
In Vitro Evaluation of Antipseudomonal Activity and Safety Profile of Peptidomimetic Furin Inhibitors


 , , , , , ,
, , , , , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Spectroscopic Studies on Protein Binding of Furin Inhibitors
2.2. MDCK Cell Culturing and Pseudomonas aeruginosa Experiments
2.3. IPEC-J2 and PHH Cell Culturing
2.4. Cytotoxicity Measurements in IPEC-J2 and PHH-Based Cell Models
2.5. Amplex Red Measurements
2.6. DCFH2-DA ROS Measurements
2.7. CYP3A4 Activity Measurement and Modelling of Its Interaction with Furin Inhibitors
2.8. Statistical Analysis
3. Results
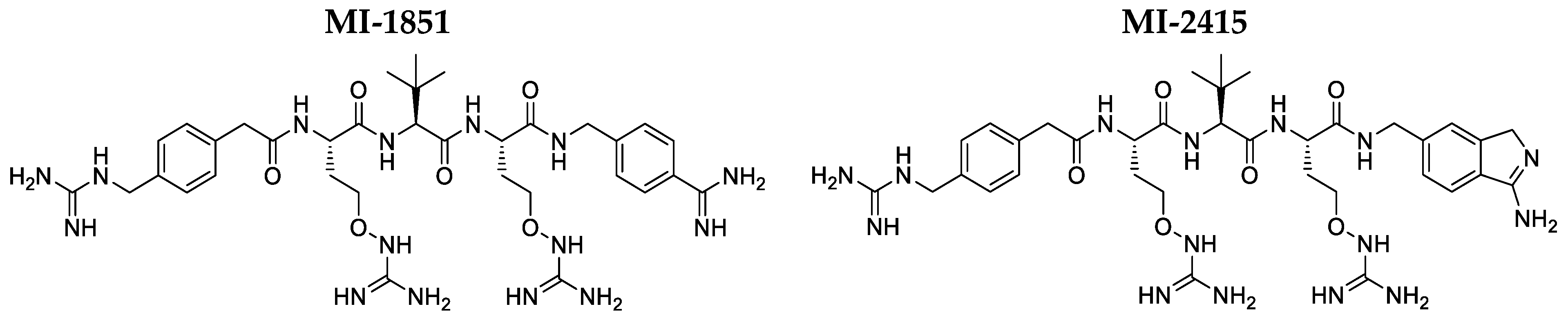
3.1. Interactions of Inhibitors MI-1851 and MI-2415 with HSA and AGP
3.2. Antipseudomonal Effect of Inhibititors MI-1851 and MI-2415
