
The Covert Side of Ascites in Cirrhosis: Cellular and Molecular Aspects

Abstract
:1. Introduction
2. Nitric Oxide (NO) and Vascular Endothelial Growth Factor (VEGF)
3. Cytokines, Signaling Molecules That Orchestrate Cells in Ascites Development
4. Hepatic Stellate Cell (HSC) and Sinusoidal Endothelial Cell (SEC) Crosstalk
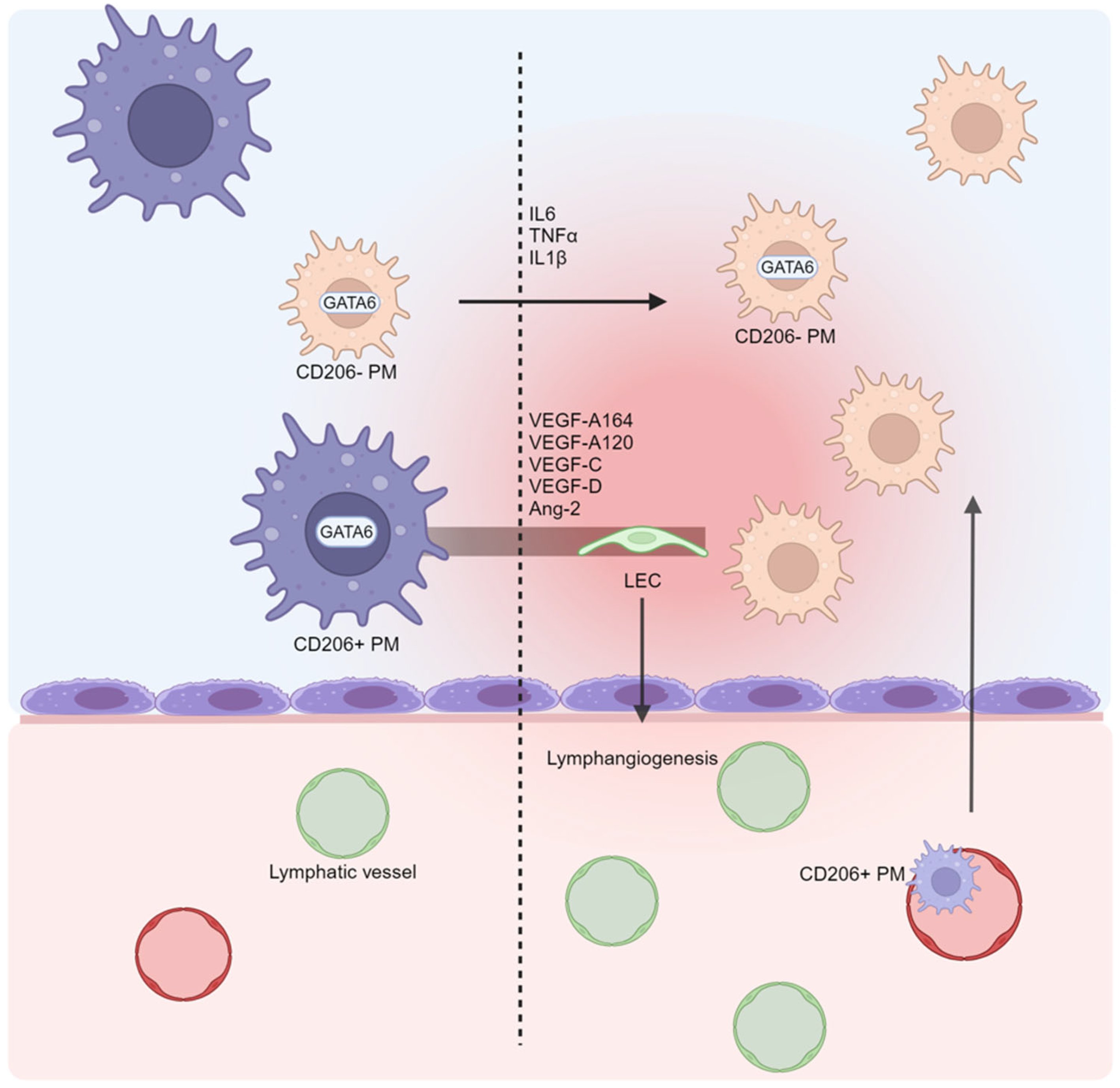
5. Macrophages: Neglected Cells in Ascites Formation?
6. The Lymphatic System and Lymphangiogenesis: Drainage Impairment in Ascites Pathogenesis
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Balcar, L.; Tonon, M.; Semmler, G.; Calvino, V.; Hartl, L.; Incicco, S.; Jachs, M.; Bauer, D.; Hofer, B.S.; Gambino, C.G.; et al. Risk of Further Decompensation/Mortality in Patients with Cirrhosis and Ascites as the First Single Decompensation Event. JHEP Rep. 2022, 4, 100513. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, V.; Angeli, P.; Moreau, R.; Jalan, R.; Clària, J.; Trebicka, J.; Fernández, J.; Gustot, T.; Caraceni, P.; Bernardi, M. The Systemic Inflammation Hypothesis: Towards a New Paradigm of Acute Decompensation and Multiorgan Failure in Cirrhosis. J. Hepatol. 2021, 74, 670–685. [Google Scholar] [CrossRef] [PubMed]
- Levitt, D.G.; Levitt, M.D. Quantitative Modeling of the Physiology of Ascites in Portal Hypertension. BMC Gastroenterol. 2012, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.S.; Bendtsen, F.; Møller, S. Management of Cirrhotic Ascites. Ther. Adv. Chronic. Dis. 2015, 6, 124–137. [Google Scholar] [CrossRef]
- Kuiper, J.J.; Boomsma, F.; Van Buren, H.; De Man, R.; Danser, A.H.J.; Van Den Meiracker, A.H. Components of the Renin-Angiotensin-Aldosterone System in Plasma and Ascites in Hepatic Cirrhosis. Eur. J. Clin. Investig. 2008, 38, 939–944. [Google Scholar] [CrossRef]
- John, S.; Thuluvath, P.J. Hyponatremia in Cirrhosis: Pathophysiology and Management. World J. Gastroenterol. 2015, 21, 3197–3205. [Google Scholar] [CrossRef]
- Dirchwolf, M.; Ruf, A.E. Role of Systemic Inflammation in Cirrhosis: From Pathogenesis to Prognosis. World J. Hepatol. 2015, 7, 1974–1981. [Google Scholar] [CrossRef]
- Starling, E.H. On the Absorption of Fluids from the Connective Tissue Spaces. J. Physiol. 1896, 19, 312–326. [Google Scholar] [CrossRef]
- Tousoulis, D.; Kampoli, A.-M.; Tentolouris Nikolaos Papageorgiou, C.; Stefanadis, C. The Role of Nitric Oxide on Endothelial Function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef]
- Moro, M.A.; Russell, R.J.; Cellek, S.; Lizasoain, I.; Su, Y.; Darley-Usmar, V.M.; Radomski, M.W.; Moncada, S. CGMP Mediates the Vascular and Platelet Actions of Nitric Oxide: Confirmation Using an Inhibitor of the Soluble Guanylyl Cyclase. Proc. Natl. Acad. Sci. USA 1996, 93, 1480–1485. [Google Scholar] [CrossRef]
- Allaire, M.; Rudler, M.; Thabut, D. Portal Hypertension and Hepatocellular Carcinoma: Des Liaisons Dangereuses…. Liver Int. 2021, 41, 1734–1743. [Google Scholar] [CrossRef] [PubMed]
