
A Multiplexed Quantitative Proteomics Approach to the Human Plasma Protein Signature


 , , , , , , and
, , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
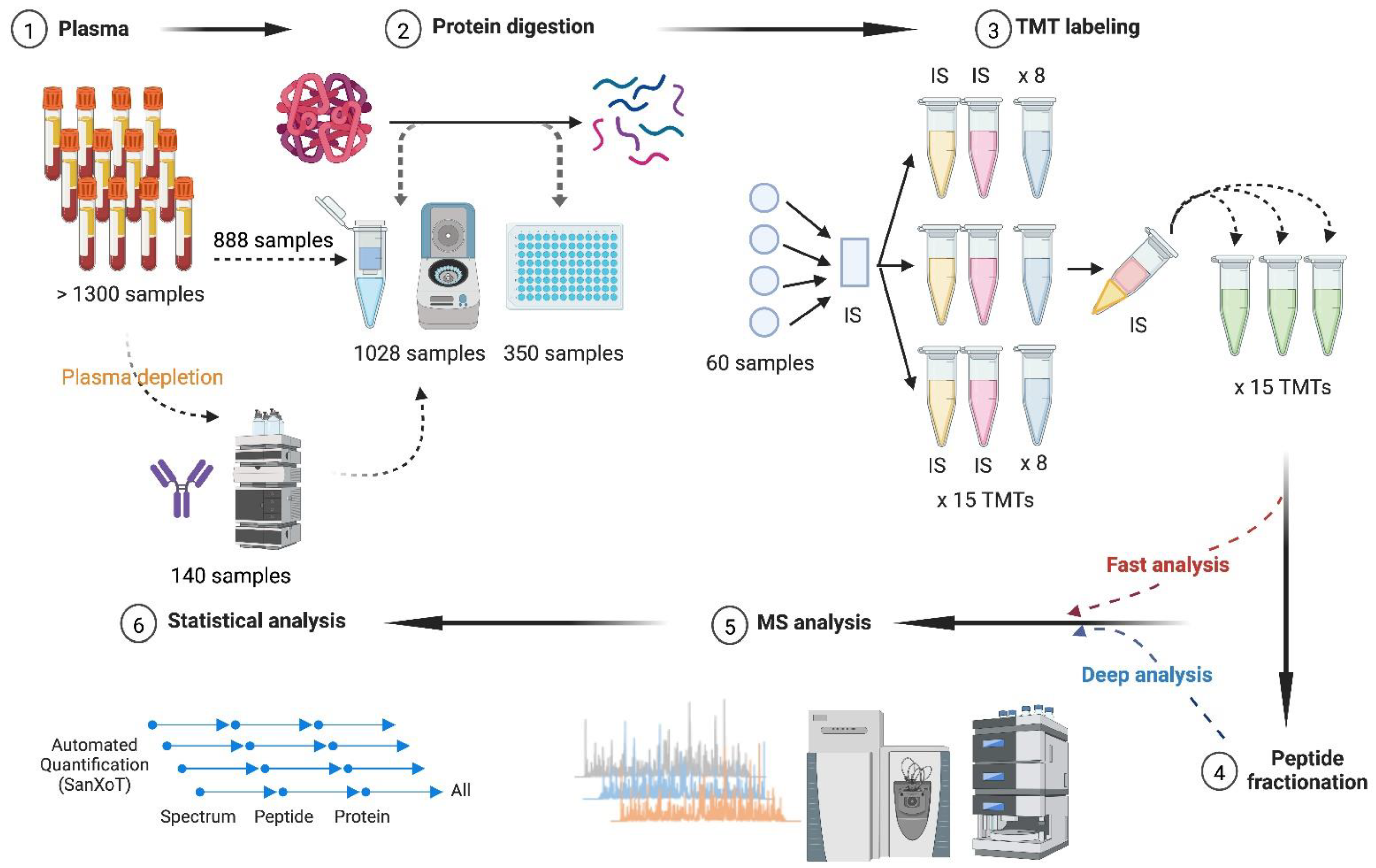
2.1. Plasma Samples
2.2. Plasma Depletion
2.3. Protein Digestion
2.4. Isobaric Labeling
2.5. Fractionation of Peptide Samples
2.6. LC-MS/MS Analysis
2.7. Protein Identification
2.8. Protein Quantification and Statistical Analysis
2.9. Functional Enrichment and Clustering Analyses
2.10. Biochemical Measurements
3. Results
3.1. Influence of Plasma Depletion on Protein Quantification
3.2. Performance of the Plasma Proteomics Workflow
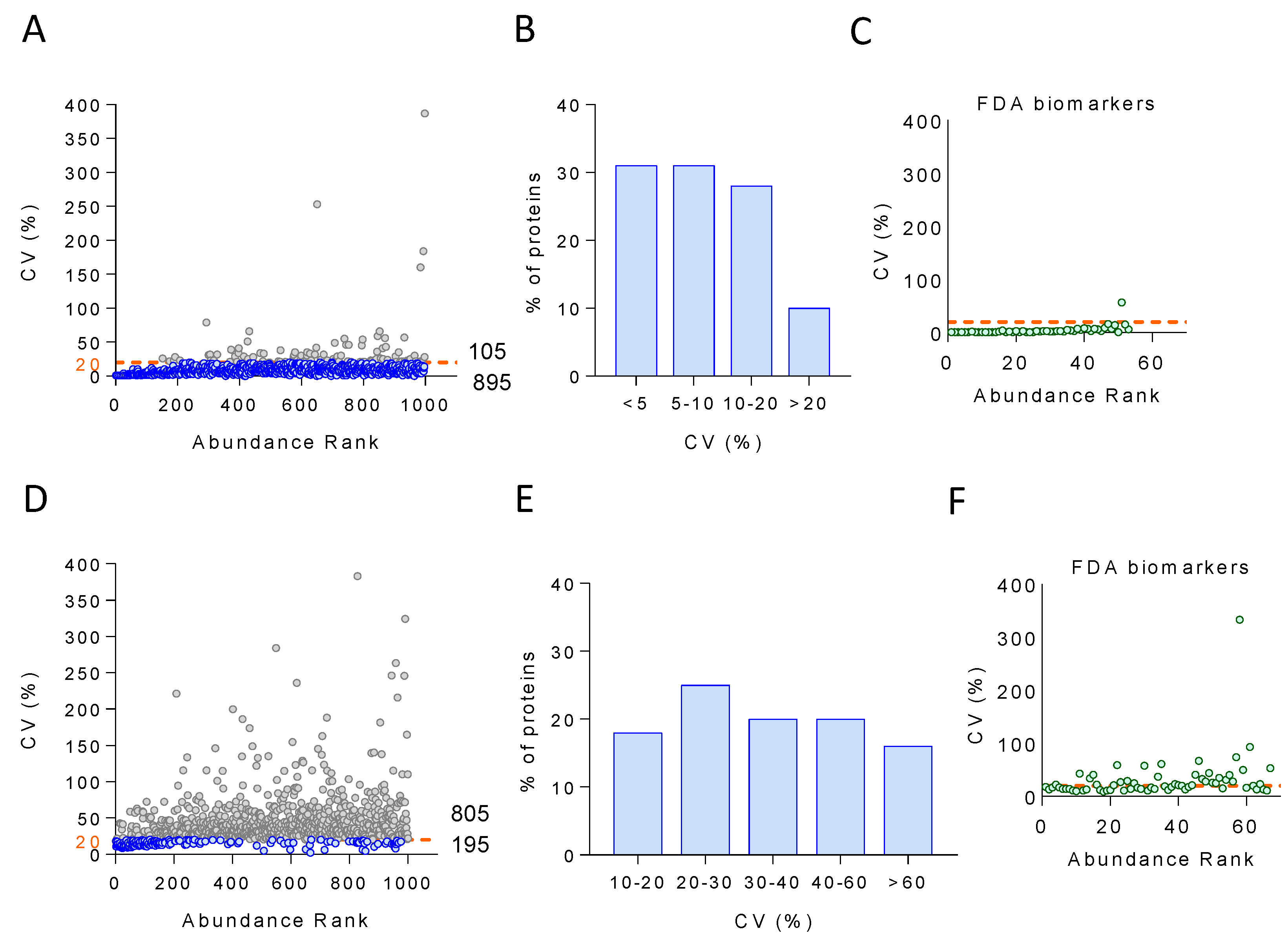
3.3. Quantification Accuracy of the Plasma Proteomics Workflow
3.4. Technical and Biological Variability of the Plasma Proteome
3.5. Long-Term Temporal Stability of the Plasma Proteome
3.6. Classification of Plasma Proteins According to Temporal Stability and Biological Variability
3.7. Performance of the On-Plate Sample Preparation Method
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arican, O.; Aral, M.; Sasmaz, S.; Ciragil, P. Serum Levels of Tnf-Alpha, Ifn-Gamma, Il-6, Il-8, Il-12, Il-17, and Il-18 in Patients with Active Psoriasis and Correlation with Disease Severity. Mediat. Inflamm. 2005, 2005, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Geyer, P.E.; Holdt, L.M.; Teupser, D.; Mann, M. Revisiting Biomarker Discovery by Plasma Proteomics. Mol. Syst. Biol. 2017, 13, 942. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Kelleher, N.L. Proteoforms as the Next Proteomics Currency. Science 2018, 359, 1106–1107. [Google Scholar] [CrossRef]
- Hellinger, R.; Sigurdsson, A.; Wu, W.; Romanova, E.V.; Li, L.; Sweedler, J.V.; Sussmuth, R.D.; Gruber, C.W. Peptidomics. Nat. Rev. Methods Primers 2023, 3, 25. [Google Scholar] [CrossRef]
- Prensner, J.R.; Abelin, J.G.; Kok, L.W.; Clauser, K.R.; Mudge, J.M.; Ruiz-Orera, J.; Bassani-Sternberg, M.; Moritz, R.L.; Deutsch, E.W.; van Heesch, S. What Can Ribo-Seq, Immunopeptidomics, and Proteomics Tell Us About the Noncanonical Proteome? Mol. Cell Proteom. 2023, 22, 100631. [Google Scholar] [CrossRef]
- Anderson, N.L.; Ptolemy, A.S.; Rifai, N. The Riddle of Protein Diagnostics: Future Bleak or Bright? Clin. Chem. 2013, 59, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, N.; Wisniewski, J.R.; Geiger, T.; Cox, J.; Kircher, M.; Kelso, J.; Paabo, S.; Mann, M. Deep Proteome and Transcriptome Mapping of a Human Cancer Cell Line. Mol. Syst. Biol. 2011, 7, 548. [Google Scholar] [CrossRef]
- Beck, M.; Schmidt, A.; Malmstroem, J.; Claassen, M.; Ori, A.; Szymborska, A.; Herzog, F.; Rinner, O.; Ellenberg, J.; Aebersold, R. The Quantitative Proteome of a Human Cell Line. Mol. Syst. Biol. 2011, 7, 549. [Google Scholar] [CrossRef]
