
Probiotic-Derived Metabolites from Lactiplantibacillus plantarum OC01 Reprogram Tumor-Associated Macrophages to an Inflammatory Anti-Tumoral Phenotype: Impact on Colorectal Cancer Cell Proliferation and Migration
 , ,
, ,  , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Probiotic Formulation
2.3. Collection of the Colorectal Cancer Cell Conditioning Medium (CRC Cell CM)
2.4. Antibodies
2.5. Western Blotting Analysis
2.6. Immunofluorescence Assay
2.7. Phagocytosis Study
2.8. Anion Superoxide Production by MitoSOX™ Fluorescence Assay
2.9. RNA Isolation and Quantitative PCR
2.10. Macrophages and Colorectal Cancer Cell Lines Co-Culture System Assay
2.11. Statistical Analysis
3. Results
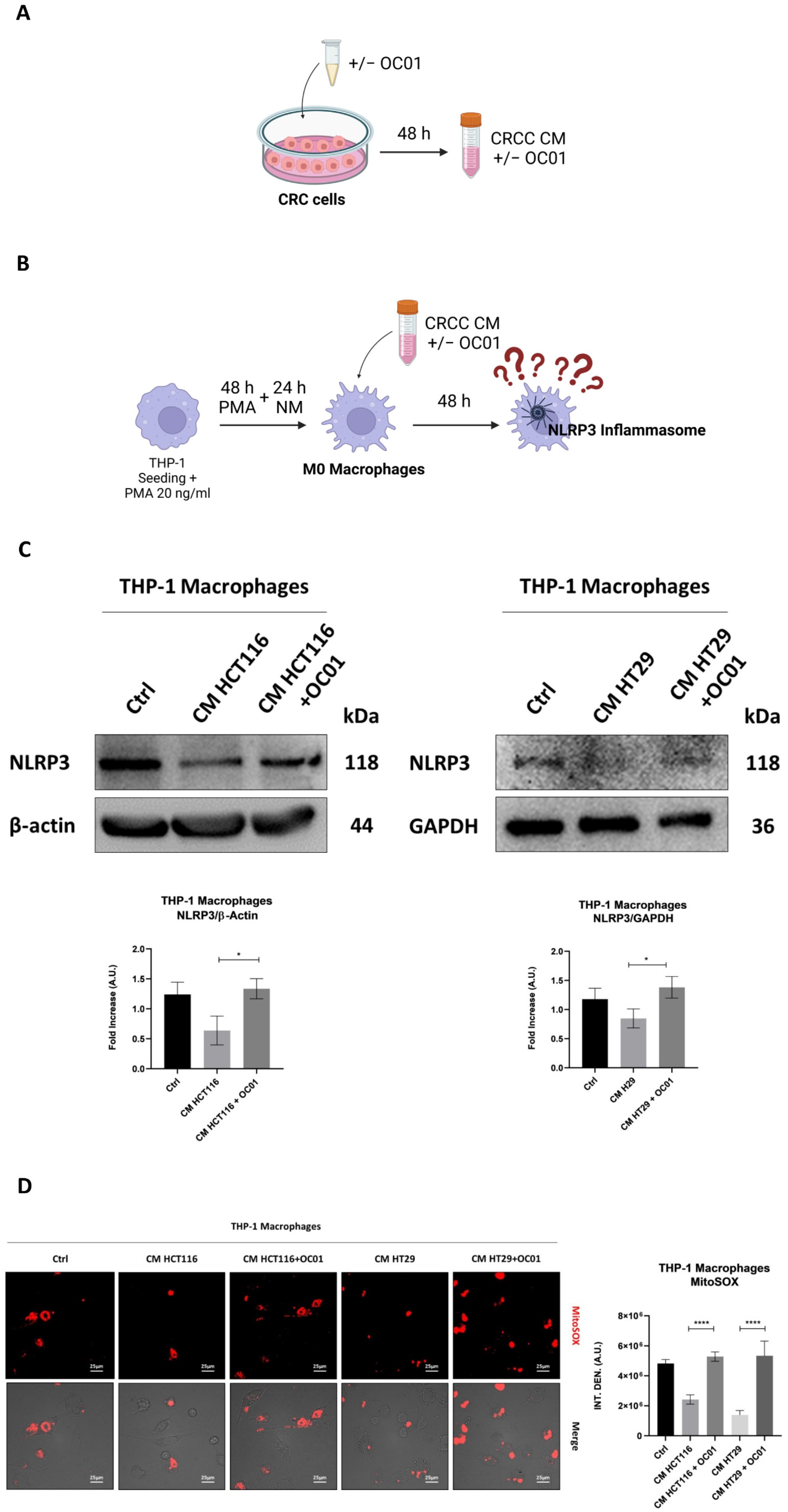
3.1. Differentiation of THP-1 into Macrophage-like Phenotype
3.2. Conditioning Medium from Lactiplantibacillus plantarum OC01-Treated Colorectal Cancer Cells Promotes Macrophage Inflammatory Phenotypes via NLRP3 Activation
3.3. Lactiplantibacillus plantarum OC01 Metabolites Reduce the Expression of TGF-β in Colorectal Cancer Cells
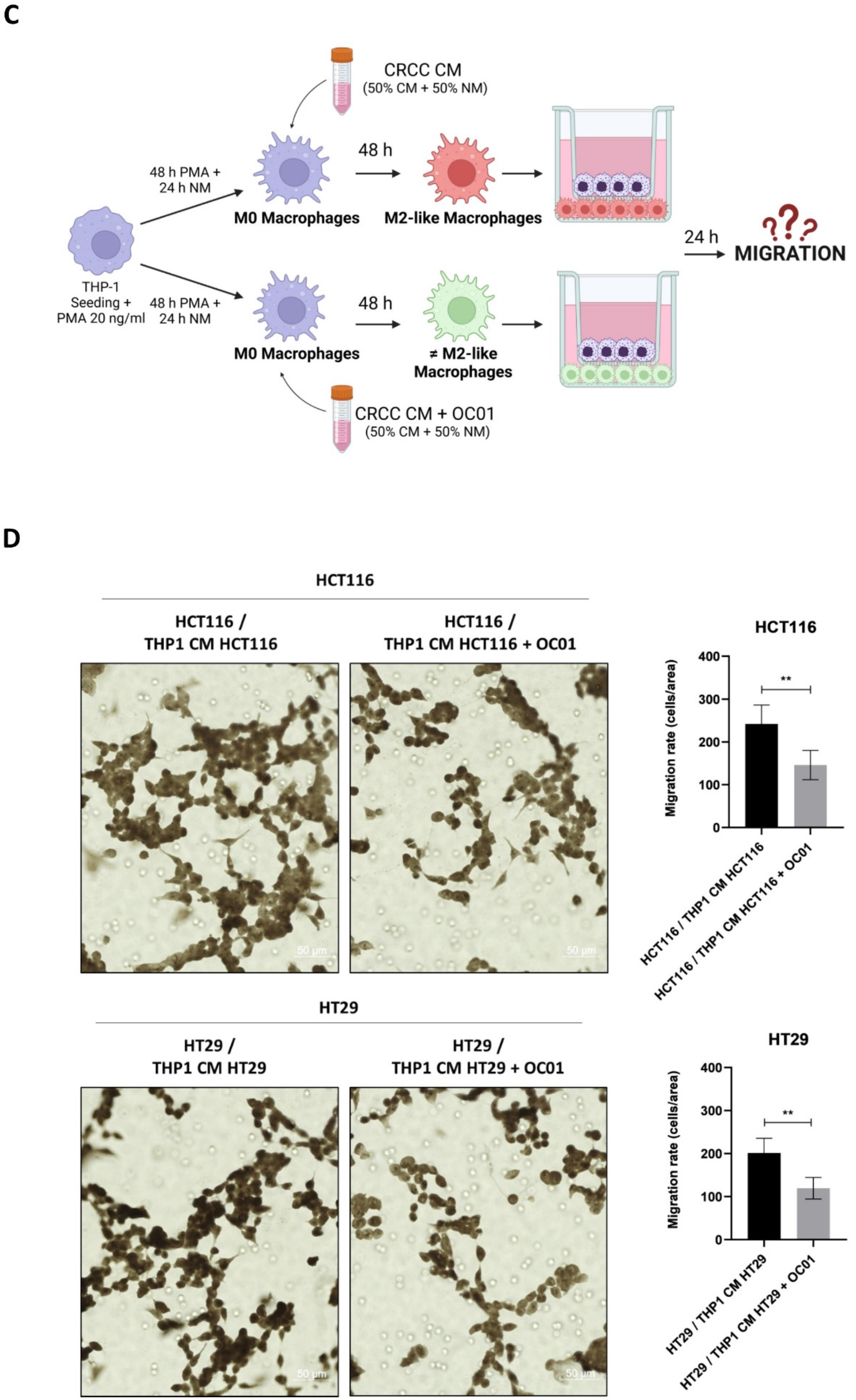
3.4. Conditioning Medium from Lactiplantibacillus plantarum OC01-Treated Colorectal Cancer Cells Reduces the Polarization of Macrophages into the M2-like Phenotype
3.5. Macrophages Reprogrammed by Conditioning Medium from LpOC01-SN-Cured CRC Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TAMs | Tumor-associated macrophages |
| CRC | Colorectal cancer |
| TME | Tumor microenvironment |
| IL | Interleukin |
| CM | Conditioning medium |
| TGF LPS | Tumor growth factor Lipopolysaccharide |
| ECM | Extracellular matrix |
| TNF | Tumor necrosis factor |
| ROS | Reactive oxygen species |
| PMA | Phorbol-12-Myristate-13-Acetate |
| COOH-NPs | Carboxy-functionalized nanoparticles |
| NM | Normal media |
| LpOC01-SN | Lactiplantibacillus plantarum OC01 supernatant |
| CSF-1 | Colony-stimulating factor-1 |
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.; Kwong, T.N.Y.; Wu, C.Y.; Yu, J. Clinical applications of gut microbiota in cancer biology. Semin. Cancer Biol. 2019, 55, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Kovács, T.; Mikó, E.; Ujlaki, G.; Sári, Z.; Bai, P. The Microbiome as a Component of the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1225, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Gagnière, J.; Raisch, J.; Veziant, J.; Barnich, N.; Bonnet, R.; Buc, E.; Bringer, M.A.; Pezet, D.; Bonnet, M. Gut microbiota imbalance and colorectal cancer. World J. Gastroenterol. 