- Abraldes, J.G.; Iwakiri, Y.; Loureiro-Silva, M.; Haq, O.; Sessa, W.C.; Groszmann, R.J. Mild Increases in Portal Pressure Upregulate Vascular Endothelial Growth Factor and Endothelial Nitric Oxide Synthase in the Intestinal Microcirculatory Bed, Leading to a Hyperdynamic State. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G980–G987. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Varo, G.; Melgar-Lesmes, P.; Casals, G.; Pauta, M.; Arroyo, V.; Morales-Ruiz, M.; Ros, J.; Jiménez, W. Inactivation of Extrahepatic Vascular Akt Improves Systemic Hemodynamics and Sodium Excretion in Cirrhotic Rats. J. Hepatol. 2010, 53, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Ribera, J.; Pauta, M.; Melgar-Lesmes, P.; Tugues, S.; Fernández-Varo, G.; Held, K.F.; Soria, G.; Tudela, R.; Planas, A.M.; Fernández-Hernando, C.; et al. Increased Nitric Oxide Production in Lymphatic Endothelial Cells Causes Impairment of Lymphatic Drainage in Cirrhotic Rats. Gut 2013, 62, 138–145. [Google Scholar] [CrossRef]
- Masoumi, A.; Ortiz, F.; Radhakrishnan, J.; Schrier, R.W.; Colombo, P.C. Mineralocorticoid Receptor Antagonists as Diuretics: Can Congestive Heart Failure Learn from Liver Failure? Heart Fail. Rev. 2015, 20, 283–290. [Google Scholar] [CrossRef]
- Albillos, A.; Lario, M.; Álvarez-Mon, M. Cirrhosis-Associated Immune Dysfunction: Distinctive Features and Clinical Relevance. J. Hepatol. 2014, 61, 1385–1396. [Google Scholar] [CrossRef]
- Norlander, A.E.; Madhur, M.S. Inflammatory Cytokines Regulate Renal Sodium Transporters: How, Where, and Why? Am. J. Physiol. Renal Physiol. 2017, 313, F141–F144. [Google Scholar] [CrossRef]
- Ruiz-del-Arbol, L.; Urman, J.; Fernández, J.; González, M.; Navasa, M.; Monescillo, A.; Albillos, A.; Jiménez, W.; Arroyo, V. Systemic, Renal, and Hepatic Hemodynamic Derangement in Cirrhotic Patients with Spontaneous Bacterial Peritonitis. Hepatology 2003, 38, 1210–1218. [Google Scholar] [CrossRef]
- Kolomeyevskaya, N.; Eng, K.H.; Khan, A.N.H.; Grzankowski, K.S.; Singel, K.L.; Moysich, K.; Segal, B.H. Cytokine Profiling of Ascites at Primary Surgery Identifies an Interaction of Tumor Necrosis Factor-α and Interleukin-6 in Predicting Reduced Progression-Free Survival in Epithelial Ovarian Cancer. Gynecol. Oncol. 2015, 138, 352–357. [Google Scholar] [CrossRef]
- Aquino-Acevedo, A.N.; Knochenhauer, H.; Castillo-Ocampo, Y.; Ortiz-León, M.; Rivera-López, Y.A.; Morales-López, C.; Cruz-Robles, M.E.; Hernández-Cordero, E.R.; Russell, S.; Whitaker, R.; et al. Stress Hormones Are Associated with Inflammatory Cytokines and Attenuation of T-Cell Function in the Ascites from Patients with High Grade Serous Ovarian Cancer. Brain Behav. Immun. Health 2022, 26, 100558. [Google Scholar] [CrossRef]
- Atta, S.; Kamel, M.; Mansour, W.; Hussein, T.; Maher, K.; Elrefaiy, M.A. Ascitic Fluid Cytokines in Chronic Liver Disease: A Possible Prognostic Tool. Dig. Dis. 2021, 39, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, A.S.; Gretzer, C.; Wallerstedt, S. Elevation of Cytokines in Peritoneal Fluid and Blood in Patients with Liver Cirrhosis. Hepatogastroenterology 2004, 51, 505–509. [Google Scholar]
- Liu, T.; Liu, F.; Peng, L.W.; Chang, L.; Jiang, Y.M. The Peritoneal Macrophages in Inflammatory Diseases and Abdominal Cancers. Oncol. Res. 2018, 26, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Mehta, G.; Gustot, T.; Mookerjee, R.P.; Garcia-Pagan, J.C.; Fallon, M.B.; Shah, V.H.; Moreau, R.; Jalan, R. Inflammation and Portal Hypertension—The Undiscovered Country. J. Hepatol. 2014, 61, 155–163. [Google Scholar] [CrossRef] [PubMed]