- Pieper, R.; Su, Q.; Gatlin, C.L.; Huang, S.T.; Anderson, N.L.; Steiner, S. Multi-Component Immunoaffinity Subtraction Chromatography: An Innovative Step Towards a Comprehensive Survey of the Human Plasma Proteome. Proteomics 2003, 3, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.J.; Kaleta, D.T.; Petritis, B.O.; Jiang, H.; Liu, T.; Zhang, X.; Mottaz, H.M.; Varnum, S.M.; Camp, D.G., 2nd; Huang, L.; et al. Enhanced Detection of Low Abundance Human Plasma Proteins Using a Tandem Igy12-Supermix Immunoaffinity Separation Strategy. Mol. Cell Proteom. 2008, 7, 1963–1973. [Google Scholar] [CrossRef]
- Luque-Garcia, J.L.; Neubert, T.A. Sample Preparation for Serum/Plasma Profiling and Biomarker Identification by Mass Spectrometry. J. Chromatogr. A 2007, 1153, 259–276. [Google Scholar] [CrossRef] [PubMed]
- Mortezai, N.; Harder, S.; Schnabel, C.; Moors, E.; Gauly, M.; Schluter, H.; Wagener, C.; Buck, F. Tandem Affinity Depletion: A Combination of Affinity Fractionation and Immunoaffinity Depletion Allows the Detection of Low-Abundance Components in the Complex Proteomes of Body Fluids. J. Proteome Res. 2010, 9, 6126–6134. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Tang, H.Y.; Wang, H.; Liu, Q.; Speicher, D.W. Systematic Comparison of Fractionation Methods for in-Depth Analysis of Plasma Proteomes. J. Proteome Res. 2012, 11, 3090–3100. [Google Scholar] [CrossRef]
- Bellei, E.; Bergamini, S.; Monari, E.; Fantoni, L.I.; Cuoghi, A.; Ozben, T.; Tomasi, A. High-Abundance Proteins Depletion for Serum Proteomic Analysis: Concomitant Removal of Non-Targeted Proteins. Amino Acids 2011, 40, 145–156. [Google Scholar] [CrossRef]
- Gundry, R.L.; White, M.Y.; Nogee, J.; Tchernyshyov, I.; Van Eyk, J.E. Assessment of Albumin Removal from an Immunoaffinity Spin Column: Critical Implications for Proteomic Examination of the Albuminome and Albumin-Depleted Samples. Proteomics 2009, 9, 2021–2028. [Google Scholar] [CrossRef]
- Addona, T.A.; Shi, X.; Keshishian, H.; Mani, D.R.; Burgess, M.; Gillette, M.A.; Clauser, K.R.; Shen, D.; Lewis, G.D.; Farrell, L.A.; et al. A Pipeline That Integrates the Discovery and Verification of Plasma Protein Biomarkers Reveals Candidate Markers for Cardiovascular Disease. Nat. Biotechnol. 2011, 29, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Bruderer, R.; Muntel, J.; Muller, S.; Bernhardt, O.M.; Gandhi, T.; Cominetti, O.; Macron, C.; Carayol, J.; Rinner, O.; Astrup, A.; et al. Analysis of 1508 Plasma Samples by Capillary-Flow Data-Independent Acquisition Profiles Proteomics of Weight Loss and Maintenance. Mol. Cell Proteom. 2019, 18, 1242–1254. [Google Scholar] [CrossRef]