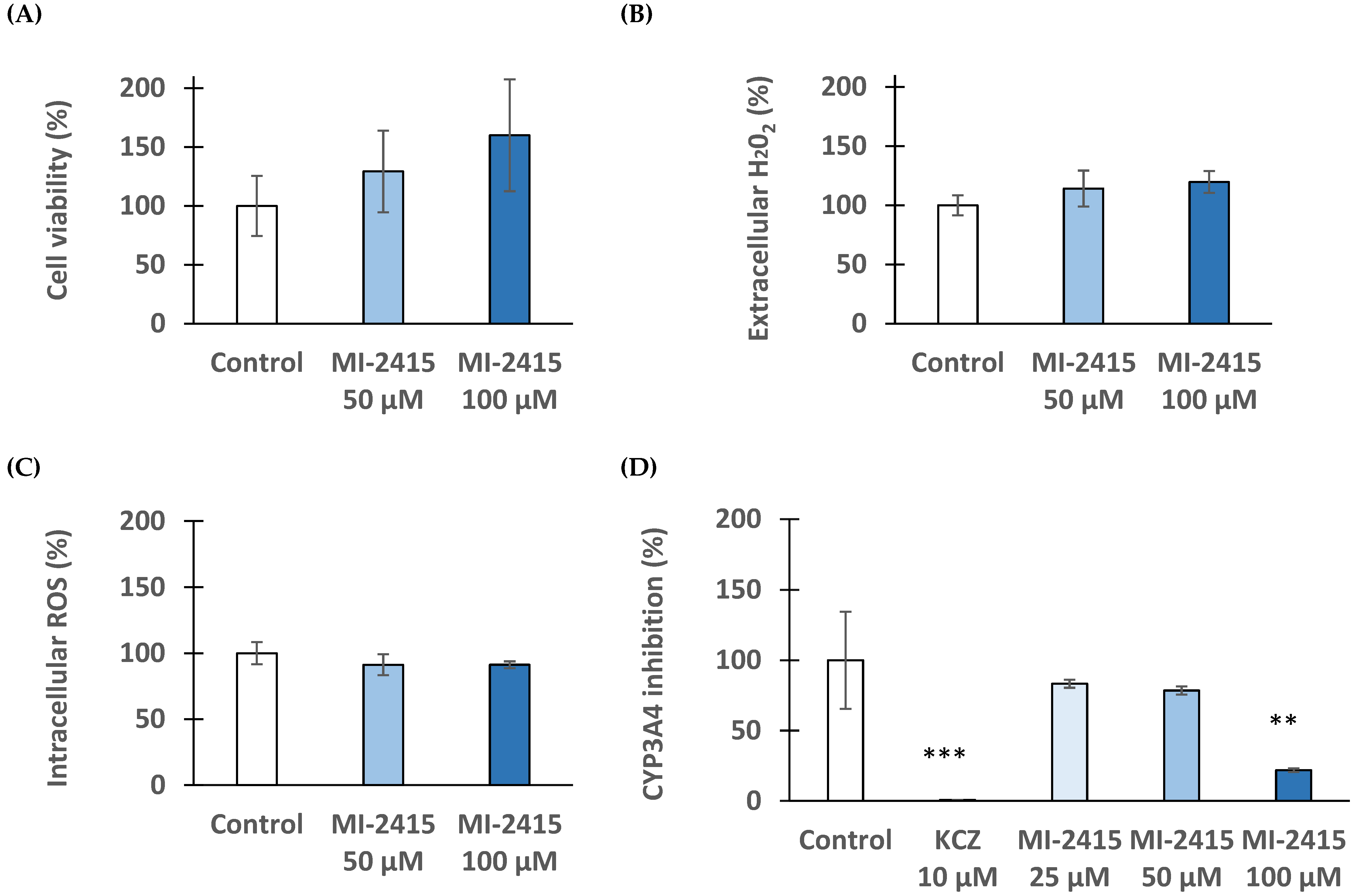
3.3. Cytotoxicity Investigations in IPEC-J2 Cells
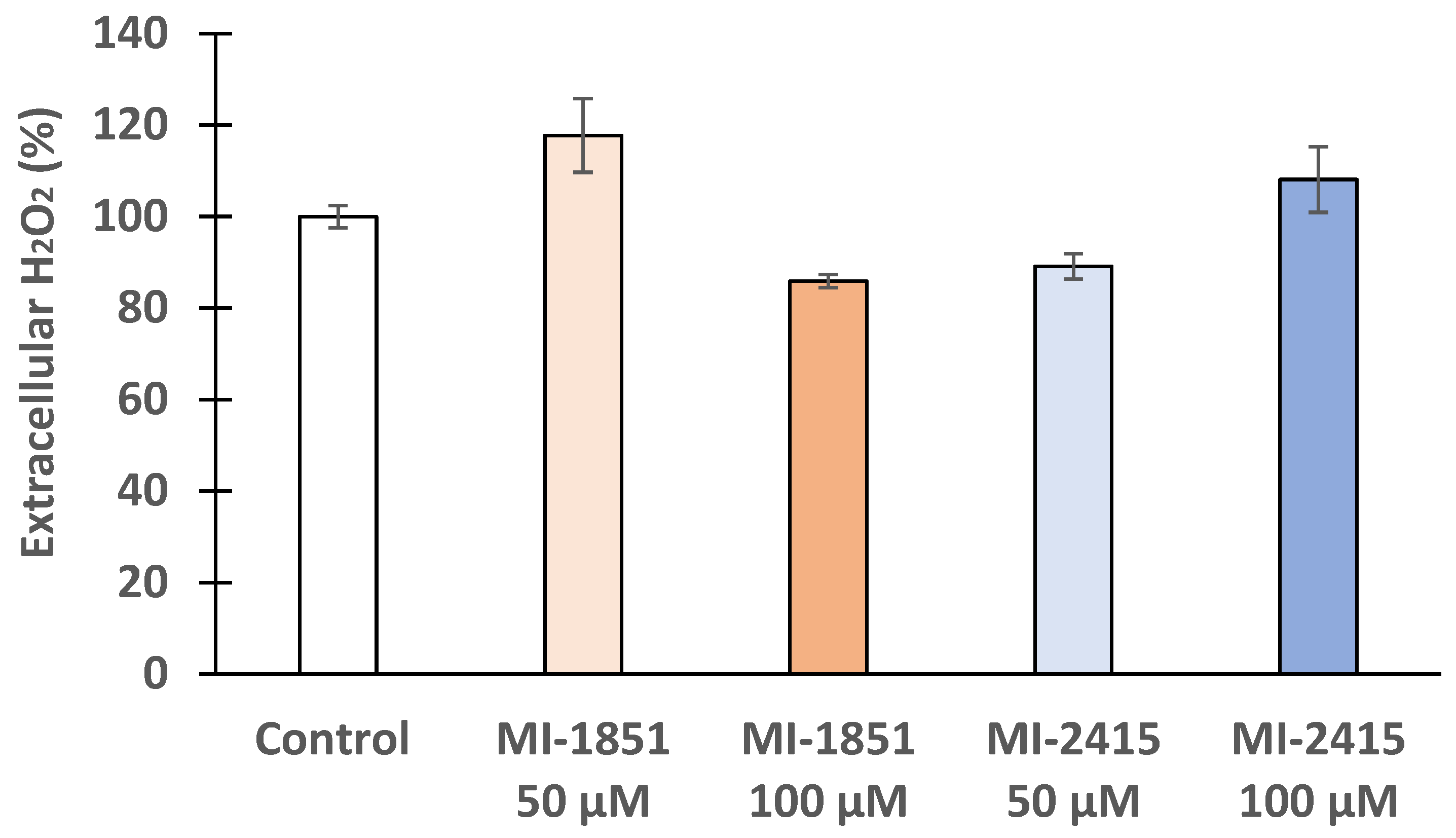
3.4. Peroxide Production in IPEC-J2 Cells
3.5. DCFH-DA Assay to Detect Peroxide Production in IPEC-J2 Cells
3.6. Viability and Redox Status of Hepatocytes Exposed to Inhibitor MI-2415
3.7. Microsomal CYP3A4 Activity Assay
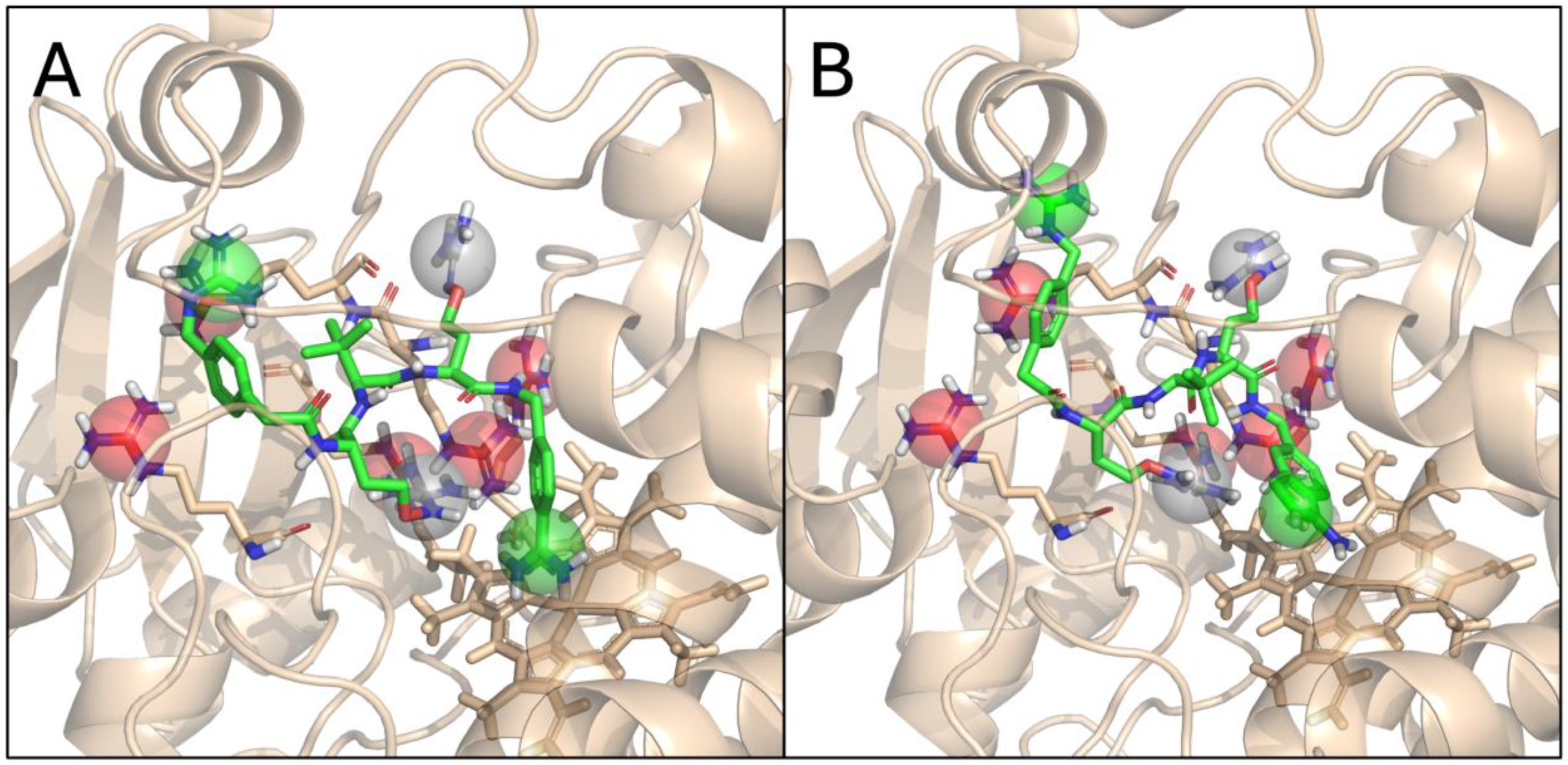
3.8. Molecular Modelling of MI-1851 and MI-2415 with CYP3A4
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marie, V.; Gordon, M.L. The (Re-)Emergence and Spread of Viral Zoonotic Disease: A Perfect Storm of Human Ingenuity and Stupidity. Viruses 2023, 15, 1638. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Prat, A. The Biology and Therapeutic Targeting of the Proprotein Convertases. Nat. Rev. Drug Discov. 2012, 11, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Devi, K.P.; Pourkarim, M.R.; Thijssen, M.; Sureda, A.; Khayatkashani, M.; Cismaru, C.A.; Neagoe, I.B.; Habtemariam, S.; Razmjouei, S.; Khayat Kashani, H.R. A Perspective on the Applications of Furin Inhibitors for the Treatment of SARS-CoV-2. Pharmacol. Rep. 2022, 74, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, X.; Bai, X.; Yao, S.; Chang, Y.-Z.; Gao, G. The Emerging Role of Furin in Neurodegenerative and Neuropsychiatric Diseases. Transl. Neurodegener. 2022, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Manzine, P.R.; Ettcheto, M.; Cano, A.; Busquets, O.; Marcello, E.; Pelucchi, S.; Di Luca, M.; Endres, K.; Olloquequi, J.; Camins, A.; et al. ADAM10 in Alzheimer’s Disease: Pharmacological Modulation by Natural Compounds and Its Role as a Peripheral Marker. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 113, 108661. [Google Scholar] [CrossRef]
- Marafie, S.K.; Al-Mulla, F. An Overview of the Role of Furin in Type 2 Diabetes. Cells 2023, 12, 2407. [Google Scholar] [CrossRef]
- Braun, E.; Sauter, D. Furin-mediated Protein Processing in Infectious Diseases and Cancer. Clin. Transl. Immunol. 2019, 8, e1073. [Google Scholar] [CrossRef]
- Global Report on Infection Prevention and Control. Available online: https://www.who.int/publications/i/item/9789240051164 (accessed on 3 July 2024).