- McConnell, M.; Iwakiri, Y. Biology of Portal Hypertension. Hepatol. Int. 2018, 12, 11–23. [Google Scholar] [CrossRef]
- Sanz-García, C.; Fernández-Iglesias, A.; Gracia-Sancho, J.; Arráez-Aybar, L.A.; Nevzorova, Y.A.; Cubero, F.J. The Space of Disse: The Liver Hub in Health and Disease. Livers 2021, 1, 3–26. [Google Scholar] [CrossRef]
- Iwaisako, K.; Brenner, D.A.; Kisseleva, T. What’s New in Liver Fibrosis? The Origin of Myofibroblasts in Liver Fibrosis. J. Gastroenterol. Hepatol. 2012, 27, 65–68. [Google Scholar] [CrossRef]
- Arriazu, E.; De Galarreta, M.R.; Cubero, F.J.; Varela-Rey, M.; De Obanos, M.P.P.; Leung, T.M.; Lopategi, A.; Benedicto, A.; Abraham-Enachescu, I.; Nieto, N. Extracellular Matrix and Liver Disease. Antioxid. Redox Signal. 2014, 21, 1078–1097. [Google Scholar] [CrossRef]
- Airola, C.; Pallozzi, M.; Cerrito, L.; Santopaolo, F.; Stella, L.; Gasbarrini, A.; Ponziani, F.R. Microvascular Thrombosis and Liver Fibrosis Progression: Mechanisms and Clinical Applications. Cells 2023, 12, 1712. [Google Scholar] [CrossRef]
- Leeming, D.J.; Karsdal, M.A.; Byrjalsen, I.; Bendtsen, F.; Trebicka, J.; Nielsen, M.J.; Christiansen, C.; Møller, S.; Krag, A. Novel Serological Neo-Epitope Markers of Extracellular Matrix Proteins for the Detection of Portal Hypertension. Aliment. Pharmacol. Ther. 2013, 38, 1086–1096. [Google Scholar] [CrossRef]
- Du, W.; Wang, L. The Crosstalk Between Liver Sinusoidal Endothelial Cells and Hepatic Microenvironment in NASH Related Liver Fibrosis. Front. Immunol. 2022, 13, 936196. [Google Scholar] [CrossRef]
- Ma, Y.N.; Wang, S.S.; Liebe, R.; Ding, H.G. Crosstalk between Hepatic Stellate Cells and Tumor Cells in the Development of Hepatocellular Carcinoma. Chin. Med. J. 2021, 134, 2544–2546. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, Y. Endothelial Dysfunction in the Regulation of Cirrhosis and Portal Hypertension. Liver Int. 2012, 32, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Liu, C.; Zhou, D.; Zhang, L. TGF-β/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J. Histochem. Cytochem. 2016, 64, 157–167. [Google Scholar] [CrossRef]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhu, J.; Ouyang, H. Recent insights into contributing factors in the pathogenesis of cirrhotic ascites. Front. Med. 2024, 11, 1376217. [Google Scholar] [CrossRef]
- Mooli, R.G.R.; Mukhi, D.; Ramakrishnan, S.K. Oxidative Stress and Redox Signaling in the Pathophysiology of Liver Diseases. Compr. Physiol. 2022, 12, 3167–3192. [Google Scholar] [CrossRef]
- Garbuzenko, D.V. Pathophysiological Mechanisms of Hepatic Stellate Cells Activation in Liver Fibrosis. World J. Clin. Cases 2022, 10, 3662–3676. [Google Scholar] [CrossRef]
- Zhang, S.; Xie, B.; Wang, L.; Yang, H.; Zhang, H.; Chen, Y.; Wang, F.; Liu, C.; He, H. Macrophage-Mediated Vascular Permeability via VLA4/VCAM1 Pathway Dictates Ascites Development in Ovarian Cancer. J. Clin. Investig. 2021, 131, e140315. [Google Scholar] [CrossRef]
- Geng, A.; Flint, E.; Bernsmeier, C. Plasticity of Monocytes and Macrophages in Cirrhosis of the Liver. Front. Netw. Physiol. 2022, 2, 937739. [Google Scholar] [CrossRef]