- Cominetti, O.; Nunez Galindo, A.; Corthesy, J.; Oller Moreno, S.; Irincheeva, I.; Valsesia, A.; Astrup, A.; Saris, W.H.; Hager, J.; Kussmann, M.; et al. Proteomic Biomarker Discovery in 1000 Human Plasma Samples with Mass Spectrometry. J. Proteome Res. 2016, 15, 389–399. [Google Scholar] [CrossRef]
- Niu, L.; Thiele, M.; Geyer, P.E.; Rasmussen, D.N.; Webel, H.E.; Santos, A.; Gupta, R.; Meier, F.; Strauss, M.; Kjaergaard, M.; et al. Noninvasive Proteomic Biomarkers for Alcohol-Related Liver Disease. Nat. Med. 2022, 28, 1277–1287. [Google Scholar] [CrossRef]
- Bonzon-Kulichenko, E.; Garcia-Marques, F.; Trevisan-Herraz, M.; Vazquez, J. Revisiting Peptide Identification by High-Accuracy Mass Spectrometry: Problems Associated with the Use of Narrow Mass Precursor Windows. J. Proteome Res. 2015, 14, 700–710. [Google Scholar] [CrossRef]
- Garcia-Marques, F.; Trevisan-Herraz, M.; Martinez-Martinez, S.; Camafeita, E.; Jorge, I.; Lopez, J.A.; Mendez-Barbero, N.; Mendez-Ferrer, S.; Del Pozo, M.A.; Ibanez, B.; et al. A Novel Systems-Biology Algorithm for the Analysis of Coordinated Protein Responses Using Quantitative Proteomics. Mol. Cell. Proteom. 2016, 15, 1740–1760. [Google Scholar] [CrossRef] [PubMed]
- Jorge, I.; Navarro, P.; Martinez-Acedo, P.; Nunez, E.; Serrano, H.; Alfranca, A.; Redondo, J.M.; Vazquez, J. Statistical Model to Analyze Quantitative Proteomics Data Obtained by 18o/16o Labeling and Linear Ion Trap Mass Spectrometry: Application to the Study of Vascular Endothelial Growth Factor-Induced Angiogenesis in Endothelial Cells. Mol. Cell. Proteom. 2009, 8, 1130–1149. [Google Scholar] [CrossRef]
- Martinez-Bartolome, S.; Navarro, P.; Martin-Maroto, F.; Lopez-Ferrer, D.; Ramos-Fernandez, A.; Villar, M.; Garcia-Ruiz, J.P.; Vazquez, J. Properties of Average Score Distributions of Sequest: The Probability Ratio Method. Mol. Cell. Proteom. 2008, 7, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Navarro, P.; Trevisan-Herraz, M.; Bonzon-Kulichenko, E.; Nunez, E.; Martinez-Acedo, P.; Perez-Hernandez, D.; Jorge, I.; Mesa, R.; Calvo, E.; Carrascal, M.; et al. General Statistical Framework for Quantitative Proteomics by Stable Isotope Labeling. J. Proteome Res. 2014, 13, 1234–1247. [Google Scholar] [CrossRef]
- Navarro, P.; Vazquez, J. A refined method to calculate false discovery rates for peptide identification using decoy databases. J. Proteome Res. 2009, 8, 1792–1796. [Google Scholar] [CrossRef]
- Baldan-Martin, M.; Mourino-Alvarez, L.; Gonzalez-Calero, L.; Moreno-Luna, R.; Sastre-Oliva, T.; Ruiz-Hurtado, G.; Segura, J.; Lopez, J.A.; Vazquez, J.; Vivanco, F.; et al. Plasma Molecular Signatures in Hypertensive Patients with Renin-Angiotensin System Suppression: New Predictors of Renal Damage and De Novo Albuminuria Indicators. Hypertension 2016, 68, 157–166. [Google Scholar] [CrossRef]
