossRef] [PubMed]
- Panera, N.; Gnani, D.; Piermarini, E.; Petrini, S.; Bertini, E.; Nobili, V.; Pastore, A.; Piemonte, F.; Alisi, A. High concentrations of H2O2 trigger hypertrophic cascade and phosphatase and tensin homologue (PTEN) glutathionylation in H9c2 cardiomyocytes. Exp. Mol. Pathol. 2016, 100, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Handayaningsih, A.E.; Iguchi, G.; Fukuoka, H.; Nishizawa, H.; Takahashi, M.; Yamamoto, M.; Herningtyas, E.H.; Okimura, Y.; Kaji, H.; Chihara, K.; et al. Reactive oxygen species play an essential role in IGF-I signaling and IGF-I-induced myocyte hypertrophy in C2C12 myocytes. Endocrinology 2011, 152, 912–921. [Google Scholar] [CrossRef]
- Adderley, S.R.; Fitzgerald, D.J. Oxidative damage of cardiomyocytes is limited by extracellular regulated kinases 1/2-mediated induction of cyclooxygenase-2. J. Biol. Chem. 1999, 274, 5038–5046. [Google Scholar] [CrossRef]
- Novak, M.L.; Billich, W.; Smith, S.M.; Sukhija, K.B.; McLoughlin, T.J.; Hornberger, T.A.; Koh, T.J. COX-2 inhibitor reduces skeletal muscle hypertrophy in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R1132–R1139. [Google Scholar] [CrossRef]
- Basic, V.T.; Jacobsen, A.; Sirsjö, A.; Abdel-Halim, S.M. TNF stimulation induces VHL overexpression and impairs angiogenic potential in skeletal muscle myocytes. Int. J. Mol. Med. 2014, 34, 228–236. [Google Scholar] [CrossRef]
- Gula, G.; Rumiński, S.; Niderla-Bielińska, J.; Jasińska, A.; Kiernozek, E.; Jankowska-Steifer, E.; Flaht-Zabost, A.; Ratajska, A. Potential functions of embryonic cardiac macrophages in angiogenesis, lymphangiogenesis and extracellular matrix remodeling. Histochem. Cell Biol. 2021, 155, 117–132. [Google Scholar] [CrossRef]
- Zhou, J.; Huang, L.; Jin, J.; Wu, X.J.; Fang, Y.Q.; Song, M.B.; Zhao, G.; Zhao, X.H.; Zhou, Y.P. The role of cyclooxygenase on angiogenesis and endothelial progenitor cell mobilization in rat ischemic myocardium. Zhonghua Xin Xue Guan Bing Za Zhi 2006, 34, 833–836. [Google Scholar]
- Arque, G.; Casanovas, A.; Dierssen, M. Dyrk1A is dynamically expressed on subsets of motor neurons and in the neuromuscular junction: Possible role in Down syndrome. PLoS ONE 2013, 8, e54285. [Google Scholar] [CrossRef]
- Young, A.; Bradley, L.A.; Farrar, E.; Bilcheck, H.O.; Tkachenko, S.; Saucerman, J.J.; Bekiranov, S.; Wolf, M.J. Inhibition of DYRK1a enhances cardiomyocyte cycling after myocardial infarction. Circ. Res. 2022, 130, 1345–1361. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Jia, K.; Chen, C.; Li, Z.; Zhao, J.; Hu, J.; Zhang, H.; Fan, Q.; Huang, C.; Xie, H.; et al. DYRK1B-STAT3 drives cardiac hypertrophy and heart failure by impairing mitochondrial bioenergetics. Circulation 2022, 145, 829–846. [Google Scholar] [CrossRef] [PubMed]
- Bhat, N.; Narayanan, A.; Fathzadeh, M.; Shah, K.; Dianatpour, M.; Abou Ziki, M.D.; Mani, A. Dyrk1b promotes autophagy during skeletal muscle differentiation by upregulating 4e-bp. Cell. Signal. 2022, 90, 110186. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Mercer, S.E.; Sun, C.J.; Friedman, E. The normal function of the cancer kinase Mirk/dyrk1B is to reduce reactive oxygen species. Genes Cancer 2014, 5, 22–30. [Google Scholar] [CrossRef]
- Rozen, E.J.; Roewenstrunk, J.; Barallobre, M.J.; Di Vona, C.; Jung, C.; Figueiredo, A.F.; Luna, J.; Fillat, C.; Arbonés, M.L.; Graupera, M.; et al. DYRK1A kinase positively regulates angiogenic responses in endothelial cells. Cell Rep. 2018, 23, 1867–1878. [Google Scholar] [CrossRef] [PubMed]
- Gams, A.; Nevarez, A.; Perkail, S.; Venegas, A.; George, S.A.; Efimova, T.; Efimov, I.R. Evidence of sex differences in cancer-related cardiac complications in mouse models of pancreatic and liver cancer. Physiol. Rep. 2023, 11, e15672. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Symbol | Amplicon Length (bp) | Cat. Nr. |
|---|---|---|---|
