

Molecular Alterations in Gastric Intestinal Metaplasia Shed Light on Alteration of Methionine Metabolism: Insight into New Diagnostic and Treatment Approaches

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Gastric Intestinal Metaplasia
3. Molecular Alterations to Shed Light on Altered Methionine Metabolism
3.1. Gut Microbiota Alteration
3.1.1. Helicobacter Pylori Abundance
3.1.2. Prevailing Abundance of Lactobacillus
3.2. Increased Gene Methylation Index and CDX2 Downregulation
3.3. Cellular Proliferation
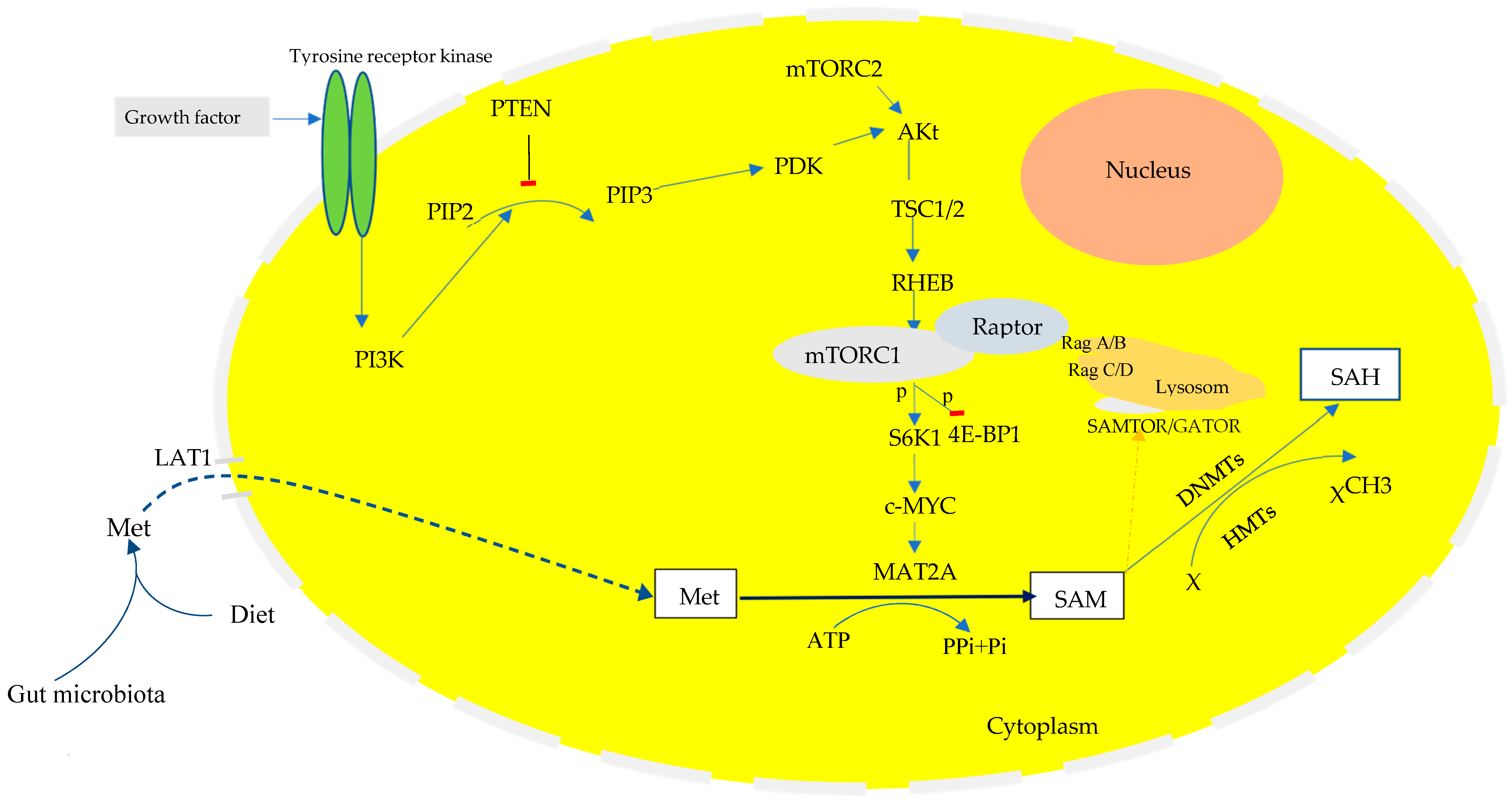
The PI3K-Akt-mTORC1 Signaling Pathway in Cellular Proliferation
3.4. The PI3K/Akt/mTORC1-c-MYC Signaling Pathway in Metabolic Reprogramming
4. Insight into New Diagnostic and Treatment Approaches
5. Future Research Directions
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Ilic, M.; Ilic, I. Epidemiology of stomach cancer. World J. Gastroenterol. 2022, 28, 1187–1203. [Google Scholar] [CrossRef]
- Zare, A.; Mahmoodi, M.; Mohammad, K.; Zeraati, H.; Hosseini, M.; Holakouie Naieni, K. Factors Affecting the Survival of Patients with Gastric Cancer Undergone Surgery at Iran Cancer Institute: Univariate and Multivariate Analyses. Iran. J. Public Health 2014, 43, 800–808. [Google Scholar]
- Liu, Y.; Baba, Y.; Ishimoto, T.; Gu, X.; Zhang, J.; Nomoto, D.; Okadome, K.; Baba, H.; Qiu, P. Gut microbiome in gastrointestinal cancer: A friend or foe? Int. J. Biol. Sci. 2022, 18, 4101–4117. [Google Scholar] [CrossRef]
- Biological Agents. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 2012; Volume 100, pp. 1–441. [Google Scholar]
- Wu, X.; Han, Z.; Liu, B.; Yu, D.; Sun, J.; Ge, L.; Tang, W.; Liu, S. Gut microbiota contributes to the methionine metabolism in host. Front. Microbiol. 2022, 13, 1065668. [Google Scholar] [CrossRef]
- Jonaitis, P.; Kupcinskas, L.; Kupcinskas, J. Molecular Alterations in Gastric Intestinal Metaplasia. Int. J. Mol. Sci. 2021, 22, 5758. [Google Scholar] [CrossRef]
- Battista, S.; Ambrosio, M.R.; Limarzi, F.; Gallo, G.; Saragoni, L. Molecular Alterations in Gastric Preneoplastic Lesions and Early Gastric Cancer. Int. J. Mol. Sci. 2021, 22, 6652. [Google Scholar] [CrossRef]
- Oue, N.; Mitani, Y.; Motoshita, J.; Matsumura, S.; Yoshida, K.; Kuniyasu, H.; Nakayama, H.; Yasui, W. Accumulation of DNA methylation is associated with tumor stage in gastric cancer. Cancer 2006, 106, 1250–1259. [Google Scholar] [CrossRef]