- Baldan-Martin, M.; Lopez, J.A.; Corbacho-Alonso, N.; Martinez, P.J.; Rodriguez-Sanchez, E.; Mourino-Alvarez, L.; Sastre-Oliva, T.; Martin-Rojas, T.; Rincon, R.; Calvo, E.; et al. Potential Role of New Molecular Plasma Signatures on Cardiovascular Risk Stratification in Asymptomatic Individuals. Sci. Rep. 2018, 8, 4802. [Google Scholar] [CrossRef] [PubMed]
- Santos-Lozano, A.; Fiuza-Luces, C.; Fernandez-Moreno, D.; Llavero, F.; Arenas, J.; Lopez, J.A.; Vazquez, J.; Escribano-Subias, P.; Zugaza, J.L.; Lucia, A. Exercise Benefits in Pulmonary Hypertension. J. Am. Coll. Cardiol. 2019, 73, 2906–2907. [Google Scholar] [CrossRef]
- Calvo, E.; Corbacho-Alonso, N.; Sastre-Oliva, T.; Nunez, E.; Baena-Galan, P.; Hernandez-Fernandez, G.; Rodriguez-Cola, M.; Jimenez-Velasco, I.; Corrales, F.J.; Gambarrutta-Malfati, C.; et al. Why Does Covid-19 Affect Patients with Spinal Cord Injury Milder? A Case-Control Study: Results from Two Observational Cohorts. J. Pers. Med. 2020, 10, 182. [Google Scholar] [CrossRef]
- Nunez, E.; Orera, I.; Carmona-Rodriguez, L.; Pano, J.R.; Vazquez, J.; Corrales, F.J. Mapping the Serum Proteome of Covid-19 Patients; Guidance for Severity Assessment. Biomedicines 2022, 10, 1690. [Google Scholar] [CrossRef]
- de la Fuente-Alonso, A.; Toral, M.; Alfayate, A.; Ruiz-Rodriguez, M.J.; Bonzon-Kulichenko, E.; Teixido-Tura, G.; Martinez-Martinez, S.; Mendez-Olivares, M.J.; Lopez-Maderuelo, D.; Gonzalez-Valdes, I.; et al. Aortic Disease in Marfan Syndrome Is Caused by Overactivation of Sgc-Prkg Signaling by No. Nat. Commun. 2021, 12, 2628. [Google Scholar] [CrossRef] [PubMed]
- Corbacho-Alonso, N.; Baldan-Martin, M.; Lopez, J.A.; Rodriguez-Sanchez, E.; Martinez, P.J.; Mourino-Alvarez, L.; Sastre-Oliva, T.; Cabrera, M.; Calvo, E.; Padial, L.R.; et al. Cardiovascular Risk Stratification Based on Oxidative Stress for Early Detection of Pathology. Antioxid. Redox Signal. 2021, 35, 602–617. [Google Scholar] [CrossRef] [PubMed]
- Nunez, E.; Fuster, V.; Gomez-Serrano, M.; Valdivielso, J.M.; Fernandez-Alvira, J.M.; Martinez-Lopez, D.; Rodriguez, J.M.; Bonzon-Kulichenko, E.; Calvo, E.; Alfayate, A.; et al. Unbiased Plasma Proteomics Discovery of Biomarkers for Improved Detection of Subclinical Atherosclerosis. EBioMedicine 2022, 76, 103874. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Ortiz, A.; Jimenez-Borreguero, L.J.; Penalvo, J.L.; Ordovas, J.M.; Mocoroa, A.; Fernandez-Friera, L.; Laclaustra, M.; Garcia, L.; Molina, J.; Mendiguren, J.M.; et al. The Progression and Early Detection of Subclinical Atherosclerosis (Pesa) Study: Rationale and Design. Am. Heart J. 2013, 166, 990–998. [Google Scholar] [CrossRef]
