
Identification and Functional Analysis of Cystathionine Beta-Synthase Gene Mutations in Chinese Families with Classical Homocystinuria

Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects and Clinical Examinations
2.2. Whole Exome Sequencing (WES)
2.3. Bioinformatics Analysis
2.4. Minigene Assays
2.5. Protein Structural Modeling
2.6. Measurement of Homocysteine (Hcy) and CBS Enzyme Levels
2.7. Construction of CBS Expression Plasmids
2.8. Cell Culture and Transfection
2.9. RNA Extraction and Quantitative Reverse Transcription-Polymerase Chain Reaction (PCR)
2.10. Western Blot
2.11. Statistical Analysis
3. Results
3.1. Clinical Presentations
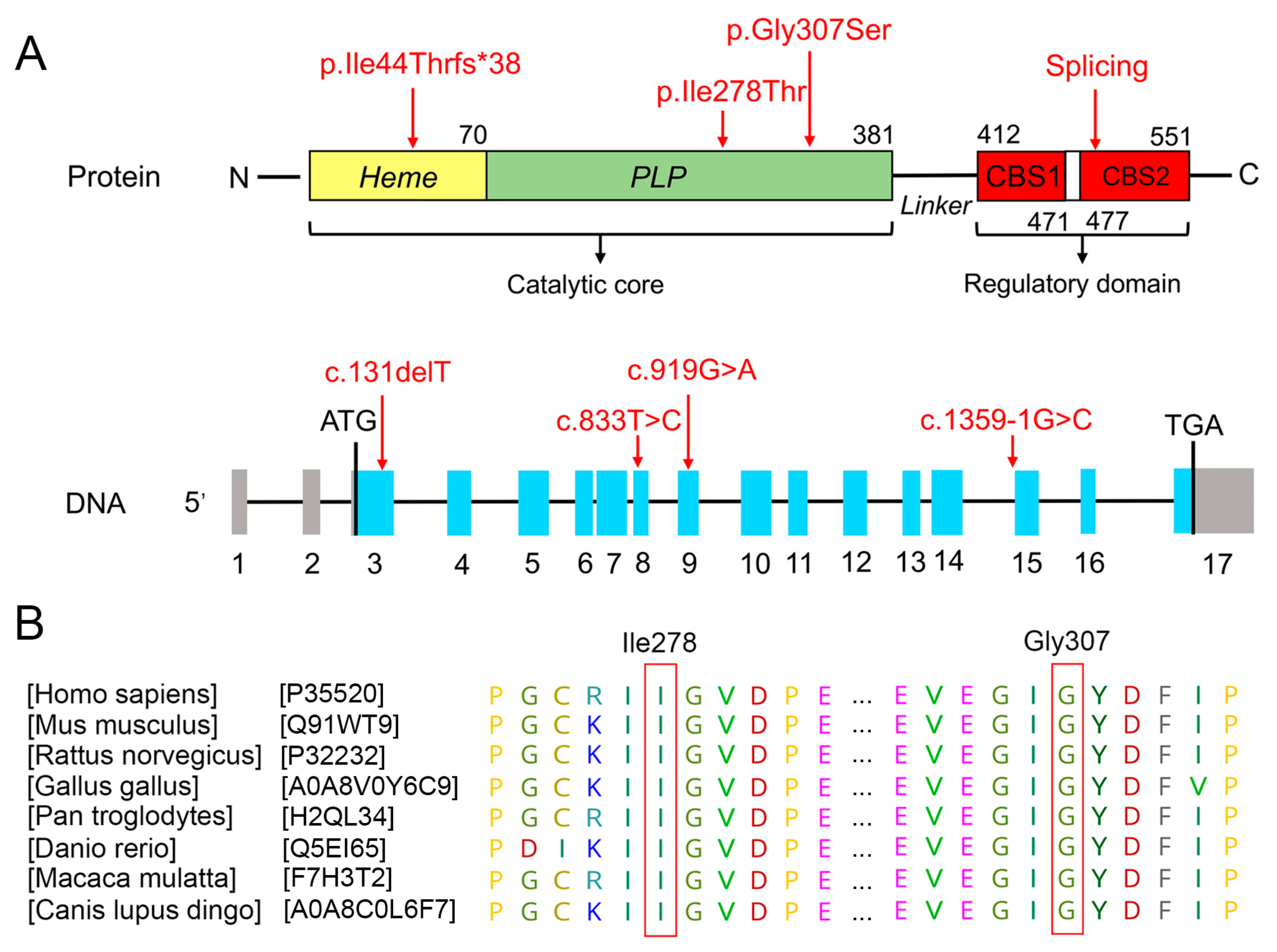
3.2. Bioinformatics Analysis of Functional Effects of Genetic Variants
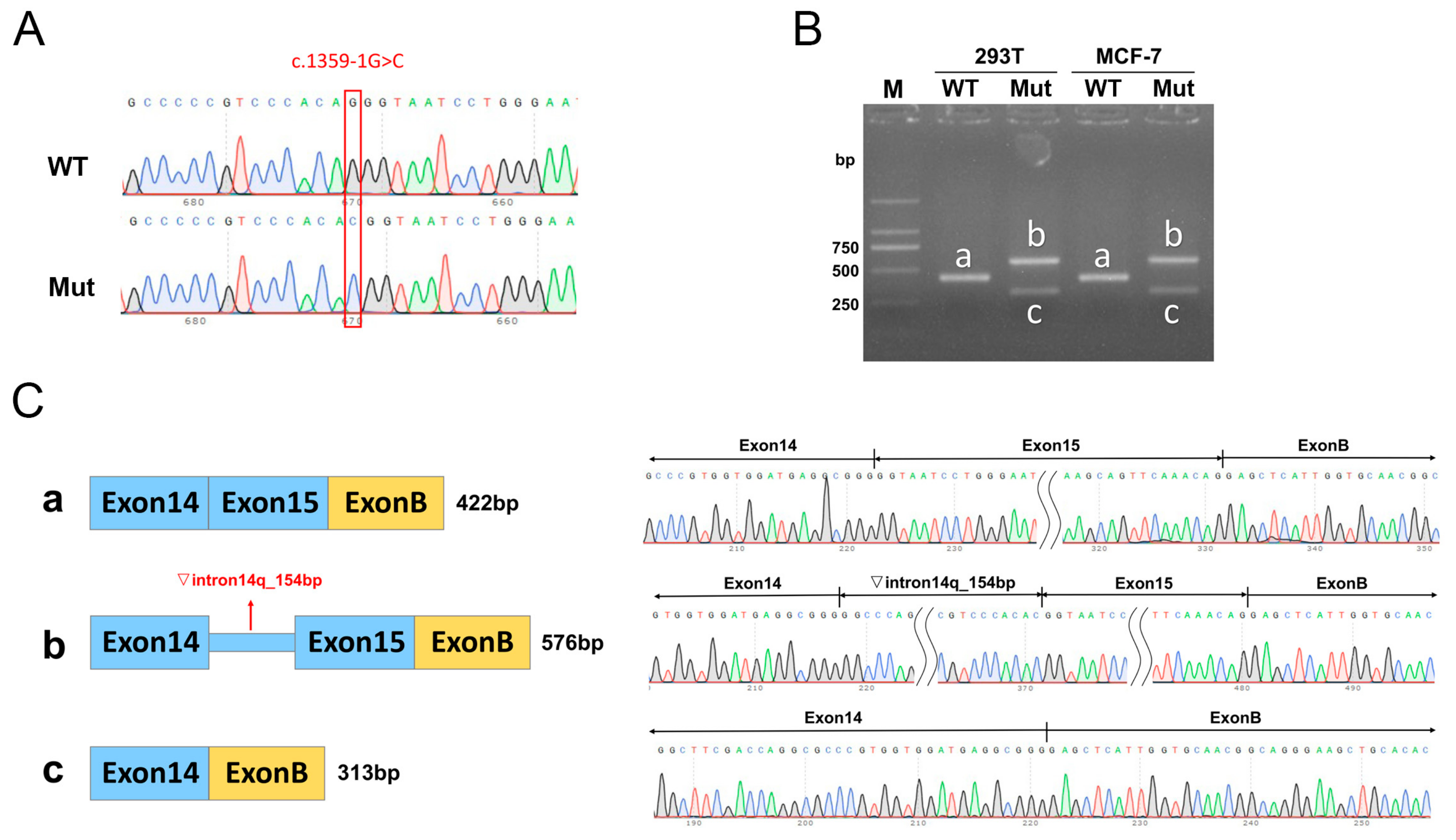
3.3. Minigene Assay
3.4. Protein Structure Analysis
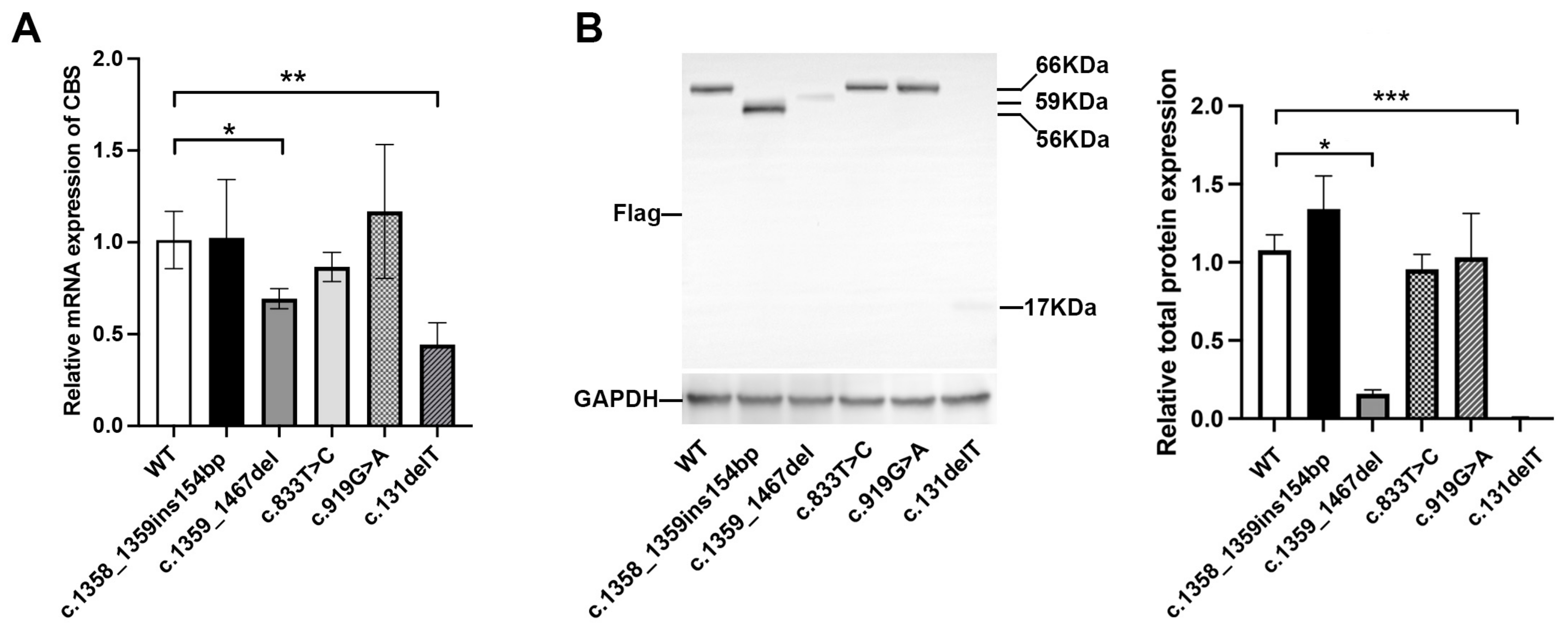
3.5. In Vitro Expression Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Faverzani, J.L.; Hammerschmidt, T.G.; Sitta, A.; Deon, M.; Wajner, M.; Vargas, C.R. Oxidative Stress in Homocystinuria Due to Cystathionine ß-Synthase Deficiency: Findings in Patients and in Animal Models. Cell. Mol. Neurobiol. 2017, 37, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Kruger, W.D.; Wang, L.; Jhee, K.H.; Singh, R.H.; Elsas, L.J., II. Cystathionine beta-synthase deficiency in Georgia (USA): Correlation of clinical and biochemical phenotype with genotype. Hum. Mutat. 2003, 22, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Škovierová, H.; Vidomanová, E.; Mahmood, S.; Sopková, J.; Drgová, A.; Červeňová, T.; Halašová, E.; Lehotský, J. The Molecular and Cellular Effect of Homocysteine Metabolism Imbalance on Human Health. Int. J. Mol. Sci. 2016, 17, 1733. [Google Scholar] [CrossRef]