- Noor, M.T.; Manoria, P. Immune Dysfunction in Cirrhosis. J. Clin. Transl. Hepatol. 2017, 5, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Grønbæk, H.; Rødgaard-Hansen, S.; Aagaard, N.K.; Arroyo, V.; Moestrup, S.K.; Garcia, E.; Solà, E.; Domenicali, M.; Piano, S.; Vilstrup, H.; et al. Macrophage Activation Markers Predict Mortality in Patients with Liver Cirrhosis without or with Acute-on-Chronic Liver Failure (ACLF). J. Hepatol. 2016, 64, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Irvine, K.M.; Ratnasekera, I.; Powell, E.E.; Hume, D.A. Casuses and Consequences of Innate Immune Dysfucntion in Cirrhosis. Front. Immunol. 2019, 10, 427136. [Google Scholar] [CrossRef]
- Rosas, M.; Davies, L.C.; Giles, P.J.; Liao, C.T.; Kharfan, B.; Stone, T.C.; O’Donnell, V.B.; Fraser, D.J.; Jones, S.A.; Taylor, P.R. The Transcription Factor Gata6 Links Tissue Macrophage Phenotype and Proliferative Renewal. Science 2014, 344, 645–648. [Google Scholar] [CrossRef]
- Heel, K.A.; Hall, J.C. Peritoneal Defences and Peritoneum-Associated Lymphoid Tissue. Br. J. Surg. 1996, 83, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Stengel, S.; Quickert, S.; Lutz, P.; Ibidapo-Obe, O.; Steube, A.; Köse-Vogel, N.; Yarbakht, M.; Reuken, P.A.; Busch, M.; Brandt, A.; et al. Peritoneal Level of CD206 Associates with Mortality and an Inflammatory Macrophage Phenotype in Patients with Decompensated Cirrhosis and Spontaneous Bacterial Peritonitis. Gastroenterology 2020, 158, 1745–1761. [Google Scholar] [CrossRef]
- Jayakumar, P.; Laganson, A.; Deng, M. GATA6+ Peritoneal Resident Macrophage: The Immune Custodian in the Peritoneal Cavity. Front. Pharmacol. 2022, 13, 866993. [Google Scholar] [CrossRef]
- Cassado, A.A.; D’Império Lima, M.R.; Bortoluci, K.R. Revisiting Mouse Peritoneal Macrophages: Heterogeneity, Development, and Function. Front. Immunol. 2015, 6, 225. [Google Scholar] [CrossRef]
- Kerjaschki, D. The Crucial Role of Macrophages in Lymphangiogenesis. J. Clin. Investig. 2005, 115, 2316–2319. [Google Scholar] [CrossRef]
- Kim, K.E.; Koh, Y.J.; Jeon, B.H.; Jang, C.; Han, J.; Kataru, R.P.; Schwendener, R.A.; Kim, J.M.; Koh, G.Y. Role of CD11b+ Macrophages in Intraperitoneal Lipopolysaccharide-Induced Aberrant Lymphangiogenesis and Lymphatic Function in the Diaphragm. Am. J. Pathol. 2009, 175, 1733–1745. [Google Scholar] [CrossRef]
- Shacter, E.; Arzadon, G.K.; Williams, J.A. Stimulation of Interleukin-6 and Prostaglandin E2 Secretion from Peritoneal Macrophages by Polymers of Albumin. Blood 1993, 82, 2853–2864. [Google Scholar] [CrossRef] [PubMed]
- Souza, M.H.L.P.; Cuncha, F.Q.; Martinelli, A.L.C. Interleukin 6 Concentration in Ascitic Fluid of Cirrhotic Patients: Relationship with Previous Episodes of Spontaneous Bacterial Peritonitis. J. Gastroenterol. 2003, 38, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.L.; Duan, W.; Su, C.Y.; Mao, F.Y.; Lv, Y.P.; Teng, Y.S.; Yu, P.W.; Zhuang, Y.; Zhao, Y.L. Interleukin 6 Induces M2 Macrophage Differentiation by STAT3 Activation That Correlates with Gastric Cancer Progression. Cancer Immunol. Immunother. 2017, 66, 1597–1608. [Google Scholar] [CrossRef] [PubMed]