2016, 22, 501–518. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gomaa, E.Z. Human gut microbiota/microbiome in health and diseases: A review. Antonie Leeuwenhoek 2020, 113, 2019–2040. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhang, B.; Zhang, Y.; Yin, T.; Cui, Y.; Liu, J.; Yang, Y.; Song, H.; Shang, D. Gut microbiome: Decision-makers in the microenvironment of colorectal cancer. Front. Cell Infect. Microbiol. 2023, 13, 1299977. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rossi, T.; Vergara, D.; Fanini, F.; Maffia, M.; Bravaccini, S.; Pirini, F. Microbiota-Derived Metabolites in Tumor Progression and Metastasis. Int. J. Mol. Sci. 2020, 21, 5786. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gomes, S.; Rodrigues, A.C.; Pazienza, V.; Preto, A. Modulation of the Tumor Microenvironment by Microbiota-Derived Short-Chain Fatty Acids: Impact in Colorectal Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 5069. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, Y.; Cao, X. The origin and function of tumor-associated macrophages. Cell Mol. Immunol. 2015, 12, 1–4. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Yang, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X.; Liu, Q.; Dou, R.; Xiong, B. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol. Cancer 2019, 18, 64. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, G.; Li, Y.; Pan, Y. Metabolic Reprogramming Induces Macrophage Polarization in the Tumor Microenvironment. Front. Immunol. 2022, 13, 840029. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, J.; Liu, X.; Wan, C.; Liu, Y.; Wang, Y.; Meng, C.; Zhang, Y.; Jiang, C. NLRP3 inflammasome mediates M1 macrophage polarization and IL-1β production in inflammatory root resorption. J. Clin. Periodontol. 2020, 47, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Xu, Y.; Wang, J.; Xue, Z.; Qiu, T.; Chen, J. Loss of NLRP3 reduces oxidative stress and polarizes intratumor macrophages to attenuate immune attack on endometrial cancer. Front. Immunol. 2023, 14, 1165602. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sun, J.; Zeng, Q.; Wu, Z.; Huang, L.; Sun, T.; Ling, C.; Zhang, B.; Chen, C.; Wang, H. Berberine inhibits NLRP3 inflammasome activation and proinflammatory macrophage M1 polarization to accelerate peripheral nerve regeneration. Neurotherapeutics 2024, 21, e00347. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wisitpongpun, P.; Potup, P.; Usuwanthim, K. Oleamide-Mediated Polarization of M1 Macrophages and IL-1β Production by Regulating NLRP3-Inflammasome Activation in Primary Human Monocyte-Derived Macrophages. Front. Immunol. 2022, 13, 856296. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, W.; Kuang, T.; Liu, L.; Deng, W. The role of innate immune cells in the colorectal cancer tumor microenvironment and advances in anti-tumor therapy research. Front. Immunol. 2024, 15, 1407449. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhou, D.; Li, Y. Gut microbiota and tumor-associated macrophages: Potential in tumor diagnosis and treatment. Gut Microbes 2023, 15, 2276314. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Deng, X.; Yang, J.; Zhang, Y.; Chen, X.; Wang, C.; Suo, H.; Song, J. An Update on the Pivotal Roles of Probiotics, Their Components, and Metabolites in Preventing Colon Cancer. Foods 2023, 12, 3706. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhao, J.; Liao, Y.; Wei, C.; Ma, Y.; Wang, F.; Chen, Y.; Zhao, B.; Ji, H.; Wang, D.; Tang, D. Potential Ability of Probiotics in the Prevention and Treatment of Colorectal Cancer. Clin. Med. Insights Oncol. 2023, 17, 11795549231188225. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vallino, L.; Garavaglia, B.; Visciglia, A.; Amoruso, A.; Pane, M.; Ferraresi, A.; Isidoro, C. Cell-free Lactiplantibacillus plantarum OC01 supernatant suppresses IL-6-induced proliferation and invasion of human colorectal cancer cells: Effect on β-Catenin degradation and induction of autophagy. J. Tradit. Complement. Med. 2023, 13, 193–206. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ferraresi, A.; Girone, C.; Maheshwari, C.; Vallino, L.; Dhanasekaran, D.N.; Isidoro, C. Ovarian Cancer Cell-Conditioning Medium Induces Cancer-Associated Fibroblast Phenoconversion through Glucose-Dependent Inhibition of Autophagy. Int. J. Mol. Sci. 2024, 25, 5691. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Garavaglia, B.; Vallino, L.; Ferraresi, A.; Esposito, A.; Salwa, A.; Vidoni, C.; Gentilli, S.; Isidoro, C. Butyrate Inhibits Colorectal Cancer Cell Proliferation through Autophagy Degradation of β-Catenin Regardless of APC and β-Catenin Mutational Status. Biomedicines 2022, 10, 1131. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ekkapongpisit, M.; Giovia, A.; Nicotra, G.; Ozzano, M.; Caputo, G.; Isidoro, C. Labeling and exocytosis of secretory compartments in RBL mastocytes by polystyrene and mesoporous silica nanoparticles. Int. J. Nanomed. 