- Mudd, S.; Levy, H.; Kraus, J.P. Disorders of transsulfuration. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C., Beaudet, A., Sly, W., Valle, D., Childs, B., Kinzler, K., Vogelstin, B., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 2007–2056. [Google Scholar]
- Rahman, M.; Sharma, M.; Aggarwal, P.; Singla, S.; Jain, N. Homocystinuria and ocular complications—A review. Indian J. Ophthalmol. 2022, 70, 2272–2278. [Google Scholar] [CrossRef]
- Mudd, S.H.; Skovby, F.; Levy, H.L.; Pettigrew, K.D.; Wilcken, B.; Pyeritz, R.E.; Andria, G.; Boers, G.H.; Bromberg, I.L.; Cerone, R.; et al. The natural history of homocystinuria due to cystathionine beta-synthase deficiency. Am. J. Hum. Genet. 1985, 37, 1–31. [Google Scholar]
- Walter, J.H.; Jahnke, N.; Remmington, T. Newborn screening for homocystinuria. Cochrane Database Syst. Rev. 2011, 8, Cd008840. [Google Scholar]
- Naughten, E.R.; Yap, S.; Mayne, P.D. Newborn screening for homocystinuria: Irish and world experience. Eur. J. Pediatr. 1998, 157 (Suppl. S2), S84–S87. [Google Scholar] [CrossRef]
- Yap, S.; Naughten, E. Homocystinuria due to cystathionine beta-synthase deficiency in Ireland: 25 years’ experience of a newborn screened and treated population with reference to clinical outcome and biochemical control. J. Inherit. Metab. Dis. 1998, 21, 738–747. [Google Scholar] [CrossRef]
- Garland, J.; Prasad, A.; Vardy, C.; Prasad, C. Homocystinuria: Challenges in diagnosis and management. Paediatr. Child Health 1999, 4, 557–562. [Google Scholar] [CrossRef]
- Poloni, S.; Sperb-Ludwig, F.; Borsatto, T.; Weber Hoss, G.; Doriqui, M.J.R.; Embiruçu, E.K.; Boa-Sorte, N.; Marques, C.; Kim, C.A.; Fischinger Moura de Souza, C.; et al. CBS mutations are good predictors for B6-responsiveness: A study based on the analysis of 35 Brazilian Classical Homocystinuria patients. Mol. Genet. Genom. Med. 2018, 6, 160–170. [Google Scholar] [CrossRef]