- Louwe, P.A.; Badiola Gomez, L.; Webster, H.; Perona-Wright, G.; Bain, C.C.; Forbes, S.J.; Jenkins, S.J. Recruited Macrophages That Colonize the Post-Inflammatory Peritoneal Niche Convert into Functionally Divergent Resident Cells. Nat. Commun. 2021, 12, 1770. [Google Scholar] [CrossRef]
- Maccauro, V.; Airola, C.; Santopaolo, F.; Gasbarrini, A.; Ponziani, F.R.; Pompili, M. Gut Microbiota and Infectious Complications in Advanced Chronic Liver Disease: Focus on Spontaneous Bacterial Peritonitis. Life 2023, 13, 991. [Google Scholar] [CrossRef]
- Tapia-Abellán, A.; Martínez-Esparza, M.; Ruiz-Alcaraz, A.J.; Hernández-Caselles, T.; Martínez-Pascual, C.; Miras-López, M.; Such, J.; Francés, R.; García-Peñarrubia, P. The Peritoneal Macrophage Inflammatory Profile in Cirrhosis Depends on the Alcoholic or Hepatitis C Viral Etiology and Is Related to ERK Phosphorylation. BMC Immunol. 2012, 13, 42. [Google Scholar] [CrossRef]
- Alba-Loureiro, T.C.; Pithon-Curi, T.C.; Curi, R. Reduced Cytokine Production by Glycogen-Elicited Peritoneal Cells from Diabetic Rats. Shock 2008, 30, 308–310. [Google Scholar] [CrossRef]
- Breuillard, C.; Bonhomme, S.; Couderc, R.; Cynober, L.; De Bandt, J.P. In Vitro Anti-Inflammatory Effects of Citrulline on Peritoneal Macrophages in Zucker Diabetic Fatty Rats. Br. J. Nutr. 2015, 113, 120–124. [Google Scholar] [CrossRef]
- Chang, H.; Ni, Y.; Shen, C.; Li, C.; He, K.; Zhu, X.; Chen, L.; Chen, L.; Qiu, J.; Ji, Y.; et al. Peritoneal GATA6+ Macrophage Drives Hepatic Immunopathogenesis and Maintains the Treg Cell Niche in the Liver. Immunology 2022, 167, 77–93. [Google Scholar] [CrossRef]
- Wen, Y.; Lambrecht, J.; Ju, C.; Tacke, F. Hepatic Macrophages in Liver Homeostasis and Diseases-Diversity, Plasticity and Therapeutic Opportunities. Cell. Mol. Immunol. 2020, 18, 45–56. [Google Scholar] [CrossRef]
- Rehermann, B. Mature Peritoneal Macrophages Take an Avascular Route into the Injured Liver and Promote Tissue Repair. Hepatology 2017, 65, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Lackey, D.E.; Olefsky, J.M. Regulation of Metabolism by the Innate Immune System. Nat. Rev. Endocrinol. 2016, 12, 15–20. [Google Scholar] [CrossRef]
- Morgantini, C.; Jager, J.; Li, X.; Levi, L.; Azzimato, V.; Sulen, A.; Barreby, E.; Xu, C.; Tencerova, M.; Näslund, E.; et al. Liver Macrophages Regulate Systemic Metabolism through Non-Inflammatory Factors. Nat. Metab. 2019, 1, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, J.; Zhu, B.; Duan, Y.; Chen, F.; Nian, W.; Sun, J.; Zhang, B.; Tong, Z.; Chen, Z.; et al. IGFBP7 Functions as a Potential Lymphangiogenesis Inducer in Non-Small Cell Lung Carcinoma. Oncol. Rep. 2016, 35, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Lutz, P.; Jeffery, H.C.; Jones, N.; Birtwistle, J.; Krämer, B.; Nattermann, J.; Spengler, U.; Strassburg, C.P.; Adams, D.H.; Oo, Y.H. NK Cells in Ascites from Liver Disease Patients Display a Particular Phenotype and Take Part in Antibacterial Immune Response. Front. Immunol. 2019, 10, 462029. [Google Scholar] [CrossRef]
- Legaz, I.; Morales, R.; Bolarín, J.M.; Collados-Ros, A.; Pons, J.A.; Muro, M. Is the Development of Ascites in Alcoholic Liver Patients Influenced by Specific KIR/HLA Gene Profiles? Biomedicines 2023, 11, 2405. [Google Scholar] [CrossRef]