- Casasnovas, J.A.; Alcaide, V.; Civeira, F.; Guallar, E.; Ibanez, B.; Borreguero, J.J.; Laclaustra, M.; Leon, M.; Penalvo, J.L.; Ordovas, J.M.; et al. Aragon Workers– Health Study–Design and Cohort Description. BMC Cardiovasc. Disord. 2012, 12, 45. [Google Scholar] [CrossRef]
- Laclaustra, M.; Casasnovas, J.A.; Fernandez-Ortiz, A.; Fuster, V.; Leon-Latre, M.; Jimenez-Borreguero, L.J.; Pocovi, M.; Hurtado-Roca, Y.; Ordovas, J.M.; Jarauta, E.; et al. Femoral and Carotid Subclinical Atherosclerosis Association with Risk Factors and Coronary Calcium: The Awhs Study. J. Am. Coll. Cardiol. 2016, 67, 1263–1274. [Google Scholar] [CrossRef]
- Rodriguez, J.M.; Jorge, I.; Martinez-Val, A.; Barrero-Rodriguez, R.; Magni, R.; Nunez, E.; Laguillo, A.; Devesa, C.A.; Lopez, J.A.; Camafeita, E.; et al. Isanxot: A Standalone Application for the Integrative Analysis of Mass Spectrometry-Based Quantitative Proteomics Data. Comput. Struct. Biotechnol. J. 2024, 23, 452–459. [Google Scholar] [CrossRef]
- Trevisan-Herraz, M.; Bagwan, N.; Garcia-Marques, F.; Rodriguez, J.M.; Jorge, I.; Ezkurdia, I.; Bonzon-Kulichenko, E.; Vazquez, J. Sanxot: A Modular and Versatile Package for the Quantitative Analysis of High-Throughput Proteomics Experiments. Bioinformatics 2019, 35, 1594–1596. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The String Database in 2023: Protein-Protein Association Networks and Functional Enrichment Analyses for Any Sequenced Genome of Interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bennike, T.B.; Bellin, M.D.; Xuan, Y.; Stensballe, A.; Moller, F.T.; Beilman, G.J.; Levy, O.; Cruz-Monserrate, Z.; Andersen, V.; Steen, J.; et al. A Cost-Effective High-Throughput Plasma and Serum Proteomics Workflow Enables Mapping of the Molecular Impact of Total Pancreatectomy with Islet Autotransplantation. J. Proteome Res. 2018, 17, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.; Yan, H.; Welinder, C.; Vegvari, A.; Hamrefors, V.; Back, M.; Sutton, R.; Fedorowski, A. Plasma Proteomic Profiling in Postural Orthostatic Tachycardia Syndrome (Pots) Reveals New Disease Pathways. Sci. Rep. 2022, 12, 20051. [Google Scholar] [CrossRef] [PubMed]
- Mc Ardle, A.; Binek, A.; Moradian, A.; Chazarin Orgel, B.; Rivas, A.; Washington, K.E.; Phebus, C.; Manalo, D.M.; Go, J.; Venkatraman, V.; et al. Standardized Workflow for Precise Mid- and High-Throughput Proteomics of Blood Biofluids. Clin. Chem. 2022, 68, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.; Zhang, Q. A Streamlined High-Throughput Plasma Proteomics Platform for Clinical Proteomics with Improved Proteome Coverage, Reproducibility, and Robustness. J. Am. Soc. Mass Spectrom. 2023, 34, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Geyer, P.E.; Kulak, N.A.; Pichler, G.; Holdt, L.M.; Teupser, D.; Mann, M. Plasma Proteome Profiling to Assess Human Health and Disease. Cell Syst. 2016, 2, 185–195. [Google Scholar] [CrossRef]