2012, 7, 1829–1840. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vidoni, C.; Secomandi, E.; Castiglioni, A.; Melone, M.A.B.; Isidoro, C. Resveratrol protects neuronal-like cells expressing mutant Huntingtin from dopamine toxicity by rescuing ATG4-mediated autophagosome formation. Neurochem. Int. 2018, 117, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Choconta, J.L.; Labi, V.; Dumbraveanu, C.; Kalpachidou, T.; Kummer, K.K.; Kress, M. Age-related neuroimmune signatures in dorsal root ganglia of a Fabry disease mouse model. Immun. Ageing 2023, 20, 22. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhong, X.; Chen, B.; Yang, Z. The Role of Tumor-Associated Macrophages in Colorectal Carcinoma Progression. Cell Physiol. Biochem. 2018, 45, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, F.; Wang, H.; Wang, X.; Jiang, G.; Liu, H.; Zhang, G.; Wang, H.; Fang, R.; Bu, X.; Cai, S.; et al. TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget 2016, 7, 52294–52306. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ruytinx, P.; Proost, P.; Van Damme, J.; Struyf, S. Chemokine-Induced Macrophage Polarization in Inflammatory Conditions. Front. Immunol. 2018, 9, 1930. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Waldner, M.J.; Neurath, M.F. TGFβ and the Tumor Microenvironment in Colorectal Cancer. Cells 2023, 12, 1139. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Morris, R.M.; Mortimer, T.O.; O’Neill, K.L. Cytokines: Can Cancer Get the Message? Cancers 2022, 14, 2178. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yang, L.; Zhang, Y. Tumor-associated macrophages: From basic research to clinical application. J. Hematol. Oncol. 2017, 10, 58. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sun, X.; Li, Y.; Deng, Q.; Hu, Y.; Dong, J.; Wang, W.; Wang, Y.; Li, C. Macrophage Polarization, Metabolic Reprogramming, and Inflammatory Effects in Ischemic Heart Disease. Front. Immunol. 2022, 13, 934040. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mola, S.; Pandolfo, C.; Sica, A.; Porta, C. The Macrophages-Microbiota Interplay in Colorectal Cancer (CRC)-Related Inflammation: Prognostic and Therapeutic Significance. Int. J. Mol. Sci. 2020, 21, 6866. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mehla, K.; Singh, P.K. Metabolic Regulation of Macrophage Polarization in Cancer. Trends Cancer 2019, 5, 822–834. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, L.X.; Zhang, S.X.; Wu, H.J.; Rong, X.L.; Guo, J. M2b macrophage polarization and its roles in diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Amplicon Length | Forward 5′ -> 3′ | Reverse 3′ -> 5′ |