- Bhardwaj, R.; Vaziri, H.; Gautam, A.; Ballesteros, E.; Karimeddini, D.; Wu, G.Y. Chylous Ascites: A Review of Pathogenesis, Diagnosis and Treatment. J. Clin. Transl. Hepatol. 2018, 6, 105–113. [Google Scholar] [CrossRef]
- Tsilibary, E.C.; Wissig, S.L. Light and Electron Microscope Observations of the Lymphatic Drainage Units of the Peritoneal Cavity of Rodents. Am. J. Anat. 1987, 180, 195–207. [Google Scholar] [CrossRef]
- Tammela, T.; Alitalo, K. Lymphangiogenesis: Molecular Mechanisms and Future Promise. Cell 2010, 140, 460–476. [Google Scholar] [CrossRef]
- Melichar, B.; Freedman, R.S. Immunology of the Peritoneal Cavity: Relevance for Host-Tumor Relation. Int. J. Gynecol. Cancer 2002, 12, 3–17. [Google Scholar] [CrossRef]
- Broche, F.; Tellado, J.M. Defense Mechanisms of the Peritoneal Cavity. Curr. Opin. Crit. Care 2001, 7, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Schmid-Schönbein, G.W. Microlymphatics and Lymph Flow. Physiol. Rev. 1990, 70, 987–1028. [Google Scholar] [CrossRef] [PubMed]
- Leak, L.V.; Burke, J.F. Ultrastructural studies on the lymphatic anchoring filaments. J. Cell Biol. 1968, 36, 129. [Google Scholar] [CrossRef]
- Yamauchi, Y.; Michitaka, K.; Onji, M. Morphometric Analysis of Lymphatic and Blood Vessels in Human Chronic Viral Liver Diseases. Am. J. Pathol. 1998, 153, 1131–1137. [Google Scholar] [CrossRef]
- Kumar, R.; Anand, U.; Priyadarshi, R.N. Lymphatic Dysfunction in Advanced Cirrhosis: Contextual Perspective and Clinical Implications. World J. Hepatol. 2021, 13, 300–314. [Google Scholar] [CrossRef]
- Henriksen, J.H. Estimation of Lymphatic Conductance. A Model Based on Protein-Kinetic Studies and Haemodynamic Measurements in Patients with Cirrhosis of the Liver and in Pigs. Scand. J. Clin. Lab. Investig. 1985, 45, 123–130. [Google Scholar] [CrossRef]
- Hos, D.; Cursiefen, C. Lymphatic Vessels in the Development of Tissue and Organ Rejection. In Developmental Aspects of the Lymphatic Vascular System; Advances in Anatomy, Embryology and Cell Biology; Springer: Vienna, Austria, 2014; Volume 214, pp. 119–141. [Google Scholar] [CrossRef]
- Ly, C.L.; Kataru, R.P.; Mehrara, B.J. Inflammatory Manifestations of Lymphedema. Int. J. Mol. Sci. 2017, 18, 171. [Google Scholar] [CrossRef]
- Nilsson, M.; Heymach, J.V. Vascular Endothelial Growth Factor (VEGF) Pathway. J. Thorac. Oncol. 2006, 1, 768–770. [Google Scholar] [CrossRef]
- Stacker, S.A.; Achen, M.G. Emerging Roles for VEGF-D in Human Disease. Biomolecules 2018, 8, 1. [Google Scholar] [CrossRef]
- Jiron Tamburini, B.A.; Finlon, J.M.; Gillen, A.E.; Kriss, M.S.; Riemondy, K.A.; Fu, R.; Schuyler, R.P.; Hesselberth, J.R.; Rosen, H.R.; Burchill, M.A. Chronic Liver Disease in Humans Causes Expansion and Differentiation of Liver Lymphatic Endothelial Cells. Front. Immunol. 2019, 10, 439079. [Google Scholar] [CrossRef]
- Wang, S.; Nie, D.; Rubin, J.P.; Kokai, L. Lymphatic Endothelial Cells under Mechanical Stress: Altered Expression of Inflammatory Cytokines and Fibrosis. Lymphat. Res. Biol. 2017, 15, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Ordóñez, N.G. Podoplanin: A Novel Diagnostic Immunohistochemical Marker. Adv. Anat. Pathol. 2006, 13, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Thurston, G. Role of Angiopoietins and Tie Receptor Tyrosine Kinases in Angiogenesis and Lymphangiogenesis. Cell Tissue Res. 2003, 314, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.P.K.H.; Chen, S.H.; Trinh, J.; Kim, H.; Coomber, B.L.; Dumont, D.J. Differential Response of Lymphatic, Venous and Arterial Endothelial Cells to Angiopoietin-1 and Angiopoietin-2. BMC Cell Biol. 2007, 8, 10. [Google Scholar] [CrossRef]