- Calcagno, D.Q.; Leal, M.F.; Demachki, S.; Araújo, M.T.; Freitas, F.W.; Oliveira e Souza, D.; Assumpção, P.P.; Ishak, G.; de Arruda Cardoso Smith, M.; Burbano, R.R. MYC in gastric carcinoma and intestinal metaplasia of young adults. Cancer Genet. Cytogenet. 2010, 202, 63–66. [Google Scholar] [CrossRef]
- Liu, J.; Feng, W.; Liu, M.; Rao, H.; Li, X.; Teng, Y.; Yang, X.; Xu, J.; Gao, W.; Li, L. Stomach-specific c-Myc overexpression drives gastric adenoma in mice through AKT/mammalian target of rapamycin signaling. Bosn. J. Basic Med. Sci. 2021, 21, 434–446. [Google Scholar] [CrossRef]
- Zhao, L.; Su, H.; Liu, X.; Wang, H.; Feng, Y.; Wang, Y.; Chen, H.; Dai, L.; Lai, S.; Xu, S.; et al. mTORC1-c-Myc pathway rewires methionine metabolism for HCC progression through suppressing SIRT4 mediated ADP ribosylation of MAT2A. Cell Biosci. 2022, 12, 183. [Google Scholar] [CrossRef]
- Erkan, G.; Gonul, I.I.; Kandilci, U.; Dursun, A. Evaluation of apoptosis along with BCL-2 and Ki-67 expression in patients with intestinal metaplasia. Pathol. Res. Pract. 2012, 208, 89–93. [Google Scholar] [CrossRef]
- Chiang, T.H.; Cheng, H.C.; Chuang, S.L.; Chen, Y.R.; Hsu, Y.H.; Hsu, T.H.; Lin, L.J.; Lin, Y.W.; Chu, C.H.; Wu, M.S.; et al. Mass screening and eradication of Helicobacter pylori as the policy recommendations for gastric cancer prevention. J. Formos. Med. Assoc. 2022, 121, 2378–2392. [Google Scholar] [CrossRef]
- Tjandra, D.; Busuttil, R.A.; Boussioutas, A. Gastric Intestinal Metaplasia: Challenges and the Opportunity for Precision Prevention. Cancers 2023, 15, 3913. [Google Scholar] [CrossRef]
- Liou, J.M.; Malfertheiner, P.; Lee, Y.C.; Sheu, B.S.; Sugano, K.; Cheng, H.C.; Yeoh, K.G.; Hsu, P.I.; Goh, K.L.; Mahachai, V.; et al. Screening and eradication of Helicobacter pylori for gastric cancer prevention: The Taipei global consensus. Gut 2020, 69, 2093–2112. [Google Scholar] [CrossRef]
- Cho, J.H.; Jin, S.Y. Current guidelines for Helicobacter pylori treatment in East Asia 2022: Differences among China, Japan, and South Korea. World J. Clin. Cases 2022, 10, 6349–6359. [Google Scholar] [CrossRef]
- Ding, S.Z.; Du, Y.Q.; Lu, H.; Wang, W.H.; Cheng, H.; Chen, S.Y.; Chen, M.H.; Chen, W.C.; Chen, Y.; Fang, J.Y.; et al. Chinese Consensus Report on Family-Based Helicobacter pylori Infection Control and Management (2021 Edition). Gut 2022, 71, 238–253. [Google Scholar] [CrossRef]
- Asaka, M.; Kato, M.; Sakamoto, N. Roadmap to eliminate gastric cancer with Helicobacter pylori eradication and consecutive surveillance in Japan. J. Gastroenterol. 2014, 49, 1–8. [Google Scholar] [CrossRef]
- Farinati, F.; Pelizzaro, F. Gastric cancer screening in Western countries: A call to action. Dig. Liver Dis. 2024, 56, 1653–1662. [Google Scholar] [CrossRef]