| Actin Beta B cell leukemia/lymphoma 2 | ACTB BCL2 | 77 104 | QT01136772 QT02392292 |
| Caspase 3 | Casp3 | 150 | QT01164779 |
| CD68 antigen | CD68 | 67 | QT00254051 |
| Atrogin-1 (MAFbx, FBX032)) | Atrogin-1 | 103 | QT00158543 |
| Interleukin-1beta | IL-1β | 150 | QT01048355 |
| Kinase insert domain protein receptor | KDR | 95 | QT02519972 |
| Monoamine oxidase A | Mao-A | 81 | QT00109326 |
| Monoamine oxidase B | Mao-B | 93 | QT00145124 |
| Notch gene homolog 1 (Drosophila) | Notch1 | 102 | QT00156982 |
| Notch gene homolog 3 | Notch3 | 104 | QT01051729 |
| Prostaglandin endoperoxide synthase 2 | Ptgs2 | 95 | QT00165347 |
| Ribosomal protein lateral stalk subunit P0 Suppressor of cytokine signaling 3 | RPLP0 SOCS3 | 22 90 | QT00075012 QT02488990 |
| Tumor necrosis factor alpha | TNF-α | 112 | QT00104006 |
| Muscle RING-finger protein-1 (TRIM63) | MuRF1 | 116 | QT00291991 |
| Vascular endothelial growth factor A | VEGFA | 117 | QT00160769 |
| WT | WT-HH | CA | CA-HH | ANOVA | |||
|---|---|---|---|---|---|---|---|
| CA | HH | Interact. | |||||
| N (m/f) | 12 (5/7) | 11 (8/3) | 10 (6/4) | 9 (5/4) | 42 | 42 | 42 |
| body weight (g) | 27.08 ± 1.61 | 27.17 ± 1.36 | 27.46 ± 1.09 | 24.02 ± 0.91 # | p = 0.301 | p = 0.220 | p = 0.197 |
| age (weeks) | 16.58 ± 0.31 | 18.82 ± 0.70 # | 23.20 ± 0.98 *** | 19.11 ± 0.48 ## | p = 0.000 | p = 0.165 | p = 0.000 |
| WT | WT-HH | CA | CA-HH | |
|---|---|---|---|---|
| N | 8 | 11 | 8 | 9 |
| Monoamino oxidase A (MAO-A) | 1.00 | 0.88 | 0.72 | 0.69 |
| Monoamino oxidase B (MAO-B) | 1.00 | 0.91 | 0.52 | 0.77 |
| Muscle atrophy F-box (MAFbx) | 1.00 | 1.09 | 0.84 | 0.99 |
| Muscle ring finger protein 1 (MURF-1) | 1.00 | 1.30 | 1.07 | 1.47 |
| Interleukine-1β (IL-1β) | 1.00 | 0.48 # | 2.47 * | 1.37 |
| Tumor necrosis factor (TNF) | 1.00 | 0.54 | 1.87 * | 0.35 # |
| Cyclooxygenase-2 (COX-2) | 1.00 | 1.00 | 1.53 * | 0.41 # |
| Cluster of differentiation 68 (CD68) | 1.00 | 1.27 | 1.26 | 0.72 |
| Suppressor of cytokine signaling 3 (SOCS-3) | 1.00 | 0.48 # | 1.64 * | 0.39 # |
| Vascular endothelial growth factor A (VEGF-A) | 1.00 | 1.1 | 1.02 | 1.05 |
| Kinase insert domain protein receptor (KDR) | 1.00 | 1.15 | 0.46 * | 0.57 |
| Notch gene homolog 1 (Notch 1) | 1.00 | 0.59 | 0.68 | 0.34 |
| Notch gene homolog 3 (Notch 3) | 1.00 | 0.89 | 0.64 | 0.38 |
| Caspase-3 (Casp3) | 1.00 | 0.56 | 1.02 | 0.49 # |
| B cell leukemia/lymphoma 2 (Bcl-2) | 1.00 | 1.15 | 0.97 | 0.62 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stelter, K.; Alabssi, A.; Bonaterra, G.A.; Schwarzbach, H.; Fendrich, V.; Slater, E.P.; Kinscherf, R.; Hildebrandt, W. Increased Myocardial MAO-A, Atrogin-1, and IL-1β Expression in Transgenic Mice with Pancreatic Carcinoma—Benefit of MAO-A Inhibition for Cardiac Cachexia. Biomedicines 2024, 12, 2009. https://doi.org/10.3390/biomedicines12092009
Stelter K, Alabssi A, Bonaterra GA, Schwarzbach H, Fendrich V, Slater EP, Kinscherf R, Hildebrandt W. Increased Myocardial MAO-A, Atrogin-1, and IL-1β Expression in Transgenic Mice with Pancreatic Carcinoma—Benefit of MAO-A Inhibition for Cardiac Cachexia. Biomedicines. 2024; 12(9):2009. https://doi.org/10.3390/biomedicines12092009
Chicago/Turabian StyleStelter, Kira, Annalena Alabssi, Gabriel Alejandro Bonaterra, Hans Schwarzbach, Volker Fendrich, Emily P. Slater, Ralf Kinscherf, and Wulf Hildebrandt. 2024. "Increased Myocardial MAO-A, Atrogin-1, and IL-1β Expression in Transgenic Mice with Pancreatic Carcinoma—Benefit of MAO-A Inhibition for Cardiac Cachexia" Biomedicines 12, no. 9: 2009. https://doi.org/10.3390/biomedicines12092009
APA StyleStelter, K., Alabssi, A., Bonaterra, G. A., Schwarzbach, H., Fendrich, V., Slater, E. P., Kinscherf, R., & Hildebrandt, W. (2024). Increased Myocardial MAO-A, Atrogin-1, and IL-1β Expression in Transgenic Mice with Pancreatic Carcinoma—Benefit of MAO-A Inhibition for Cardiac Cachexia. Biomedicines, 12(9), 2009. https://doi.org/10.3390/biomedicines12092009