- Carlsson, L.; Lind, L.; Larsson, A. Reference Values for 27 Clinical Chemistry Tests in 70-Year-Old Males and Females. Gerontology 2010, 56, 259–265. [Google Scholar] [CrossRef]
- Crawford, P.T.; Elisens, W.J. Genetic Variation and Reproductive System among North American Species of Nuttallanthus (Plantaginaceae). Am. J. Bot. 2006, 93, 582–591. [Google Scholar] [CrossRef]
- Geyer, P.E.; Wewer Albrechtsen, N.J.; Tyanova, S.; Grassl, N.; Iepsen, E.W.; Lundgren, J.; Madsbad, S.; Holst, J.J.; Torekov, S.S.; Mann, M. Proteomics Reveals the Effects of Sustained Weight Loss on the Human Plasma Proteome. Mol. Syst. Biol. 2016, 12, 901. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Benn, M.; Tybjaerg-Hansen, A.; Nordestgaard, B.G. Extreme Lipoprotein(a) Levels and Risk of Myocardial Infarction in the General Population: The Copenhagen City Heart Study. Circulation 2008, 117, 176–184. [Google Scholar] [CrossRef]
- Liu, Y.; Buil, A.; Collins, B.C.; Gillet, L.C.; Blum, L.C.; Cheng, L.Y.; Vitek, O.; Mouritsen, J.; Lachance, G.; Spector, T.D.; et al. Quantitative Variability of 342 Plasma Proteins in a Human Twin Population. Mol. Syst. Biol. 2015, 11, 786. [Google Scholar] [CrossRef]
- Anderson, L. Six Decades Searching for Meaning in the Proteome. J. Proteom. 2014, 107, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.L. The Clinical Plasma Proteome: A Survey of Clinical Assays for Proteins in Plasma and Serum. Clin. Chem. 2010, 56, 177–185. [Google Scholar] [CrossRef]
- He, T. Implementation of Proteomics in Clinical Trials. Proteom. Clin. Appl. 2019, 13, e1800198. [Google Scholar] [CrossRef]
- DeMarco, M.L.; Nguyen, Q.; Fok, A.; Hsiung, G.R.; van der Gugten, J.G. An Automated Clinical Mass Spectrometric Method for Identification and Quantification of Variant and Wild-Type Amyloid-Beta 1-40 and 1-42 Peptides in Csf. Alzheimers Dement. 2020, 12, e12036. [Google Scholar] [CrossRef]
- Banerjee, S. Empowering Clinical Diagnostics with Mass Spectrometry. ACS Omega 2020, 5, 2041–2048. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, S.M.; Lee-McMullen, B.; Abbott, C.W.; Quijada, J.V.; Hornburg, D.; Park, H.; Perelman, D.; Peterson, D.J.; Tang, M.; Robinson, A.; et al. Global, Distinctive, and Personal Changes in Molecular and Microbial Profiles by Specific Fibers in Humans. Cell Host Microbe 2022, 30, 848–862.e847. [Google Scholar] [CrossRef] [PubMed]