- Chen, H.N.; Wang, Z.; Li, X.; Zhou, Z.G. Helicobacter pylori eradication cannot reduce the risk of gastric cancer in patients with intestinal metaplasia and dysplasia: Evidence from a meta-analysis. Gastric Cancer 2016, 19, 166–175. [Google Scholar] [CrossRef]
- Wu, S.R.; Liu, Y.H.; Shi, Y.Q. Is intestinal metaplasia the point of no return in the progression of gastric carcinogenesis? Chin. Med. J. 2021, 134, 2965–2967. [Google Scholar] [CrossRef]
- Castaño-Llano, R.; Piñeres, A.; Jaramillo, R.; Molina, S.; Aristizábal, F.; Puerta, J.E. Interval gastric cancer: A call to attentiveness and action. Rev. Gastroenterol. Mex. Engl. Ed. 2023, 88, 91–99. [Google Scholar] [CrossRef]
- Saad, R.S.; Ghorab, Z.; Khalifa, M.A.; Xu, M. CDX2 as a marker for intestinal differentiation: Its utility and limitations. World J. Gastrointest. Surg. 2011, 3, 159–166. [Google Scholar] [CrossRef]
- Yuasa, Y.; Nagasaki, H.; Akiyama, Y.; Sakai, H.; Nakajima, T.; Ohkura, Y.; Takizawa, T.; Koike, M.; Tani, M.; Iwai, T.; et al. Relationship between CDX2 gene methylation and dietary factors in gastric cancer patients. Carcinogenesis 2005, 26, 193–200. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Z.; Li, W.; Liu, S.; Han, B. Methylation of promoter region of CDX2 gene in colorectal cancer. Oncol. Lett. 2016, 12, 3229–3233. [Google Scholar] [CrossRef]
- Liu, Q.; Teh, M.; Ito, K.; Shah, N.; Ito, Y.; Yeoh, K.G. CDX2 expression is progressively decreased in human gastric intestinal metaplasia, dysplasia and cancer. Mod. Pathol. 2007, 20, 1286–1297. [Google Scholar] [CrossRef]
- Gutierrez-Angulo, M.; Ayala-Madrigal, M.L.; Moreno-Ortiz, J.M.; Peregrina-Sandoval, J.; Garcia-Ayala, F.D. Microbiota composition and its impact on DNA methylation in colorectal cancer. Front. Genet. 2023, 14, 1037406. [Google Scholar] [CrossRef]
- Sedillo, J.C.; Cryns, V.L. Targeting the methionine addiction of cancer. Am. J. Cancer Res. 2022, 12, 2249–2276. [Google Scholar]
- Sanderson, S.M.; Gao, X.; Dai, Z.; Locasale, J.W. Methionine metabolism in health and cancer: A nexus of diet and precision medicine. Nat. Rev. Cancer 2019, 19, 625–637. [Google Scholar] [CrossRef]
- Jencks, D.S.; Adam, J.D.; Borum, M.L.; Koh, J.M.; Stephen, S.; Doman, D.B. Overview of Current Concepts in Gastric Intestinal Metaplasia and Gastric Cancer. Gastroenterol. Hepatol. 2018, 14, 92–101. [Google Scholar]
- Shah, S.C.; Gawron, A.J.; Mustafa, R.A.; Piazuelo, M.B. Histologic Subtyping of Gastric Intestinal Metaplasia: Overview and Considerations for Clinical Practice. Gastroenterology 2020, 158, 745–750. [Google Scholar] [CrossRef]