- Sharma, G.; Rao, S.; Bansal, A.; Dang, S.; Gupta, S.; Gabrani, R. Pseudomonas aeruginosa Biofilm: Potential Therapeutic Targets. Biol. J. Int. Assoc. Biol. Stand. 2014, 42, 1–7. [Google Scholar] [CrossRef]
- Horcajada, J.P.; Montero, M.; Oliver, A.; Sorlí, L.; Luque, S.; Gómez-Zorrilla, S.; Benito, N.; Grau, S. Epidemiology and Treatment of Multidrug-Resistant and Extensively Drug-Resistant Pseudomonas aeruginosa Infections. Clin. Microbiol. Rev. 2019, 32, e00031-19. [Google Scholar] [CrossRef]
- Sati, H.; Tacconelli, E.; Carrara, E.; Savoldi, A.; Unit, W.; AG, W.; Zignol, M.; Cameron, A. WHO Bacterial Priority Pathogens List; World Health Organization: Geneva, Switzerland, 2024. [Google Scholar]
- Pászti-Gere, E.; Szentkirályi, A.; Fedor, Z.; Nagy, G.; Szimrók, Z.; Pászti, Z.; Pászti, A.; Pilgram, O.; Steinmetzer, T.; Bodnárová, S.; et al. In Vitro Interaction of Potential Antiviral TMPRSS2 Inhibitors with Human Serum Albumin and Cytochrome P 450 Isoenzymes. Biomed. Pharmacother. 2022, 146, 112513. [Google Scholar] [CrossRef]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Van Lam Van, T.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; et al. TMPRSS2 and Furin Are Both Essential for Proteolytic Activation of SARS-CoV-2 in Human Airway Cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef] [PubMed]
- Papa, G.; Mallery, D.L.; Albecka, A.; Welch, L.G.; Cattin-Ortolá, J.; Luptak, J.; Paul, D.; McMahon, H.T.; Goodfellow, I.G.; Carter, A.; et al. Furin Cleavage of SARS-CoV-2 Spike Promotes but Is Not Essential for Infection and Cell-Cell Fusion. PLOS Pathog. 2021, 17, e1009246. [Google Scholar] [CrossRef] [PubMed]
- Jiao, G.-S.; Cregar, L.; Wang, J.; Millis, S.Z.; Tang, C.; O’Malley, S.; Johnson, A.T.; Sareth, S.; Larson, J.; Thomas, G. Synthetic Small Molecule Furin Inhibitors Derived from 2,5-Dideoxystreptamine. Proc. Natl. Acad. Sci. USA 2006, 103, 19707–19712. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Molina, B.; Lick, A.N.; Blanco, E.H.; Posada-Salgado, J.A.; Martinez-Mayorga, K.; Johnson, A.T.; Jiao, G.-S.; Lindberg, I. Identification of Potent and Compartment-Selective Small Molecule Furin Inhibitors Using Cell-Based Assays. Biochem. Pharmacol. 2015, 96, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Douglas, L.E.J.; Reihill, J.A.; Ho, M.W.Y.; Axten, J.M.; Campobasso, N.; Schneck, J.L.; Rendina, A.R.; Wilcoxen, K.M.; Martin, S.L. A Highly Selective, Cell-Permeable Furin Inhibitor BOS-318 Rescues Key Features of Cystic Fibrosis Airway Disease. Cell Chem. Biol. 2022, 29, 947–957.e8. [Google Scholar] [CrossRef]
- Ferguson, T.E.G.; Reihill, J.A.; Walker, B.; Hamilton, R.A.; Martin, S.L. A Selective Irreversible Inhibitor of Furin Does Not Prevent Pseudomonas aeruginosa Exotoxin A-Induced Airway Epithelial Cytotoxicity. PLoS ONE 2016, 11, e0159868. [Google Scholar] [CrossRef]
- Sarac, M.S.; Cameron, A.; Lindberg, I. The Furin Inhibitor Hexa-d-Arginine Blocks the Activation of Pseudomonas aeruginosa Exotoxin A In Vivo. Infect. Immun. 2002, 70, 7136–7139. [Google Scholar] [CrossRef]