- Saba, N.; Irshad, S. Congenital cataract: An ocular manifestation of classical homocystinuria. Mol. Genet. Genom. Med. 2021, 9, e1742. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, J.D. Methionine metabolism in mammals: The biochemical basis for homocystinuria. Metabolism 1974, 23, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.K.; Sharma, V. Commentary: Ophthalmic manifestations of homocystinuria. Indian J. Ophthalmol. 2022, 70, 2278–2279. [Google Scholar] [CrossRef] [PubMed]
- Hubmacher, D.; Cirulis, J.T.; Miao, M.; Keeley, F.W.; Reinhardt, D.P. Functional consequences of homocysteinylation of the elastic fiber proteins fibrillin-1 and tropoelastin. J. Biol. Chem. 2010, 285, 1188–1198. [Google Scholar] [CrossRef]
- Petrosino, M.; Zuhra, K.; Kopec, J.; Hutchin, A.; Szabo, C.; Majtan, T. H(2)S biogenesis by cystathionine beta-synthase: Mechanism of inhibition by aminooxyacetic acid and unexpected role of serine. Cell. Mol. Life Sci. 2022, 79, 438. [Google Scholar] [CrossRef]
- Taoka, S.; West, M.; Banerjee, R. Characterization of the heme and pyridoxal phosphate cofactors of human cystathionine beta-synthase reveals nonequivalent active sites. Biochemistry 1999, 38, 2738–2744. [Google Scholar] [CrossRef]
- Taoka, S.; Widjaja, L.; Banerjee, R. Assignment of enzymatic functions to specific regions of the PLP-dependent heme protein cystathionine beta-synthase. Biochemistry 1999, 38, 13155–13161. [Google Scholar] [CrossRef]
- Bateman, A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem. Sci. 1997, 22, 12–13. [Google Scholar] [CrossRef]
- Kraus, J.P.; Janosík, M.; Kozich, V.; Mandell, R.; Shih, V.; Sperandeo, M.P.; Sebastio, G.; de Franchis, R.; Andria, G.; Kluijtmans, L.A.; et al. Cystathionine beta-synthase mutations in homocystinuria. Hum. Mutat. 1999, 13, 362–375. [Google Scholar] [CrossRef]