|---|---|---|---|
| IL-8 | 455 bp | GGACAAGAGCCAGGAAGAAA | CCTACAACAGACCCACACAATA |
| IL-1β | 567 bp | ATGACCTGAGCACCTTCTTTC | TCTCTGGGTACAGCTCTCTTTA |
| IL-10 | 677 bp | GAACCAAGACCCAGACATCAA | CCAAGCCCAGAGACAAGATAAA |
| IL-6 | 427 bp | CAGCTATGAACTCCTTCTCCAC | CTGGCTTGTTCCTCACTACTC |
| IL-18 | 505 bp | CCAAGGAAATCGGCCTCTATT | GTCTTGAACACCTGACCTCTG |
| TNF-α | 828 bp | ATCTACTCCCAGGTCCTCTTC | CCCGGTCTCCCAAATAAATACA |
| TGF-β | 662 bp | GTGGAAACCCACAACGAAATC | GTGTCCAGGCTCCAAATGTA |
| β-actin | 719 bp | GATCAAGATCATTGCTCCTCCTGAGCGCA | GTCTCAAGTCAGTGTACAGGTAAGCCCT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garavaglia, B.; Vallino, L.; Ferraresi, A.; Amoruso, A.; Pane, M.; Isidoro, C. Probiotic-Derived Metabolites from Lactiplantibacillus plantarum OC01 Reprogram Tumor-Associated Macrophages to an Inflammatory Anti-Tumoral Phenotype: Impact on Colorectal Cancer Cell Proliferation and Migration. Biomedicines 2025, 13, 339. https://doi.org/10.3390/biomedicines13020339
Garavaglia B, Vallino L, Ferraresi A, Amoruso A, Pane M, Isidoro C. Probiotic-Derived Metabolites from Lactiplantibacillus plantarum OC01 Reprogram Tumor-Associated Macrophages to an Inflammatory Anti-Tumoral Phenotype: Impact on Colorectal Cancer Cell Proliferation and Migration. Biomedicines. 2025; 13(2):339. https://doi.org/10.3390/biomedicines13020339
Chicago/Turabian StyleGaravaglia, Beatrice, Letizia Vallino, Alessandra Ferraresi, Angela Amoruso, Marco Pane, and Ciro Isidoro. 2025. "Probiotic-Derived Metabolites from Lactiplantibacillus plantarum OC01 Reprogram Tumor-Associated Macrophages to an Inflammatory Anti-Tumoral Phenotype: Impact on Colorectal Cancer Cell Proliferation and Migration" Biomedicines 13, no. 2: 339. https://doi.org/10.3390/biomedicines13020339
APA StyleGaravaglia, B., Vallino, L., Ferraresi, A., Amoruso, A., Pane, M., & Isidoro, C. (2025). Probiotic-Derived Metabolites from Lactiplantibacillus plantarum OC01 Reprogram Tumor-Associated Macrophages to an Inflammatory Anti-Tumoral Phenotype: Impact on Colorectal Cancer Cell Proliferation and Migration. Biomedicines, 13(2), 339. https://doi.org/10.3390/biomedicines13020339