- Thomas, G.; Couture, F.; Kwiatkowska, A. The Path to Therapeutic Furin Inhibitors: From Yeast Pheromones to SARS-CoV-2. Int. J. Mol. Sci. 2022, 23, 3435. [Google Scholar] [CrossRef]
- Lam van, T.V.; Heindl, M.R.; Schlutt, C.; Böttcher-Friebertshäuser, E.; Bartenschlager, R.; Klebe, G.; Brandstetter, H.; Dahms, S.O.; Steinmetzer, T. The Basicity Makes the Difference: Improved Canavanine-Derived Inhibitors of the Proprotein Convertase Furin. ACS Med. Chem. Lett. 2021, 12, 426–432. [Google Scholar] [CrossRef]
- Lange, R.W.; Bloch, K.; Heindl, M.R.; Wollenhaupt, J.; Weiss, M.S.; Brandstetter, H.; Klebe, G.; Falcone, F.H.; Böttcher-Friebertshäuser, E.; Dahms, S.O.; et al. Fragment-Based Design, Synthesis, and Characterization of Aminoisoindole-Derived Furin Inhibitors. ChemMedChem 2024, 19, e202400057. [Google Scholar] [CrossRef]
- Poór, M.; Dombi, Á.; Fliszár-Nyúl, E.; Pedroni, L.; Dellafiora, L. Effects of Chrysin and Chrysin-7-Sulfate on Ochratoxin A-Albumin Interactions and on the Plasma and Kidney Levels of the Mycotoxin in Rats. ACS Omega 2024, 9, 17655–17666. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zeng, L.-H.; Li, D.-L. A Review on the Methods for Correcting the Fluorescence Inner-Filter Effect of Fluorescence Spectrum. Appl. Spectrosc. Rev. 2017, 52, 883–908. [Google Scholar] [CrossRef]
- Van Eijk, N.; Schmacke, L.C.; Steinmetzer, T.; Pilgram, O.; Poór, M.; Pászti-Gere, E. In Vitro Testing of Host-Targeting Small Molecule Antiviral Matriptase/TMPRSS2 Inhibitors in 2D and 3D Cell-Based Assays. Biomed. Pharmacother. 2023, 168, 115761. [Google Scholar] [CrossRef]
- Sevrioukova, I.F.; Poulos, T.L. Dissecting Cytochrome P450 3A4–Ligand Interactions Using Ritonavir Analogues. Biochemistry 2013, 52, 4474–4481. [Google Scholar] [CrossRef] [PubMed]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Perrin, D.D.; Dempsey, B.; Serjeant, E.P. pKa Prediction for Organic Acids and Bases; Springer: Dordrecht, The Netherlands, 1981; ISBN 978-94-009-5885-2. [Google Scholar]
- Gamba, D.; van Eijk, N.; Lányi, K.; Monostory, K.; Steinmetzer, T.; Marosi, A.; Rácz, A.; Bajusz, D.; Kruhl, D.; Böttcher-Friebertshäuser, E.; et al. PK/PD Investigation of Antiviral Host Matriptase/TMPRSS2 Inhibitors in Cell Models. Sci. Rep. 2024, 14, 16621. [Google Scholar] [CrossRef]
- Silberberg, M.; Amateis, P. Chemistry: The Molecular Nature of Matter and Change; McGraw-Hill Higher Education: New York, NY, USA, 2023; ISBN 978-1-266-19923-3. [Google Scholar]
- Vergauwen, H. The IPEC-J2 Cell Line. In The Impact of Food Bioactives on Health; Verhoeckx, K., Cotter, P., López-Expósito, I., Kleiveland, C., Lea, T., Mackie, A., Requena, T., Swiatecka, D., Wichers, H., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 125–134. ISBN 978-3-319-15791-7. [Google Scholar]
- Schierack, P.; Nordhoff, M.; Pollmann, M.; Weyrauch, K.D.; Amasheh, S.; Lodemann, U.; Jores, J.; Tachu, B.; Kleta, S.; Blikslager, A.; et al. Characterization of a Porcine Intestinal Epithelial Cell Line for in Vitro Studies of Microbial Pathogenesis in Swine. Histochem. Cell Biol. 2006, 125, 293–305. [Google Scholar] [CrossRef]