- Mendes, M.I.; Santos, A.S.; Smith, D.E.; Lino, P.R.; Colaço, H.G.; de Almeida, I.T.; Vicente, J.B.; Salomons, G.S.; Rivera, I.; Blom, H.J.; et al. Insights into the regulatory domain of cystathionine Beta-synthase: Characterization of six variant proteins. Hum. Mutat. 2014, 35, 1195–1202. [Google Scholar] [CrossRef]
- Roman, J.V.; Mascarenhas, R.; Ceric, K.; Ballou, D.P.; Banerjee, R. Disease-causing cystathionine β-synthase linker mutations impair allosteric regulation. J. Biol. Chem. 2023, 299, 105449. [Google Scholar] [CrossRef] [PubMed]
- Kozich, V.; Kraus, J.P. Screening for mutations by expressing patient cDNA segments in E. coli: Homocystinuria due to cystathionine beta-synthase deficiency. Hum. Mutat. 1992, 1, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Silao, C.L.; Fabella, T.D.; Rama, K.I.; Estrada, S.C. Novel cystathionine β-synthase gene mutations in a Filipino patient with classic homocystinuria. Pediatr. Int. 2015, 57, 884–887. [Google Scholar] [CrossRef]
- Gong, B.; Liu, L.; Li, Z.; Ye, Z.; Xiao, Y.; Zeng, G.; Shi, Y.; Wang, Y.; Feng, X.; Li, X.; et al. Novel Compound Heterozygous CBS Mutations Cause Homocystinuria in a Han Chinese Family. Sci. Rep. 2015, 5, 17947. [Google Scholar] [CrossRef]
- Kaur, R.; Attri, S.V.; Saini, A.G.; Sankhyan, N.; Singh, S.; Faruq, M.; Ramprasad, V.L.; Sharda, S.; Murugan, S. Seven novel genetic variants in a North Indian cohort with classical homocystinuria. Sci. Rep. 2020, 10, 17299. [Google Scholar] [CrossRef]
- Li, D.X.; Li, X.Y.; Dong, H.; Liu, Y.P.; Ding, Y.; Song, J.Q.; Jin, Y.; Zhang, Y.; Wang, Q.; Yang, Y.L. Eight novel mutations of CBS gene in nine Chinese patients with classical homocystinuria. World J. Pediatr. 2018, 14, 197–203. [Google Scholar] [CrossRef]