- Geyer, P.E.; Voytik, E.; Treit, P.V.; Doll, S.; Kleinhempel, A.; Niu, L.; Muller, J.B.; Buchholtz, M.L.; Bader, J.M.; Teupser, D.; et al. Plasma Proteome Profiling to Detect and Avoid Sample-Related Biases in Biomarker Studies. EMBO Mol. Med. 2019, 11, e10427. [Google Scholar] [CrossRef]
- Hortin, G.L.; Sviridov, D.; Anderson, N.L. High-Abundance Polypeptides of the Human Plasma Proteome Comprising the Top 4 Logs of Polypeptide Abundance. Clin. Chem. 2008, 54, 1608–1616. [Google Scholar] [CrossRef]
- Millioni, R.; Tolin, S.; Puricelli, L.; Sbrignadello, S.; Fadini, G.P.; Tessari, P.; Arrigoni, G. High Abundance Proteins Depletion Vs Low Abundance Proteins Enrichment: Comparison of Methods to Reduce the Plasma Proteome Complexity. PLoS ONE 2011, 6, e19603. [Google Scholar] [CrossRef]
- Pernemalm, M.; Orre, L.M.; Lengqvist, J.; Wikstrom, P.; Lewensohn, R.; Lehtio, J. Evaluation of Three Principally Different Intact Protein Prefractionation Methods for Plasma Biomarker Discovery. J. Proteome Res. 2008, 7, 2712–2722. [Google Scholar] [CrossRef]
- Ekdahl, K.N.; Persson, B.; Mohlin, C.; Sandholm, K.; Skattum, L.; Nilsson, B. Interpretation of Serological Complement Biomarkers in Disease. Front. Immunol. 2018, 9, 2237. [Google Scholar] [CrossRef] [PubMed]
- Skattum, L.; van Deuren, M.; van der Poll, T.; Truedsson, L. Complement Deficiency States and Associated Infections. Mol. Immunol. 2011, 48, 1643–1655. [Google Scholar] [CrossRef] [PubMed]
- Banfi, G.; Del Fabbro, M. Biological Variation in Tests of Hemostasis. Semin. Thromb. Hemost. 2009, 35, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Crawford, D.C.; Peng, Z.; Cheng, J.F.; Boffelli, D.; Ahearn, M.; Nguyen, D.; Shaffer, T.; Yi, Q.; Livingston, R.J.; Rieder, M.J.; et al. Lpa and Plg Sequence Variation and Kringle Iv-2 Copy Number in Two Populations. Hum. Hered. 2008, 66, 199–209. [Google Scholar] [CrossRef]
- Maranhao, R.C.; Carvalho, P.O.; Strunz, C.C.; Pileggi, F. Lipoprotein (a): Structure, Pathophysiology and Clinical Implications. Arq. Bras. Cardiol. 2014, 103, 76–84. [Google Scholar] [CrossRef]
- Tada, H.; Won, H.H.; Melander, O.; Yang, J.; Peloso, G.M.; Kathiresan, S. Multiple Associated Variants Increase the Heritability Explained for Plasma Lipids and Coronary Artery Disease. Circ. Cardiovasc. Genet. 2014, 7, 583–587. [Google Scholar] [CrossRef]
- Schmidt, E.M.; Willer, C.J. Insights into Blood Lipids from Rare Variant Discovery. Curr. Opin. Genet. Dev. 2015, 33, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Cole, C.B.; Nikpay, M.; McPherson, R. Gene-Environment Interaction in Dyslipidemia. Curr. Opin. Lipidol. 2015, 26, 133–138. [Google Scholar] [CrossRef]