- Tzeng, H.-E.; Chang, A.-C.; Tsai, C.-H.; Wang, S.-W.; Tang, C.-H.; Tzeng, H.-E.; Chang, A.-C.; Tsai, C.-H.; Wang, S.-W.; Tang, C.-H. Basic Fibroblast Growth Factor Promotes VEGF-C-Dependent Lymphangiogenesis via Inhibition of MiR-381 in Human Chondrosarcoma Cells. Oncotarget 2016, 7, 38566–38578. [Google Scholar] [CrossRef]
- Oka, M.; Iwata, C.; Suzuki, H.I.; Kiyono, K.; Morishita, Y.; Watabe, T.; Komuro, A.; Kano, M.R.; Miyazono, K. Inhibition of Endogenous TGF-β Signaling Enhances Lymphangiogenesis. Blood 2008, 111, 4571–4579. [Google Scholar] [CrossRef]
- Karlsen, T.V.; Reikvam, T.; Tofteberg, A.; Nikpey, E.; Skogstrand, T.; Wagner, M.; Tenstad, O.; Wiig, H. Lymphangiogenesis Facilitates Initial Lymph Formation and Enhances the Dendritic Cell Mobilizing Chemokine CCL21 without Affecting Migration. Arter. Thromb. Vasc. Biol. 2017, 37, 2128–2135. [Google Scholar] [CrossRef]
- Zhuo, W.; Jia, L.; Song, N.; Lu, X.A.; Ding, Y.; Wang, X.; Song, X.; Fu, Y.; Luo, Y. The CXCL12–CXCR4 Chemokine Pathway: A Novel Axis Regulates Lymphangiogenesis. Clin. Cancer Res. 2012, 18, 5387–5398. [Google Scholar] [CrossRef]
- Cao, Y. Direct Role of PDGF-BB in Lymphangiogenesis and Lymphatic Metastasis. Cell Cycle 2005, 4, 231–233. [Google Scholar] [CrossRef]
- Roberts, L.R.; Kamath, P.S. Ascites and Hepatorenal Syndrome: Pathophysiology and Management. Mayo Clin. Proc. 1996, 71, 874–881. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Study Reference | Study Type and Model | Mechanism Demonstrated |
|---|---|---|
| Abraldes et al. (2006) [12] | Preclinical, rat model | Moderate portal hypertension increases VEGF synthesis and eNOS expression, contributing to vascular permeability and ascites formation. |
| Fernández-Varo et al. (2010) [13] | Gene transfer in cirrhotic rats | Inhibition of eNOS activity reduces ascites volume and improves renal excretion. |
| Ribera et al. (2013) [14] | In vivo, cirrhotic rat model | NO production in LECs impairs lymphatic drainage, contributing to fluid accumulation. |
| Masoumi et al. (2015) [15] | Review of clinical and preclinical studies | NO inhibits aldosterone production, promoting natriuresis and potentially alleviating ascites. |
| Study Reference | Study Type and Model | Mechanism Demonstrated |
|---|---|---|
| Albillos et al. (2014) [16] | Clinical study, cirrhotic patients | Cytokines disrupt endothelial barriers, increasing vascular permeability and ascites. |
| Norlander & Madhur (2017) [17] | Review of molecular pathways | IL-6 and IFN-γ activate the RAAS system, worsening fluid retention. |
| Kolomeyevskaya et al. (2015) [19] | Clinical study, ovarian cancer patients | Interaction of TNF-α and IL-6 predicts increased vascular permeability and reduced progression-free survival, suggesting a role in ascites formation. |