- Kwok, J.S.; Fung, S.L.; Lui, G.C.; Law, E.L.; Chan, M.H.; Leung, C.B.; Tang, N.L. CBS gene mutations found in a Chinese pyridoxine-responsive homocystinuria patient. Pathology 2011, 43, 81–83. [Google Scholar] [CrossRef]
- Hua, N.; Ning, Y.; Zheng, H.; Zhao, L.; Qian, X.; Wormington, C.; Wang, J. Recurrent dislocation of binocular crystal lenses in a patient with cystathionine beta-synthase deficiency. BMC Ophthalmol. 2021, 21, 212. [Google Scholar] [CrossRef]
- Shih, V.E.; Fringer, J.M.; Mandell, R.; Kraus, J.P.; Berry, G.T.; Heidenreich, R.A.; Korson, M.S.; Levy, H.L.; Ramesh, V. A missense mutation (I278T) in the cystathionine beta-synthase gene prevalent in pyridoxine-responsive homocystinuria and associated with mild clinical phenotype. Am. J. Hum. Genet. 1995, 57, 34–39. [Google Scholar]
- Moat, S.J.; Bao, L.; Fowler, B.; Bonham, J.R.; Walter, J.H.; Kraus, J.P. The molecular basis of cystathionine beta-synthase (CBS) deficiency in UK and US patients with homocystinuria. Hum. Mutat. 2004, 23, 206. [Google Scholar] [CrossRef]
- Gallagher, P.M.; Ward, P.; Tan, S.; Naughten, E.; Kraus, J.P.; Sellar, G.C.; McConnell, D.J.; Graham, I.; Whitehead, A.S. High frequency (71%) of cystathionine beta-synthase mutation G307S in Irish homocystinuria patients. Hum. Mutat. 1995, 6, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kelow, S.; Wang, L.; Andrake, M.D.; Dunbrack, R.L., Jr.; Kruger, W.D. Mouse modeling and structural analysis of the p.G307S mutation in human cystathionine β-synthase (CBS) reveal effects on CBS activity but not stability. J. Biol. Chem. 2018, 293, 13921–13931. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Wang, L.; Slifker, M.J.; Cai, K.Q.; Maclean, K.N.; Wasek, B.; Bottiglieri, T.; Kruger, W.D. Analysis of differential neonatal lethality in cystathionine β-synthase deficient mouse models using metabolic profiling. FASEB J. 2021, 35, e21629. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasma Measure | Proband 1 | Proband 2 | Reference Range | Unit |