- Langerholc, T.; Maragkoudakis, P.A.; Wollgast, J.; Gradisnik, L.; Cencic, A. Novel and Established Intestinal Cell Line Models—An Indispensable Tool in Food Science and Nutrition. Trends Food Sci. Technol. 2011, 22, S11–S20. [Google Scholar] [CrossRef]
- Delos, M. Cell Culture Models as an In Vitro Alternative to Study the Absorption and Biotransformation of Drugs Mycotoxins in Humans and Animals. Master’s Thesis, University of Ghent, Ghent, Belgium, 2021. [Google Scholar]
- Hardes, K.; Becker, G.L.; Lu, Y.; Dahms, S.O.; Köhler, S.; Beyer, W.; Sandvig, K.; Yamamoto, H.; Lindberg, I.; Walz, L.; et al. Novel Furin Inhibitors with Potent Anti-Infectious Activity. ChemMedChem 2015, 10, 1218–1231. [Google Scholar] [CrossRef] [PubMed]
- Kouretova, J.; Hammamy, M.Z.; Epp, A.; Hardes, K.; Kallis, S.; Zhang, L.; Hilgenfeld, R.; Bartenschlager, R.; Steinmetzer, T. Effects of NS2B-NS3 Protease and Furin Inhibition on West Nile and Dengue Virus Replication. J. Enzyme Inhib. Med. Chem. 2017, 32, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Van Lam van, T.; Ivanova, T.; Hardes, K.; Heindl, M.R.; Morty, R.E.; Böttcher-Friebertshäuser, E.; Lindberg, I.; Than, M.E.; Dahms, S.O.; Steinmetzer, T. Design, Synthesis, and Characterization of Macrocyclic Inhibitors of the Proprotein Convertase Furin. ChemMedChem 2019, 14, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Krüger, N.; Sauder, C.; Hüttl, S.; Papies, J.; Voigt, K.; Herrler, G.; Hardes, K.; Steinmetzer, T.; Örvell, C.; Drexler, J.F.; et al. Entry, Replication, Immune Evasion, and Neurotoxicity of Synthetically Engineered Bat-Borne Mumps Virus. Cell Rep. 2018, 25, 312–320.e7. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, T.; Hardes, K.; Kallis, S.; Dahms, S.O.; Than, M.E.; Künzel, S.; Böttcher-Friebertshäuser, E.; Lindberg, I.; Jiao, G.-S.; Bartenschlager, R.; et al. Optimization of Substrate-Analogue Furin Inhibitors. ChemMedChem 2017, 12, 1953–1968. [Google Scholar] [CrossRef]
- Pászti-Gere, E.; Szentkirályi-Tóth, A.; Szabó, P.; Steinmetzer, T.; Fliszár-Nyúl, E.; Poór, M. In Vitro Characterization of the Furin Inhibitor MI-1851: Albumin Binding, Interaction with Cytochrome P450 Enzymes and Cytotoxicity. Biomed. Pharmacother. 2022, 151, 113124. [Google Scholar] [CrossRef]
- Hardes, K.; Ivanova, T.; Thaa, B.; McInerney, G.M.; Klokk, T.I.; Sandvig, K.; Künzel, S.; Lindberg, I.; Steinmetzer, T. Elongated and Shortened Peptidomimetic Inhibitors of the Proprotein Convertase Furin. ChemMedChem 2017, 12, 613–620. [Google Scholar] [CrossRef]
- Gain, C.; Song, S.; Angtuaco, T.; Satta, S.; Kelesidis, T. The Role of Oxidative Stress in the Pathogenesis of Infections with Coronaviruses. Front. Microbiol. 2023, 13, 1111930. [Google Scholar] [CrossRef]
- Da Cruz Nizer, W.S.; Inkovskiy, V.; Versey, Z.; Strempel, N.; Cassol, E.; Overhage, J. Oxidative Stress Response in Pseudomonas aeruginosa. Pathogens 2021, 10, 1187. [Google Scholar] [CrossRef]