| Eriksson et al. (2004) [22] | Clinical study, plasma and ascites analysis | Elevated IL-1α, IL-6, and TNF-α in ascitic fluid correlate with increased vascular permeability. |
| Study Reference | Study Type and Model | Mechanism Demonstrated |
|---|---|---|
| Leeming et al. (2013) [30] | Clinical biomarkers study | ECM deposition by activated HSCs correlates with portal hypertension, contributing to ascites. |
| Du & Wang (2022) [31] | Review of cellular interactions | HSCs release TGF-β and PDGF, enhancing SECs’ ET-1 production, increasing intrahepatic resistance. |
| Roehlen et al. (2020) [35] | Review of molecular pathways | TGF-β/Smad signaling promotes pro-fibrotic phenotype in SECs, aggravating ascites. |
| Garbuzenko (2022) [38] | Review of oxidative stress mechanisms | ROS-mediated signaling between HSCs and SECs perpetuates inflammation and fibrosis, worsening fluid accumulation. |
| Study Reference | Study Type and Model | Mechanism Demonstrated |
|---|---|---|
| Kerjaschki (2005) [49] | Review of lymphangiogenesis | PMs transdifferentiate into LECs, enhancing abnormal lymphangiogenesis and impairing drainage. |
| Kim et al. (2009) [50] | Preclinical, mouse model | Inflammatory stimuli induce PMs’ production of lymphangiogenic VEGFs, worsening ascites. |
| Tapia-Abellán et al. (2012) [56] | Clinical observation, alcohol-related cirrhosis | Pro-inflammatory cytokine production by PMs in alcohol-related cirrhosis activates ERK1/2 signaling. |
| Chang et al. (2022) [59] | Preclinical, mouse model | Migration of PMs to the liver influences hepatic macrophage activity, contributing to ascites formation. |
| Study Reference | Study Type and Model | Mechanism Demonstrated |
|---|---|---|
| Yamauchi et al. (1998) [74] | Clinical, morphometric analysis | Enlarged hepatic lymphatic vessels in cirrhosis lead to increased lymphatic pressure and ascites. |
| Henriksen (1985) [76] | Clinical study, thoracic duct conductivity | Reduced lymphatic conductivity in cirrhotic patients with ascites impairs fluid drainage. |
| Kumar et al. (2021) [75] | Review of lymphatic dysfunction | Dysregulated lymphangiogenesis in cirrhosis impairs lymphatic outflow, contributing to ascites. |
| Stacker & Achen (2018) [80] | Review of VEGF signaling | VEGF-C and VEGF-D mediated lymphangiogenesis exacerbates fluid accumulation. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Airola, C.; Varca, S.; Del Gaudio, A.; Pizzolante, F. The Covert Side of Ascites in Cirrhosis: Cellular and Molecular Aspects. Biomedicines 2025, 13, 680. https://doi.org/10.3390/biomedicines13030680
Airola C, Varca S, Del Gaudio A, Pizzolante F. The Covert Side of Ascites in Cirrhosis: Cellular and Molecular Aspects. Biomedicines. 2025; 13(3):680. https://doi.org/10.3390/biomedicines13030680
Chicago/Turabian StyleAirola, Carlo, Simone Varca, Angelo Del Gaudio, and Fabrizio Pizzolante. 2025. "The Covert Side of Ascites in Cirrhosis: Cellular and Molecular Aspects" Biomedicines 13, no. 3: 680. https://doi.org/10.3390/biomedicines13030680
APA StyleAirola, C., Varca, S., Del Gaudio, A., & Pizzolante, F. (2025). The Covert Side of Ascites in Cirrhosis: Cellular and Molecular Aspects. Biomedicines, 13(3), 680. https://doi.org/10.3390/biomedicines13030680