|---|---|---|---|---|
| Vitamin B2 | 3.35 | 2.97 | 2.3–14.6 | ng/mL |
| Vitamin B6 | 1.85 | 1.03 | 3–30 | ng/mL |
| Vitamin B9 | 3.97 | 4.31 | >4 | ng/mL |
| Vitamin B12 | 51.49 | 24.58 | <52.91 | ng/mL |
| 5-methyltetrahydrofolate | 3.7 | 2.46 | >4 | ng/mL |
| Methionine | 95.69 | 73.35 | 4–44 | μmol/L |
| Cystathionine | 0.16 | 1.27 | <5 | μmol/L |
| Splicing prediction | c.1359-1G>C | c.131delT | |||
| Software | ΔScore | Effect | ΔScore | Effect | |
| HSF (Version 3.1) | / | + | / | - | |
| Varseak | / | + | / | - | |
| SpliceAI | 0.98 | + | 0.00 | - | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Liu, X.; Liu, J.; Guo, J.; Nie, D.; Wang, J. Identification and Functional Analysis of Cystathionine Beta-Synthase Gene Mutations in Chinese Families with Classical Homocystinuria. Biomedicines 2025, 13, 919. https://doi.org/10.3390/biomedicines13040919
Liu X, Liu X, Liu J, Guo J, Nie D, Wang J. Identification and Functional Analysis of Cystathionine Beta-Synthase Gene Mutations in Chinese Families with Classical Homocystinuria. Biomedicines. 2025; 13(4):919. https://doi.org/10.3390/biomedicines13040919
Chicago/Turabian StyleLiu, Xin, Xinhua Liu, Jinfeng Liu, Junhong Guo, Danyao Nie, and Jiantao Wang. 2025. "Identification and Functional Analysis of Cystathionine Beta-Synthase Gene Mutations in Chinese Families with Classical Homocystinuria" Biomedicines 13, no. 4: 919. https://doi.org/10.3390/biomedicines13040919
APA StyleLiu, X., Liu, X., Liu, J., Guo, J., Nie, D., & Wang, J. (2025). Identification and Functional Analysis of Cystathionine Beta-Synthase Gene Mutations in Chinese Families with Classical Homocystinuria. Biomedicines, 13(4), 919. https://doi.org/10.3390/biomedicines13040919