- Wong, W.M.; Hawe, E.; Li, L.K.; Miller, G.J.; Nicaud, V.; Pennacchio, L.A.; Humphries, S.E.; Talmud, P.J. Apolipoprotein Aiv Gene Variant S347 Is Associated with Increased Risk of Coronary Heart Disease and Lower Plasma Apolipoprotein Aiv Levels. Circ. Res. 2003, 92, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Schenk, M.; Reichmann, R.; Koelman, L.; Pfeiffer, A.F.H.; Rudovich, N.N.; Aleksandrova, K. Intra-Individual Reproducibility of Galectin-1, Haptoglobin, and Nesfatin-1 as Promising New Biomarkers of Immunometabolism. Metab. Open 2020, 6, 100034. [Google Scholar] [CrossRef]
- Hosogaya, S.; Naito, K.; Sakamoto, M.; Osada, M.; Yatomi, Y.; Ozaki, Y. Biological Inter- and Intra-Individual Variations of Serum Immunochemical Constituents and Their Allowable Limits of Analytical Error. Rinsho Byori 1999, 47, 875–880. [Google Scholar] [PubMed]
- Doran, S.; Arif, M.; Lam, S.; Bayraktar, A.; Turkez, H.; Uhlen, M.; Boren, J.; Mardinoglu, A. Multi-Omics Approaches for Revealing the Complexity of Cardiovascular Disease. Brief. Bioinform. 2021, 22, bbab061. [Google Scholar] [CrossRef] [PubMed]
- Ahadi, S.; Zhou, W.; Schussler-Fiorenza Rose, S.M.; Sailani, M.R.; Contrepois, K.; Avina, M.; Ashland, M.; Brunet, A.; Snyder, M. Personal Aging Markers and Ageotypes Revealed by Deep Longitudinal Profiling. Nat. Med. 2020, 26, 83–90. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Núñez, E.; Gómez-Serrano, M.; Calvo, E.; Bonzon-Kulichenko, E.; Trevisan-Herraz, M.; Rodríguez, J.M.; García-Marqués, F.; Magni, R.; Lara-Pezzi, E.; Martín-Ventura, J.L.; et al. A Multiplexed Quantitative Proteomics Approach to the Human Plasma Protein Signature. Biomedicines 2024, 12, 2118. https://doi.org/10.3390/biomedicines12092118
Núñez E, Gómez-Serrano M, Calvo E, Bonzon-Kulichenko E, Trevisan-Herraz M, Rodríguez JM, García-Marqués F, Magni R, Lara-Pezzi E, Martín-Ventura JL, et al. A Multiplexed Quantitative Proteomics Approach to the Human Plasma Protein Signature. Biomedicines. 2024; 12(9):2118. https://doi.org/10.3390/biomedicines12092118
Chicago/Turabian StyleNúñez, Estefanía, María Gómez-Serrano, Enrique Calvo, Elena Bonzon-Kulichenko, Marco Trevisan-Herraz, José Manuel Rodríguez, Fernando García-Marqués, Ricardo Magni, Enrique Lara-Pezzi, José Luis Martín-Ventura, and et al. 2024. "A Multiplexed Quantitative Proteomics Approach to the Human Plasma Protein Signature" Biomedicines 12, no. 9: 2118. https://doi.org/10.3390/biomedicines12092118
APA StyleNúñez, E., Gómez-Serrano, M., Calvo, E., Bonzon-Kulichenko, E., Trevisan-Herraz, M., Rodríguez, J. M., García-Marqués, F., Magni, R., Lara-Pezzi, E., Martín-Ventura, J. L., Camafeita, E., & Vázquez, J. (2024). A Multiplexed Quantitative Proteomics Approach to the Human Plasma Protein Signature. Biomedicines, 12(9), 2118. https://doi.org/10.3390/biomedicines12092118