- Tavassolifar, M.J.; Aghdaei, H.A.; Sadatpour, O.; Maleknia, S.; Fayazzadeh, S.; Mohebbi, S.R.; Montazer, F.; Rabbani, A.; Zali, M.R.; Izad, M.; et al. New Insights into Extracellular and Intracellular Redox Status in COVID-19 Patients. Redox Biol. 2022, 59, 102563. [Google Scholar] [CrossRef]
- Fanali, G.; Di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human Serum Albumin: From Bench to Bedside. Mol. Aspects Med. 2012, 33, 209–290. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; Waters, N.J. Pharmacokinetic and Pharmacodynamic Considerations for Drugs Binding to Alpha-1-Acid Glycoprotein. Pharm. Res. 2019, 36, 30. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Liu, Y. Probing the Interaction of Cefodizime with Human Serum Albumin Using Multi-Spectroscopic and Molecular Docking Techniques. J. Pharm. Biomed. Anal. 2015, 107, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.-L.; Kou, S.-B.; Lin, Z.-Y.; Shi, J.-H.; Liu, Y.-X. Insights on the Interaction Mechanism of Brigatinib to Human α-1-Acid Glycoprotein: Experimental and Computational Approaches. Int. J. Biol. Macromol. 2020, 157, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Van De Weert, M.; Stella, L. Fluorescence Quenching and Ligand Binding: A Critical Discussion of a Popular Methodology. J. Mol. Struct. 2011, 998, 144–150. [Google Scholar] [CrossRef]
- Karicherla, P.; Hobden, J.A. Nona-D-Arginine Amide for Prophylaxis and Treatment of Experimental Pseudomonas aeruginosa Keratitis. Curr. Eye Res. 2010, 35, 220–224. [Google Scholar] [CrossRef]
- Jiang, X.; Li, D.; Maghsoudloo, M.; Zhang, X.; Ma, W.; Fu, J. Targeting Furin, a Cellular Proprotein Convertase, for COVID-19 Prevention and Therapeutics. Drug Discov. Today 2024, 29, 104026. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maluck, S.; Bobrovsky, R.; Poór, M.; Lange, R.W.; Steinmetzer, T.; Jerzsele, Á.; Adorján, A.; Bajusz, D.; Rácz, A.; Pászti-Gere, E. In Vitro Evaluation of Antipseudomonal Activity and Safety Profile of Peptidomimetic Furin Inhibitors. Biomedicines 2024, 12, 2075. https://doi.org/10.3390/biomedicines12092075
Maluck S, Bobrovsky R, Poór M, Lange RW, Steinmetzer T, Jerzsele Á, Adorján A, Bajusz D, Rácz A, Pászti-Gere E. In Vitro Evaluation of Antipseudomonal Activity and Safety Profile of Peptidomimetic Furin Inhibitors. Biomedicines. 2024; 12(9):2075. https://doi.org/10.3390/biomedicines12092075
Chicago/Turabian StyleMaluck, Sara, Rivka Bobrovsky, Miklós Poór, Roman W. Lange, Torsten Steinmetzer, Ákos Jerzsele, András Adorján, Dávid Bajusz, Anita Rácz, and Erzsébet Pászti-Gere. 2024. "In Vitro Evaluation of Antipseudomonal Activity and Safety Profile of Peptidomimetic Furin Inhibitors" Biomedicines 12, no. 9: 2075. https://doi.org/10.3390/biomedicines12092075
APA StyleMaluck, S., Bobrovsky, R., Poór, M., Lange, R. W., Steinmetzer, T., Jerzsele, Á., Adorján, A., Bajusz, D., Rácz, A., & Pászti-Gere, E. (2024). In Vitro Evaluation of Antipseudomonal Activity and Safety Profile of Peptidomimetic Furin Inhibitors. Biomedicines, 12(9), 2075. https://doi.org/10.3390/biomedicines12092075