- Shao, L.; Li, P.; Ye, J.; Chen, J.; Han, Y.; Cai, J.; Lu, X. Risk of gastric cancer among patients with gastric intestinal metaplasia. Int. J. Cancer 2018, 143, 1671–1677. [Google Scholar] [CrossRef]
- González, C.A.; Pardo, M.L.; Liso, J.M.; Alonso, P.; Bonet, C.; Garcia, R.M.; Sala, N.; Capella, G.; Sanz-Anquela, J.M. Gastric cancer occurrence in preneoplastic lesions: A long-term follow-up in a high-risk area in Spain. Int. J. Cancer 2010, 127, 2654–2660. [Google Scholar] [CrossRef]
- Whiting, J.L.; Sigurdsson, A.; Rowlands, D.C.; Hallissey, M.T.; Fielding, J.W. The long term results of endoscopic surveillance of premalignant gastric lesions. Gut 2002, 50, 378–381. [Google Scholar] [CrossRef]
- Tava, F.; Luinetti, O.; Ghigna, M.R.; Alvisi, C.; Perego, M.; Trespi, E.; Klersy, C.; Fratti, C.; Fiocca, R.; Solcia, E. Type or extension of intestinal metaplasia and immature/atypical "indefinite-for-dysplasia" lesions as predictors of gastric neoplasia. Hum. Pathol. 2006, 37, 1489–1497. [Google Scholar] [CrossRef]
- Rakici, H.; Uyanik, E.; Rakici, I.M.; Polat, H.B.; Akdogan, R.A.; Aydin, G.; Ayvaz, M.A.; Bedir, R. Gastric intestinal metaplasia: Long-term follow-up results. Niger. J. Clin. Pract. 2022, 25, 315–324. [Google Scholar] [CrossRef]
- Banks, M.; Graham, D.; Jansen, M.; Gotoda, T.; Coda, S.; di Pietro, M.; Uedo, N.; Bhandari, P.; Pritchard, D.M.; Kuipers, E.J.; et al. British Society of Gastroenterology guidelines on the diagnosis and management of patients at risk of gastric adenocarcinoma. Gut 2019, 68, 1545–1575. [Google Scholar] [CrossRef]
- Li, D.; Bautista, M.C.; Jiang, S.F.; Daryani, P.; Brackett, M.; Armstrong, M.A.; Hung, Y.Y.; Postlethwaite, D.; Ladabaum, U. Risks and Predictors of Gastric Adenocarcinoma in Patients with Gastric Intestinal Metaplasia and Dysplasia: A Population-Based Study. Am. J. Gastroenterol. 2016, 111, 1104–1113. [Google Scholar] [CrossRef]
- Kwon, S.-K.; Park, J.C.; Kim, K.H.; Yoon, J.; Cho, Y.; Lee, B.; Lee, J.-J.; Jeong, H.; Oh, Y.; Kim, S.-H. Human gastric microbiota transplantation recapitulates premalignant lesions in germ-free mice. Gut 2022, 71, 1266–1276. [Google Scholar]
- D’Aquila, P.; Carelli, L.L.; De Rango, F.; Passarino, G.; Bellizzi, D. Gut Microbiota as Important Mediator Between Diet and DNA Methylation and Histone Modifications in the Host. Nutrients 2020, 12, 597. [Google Scholar] [CrossRef]
- Vernocchi, P.; Del Chierico, F.; Putignani, L. Gut Microbiota Metabolism and Interaction with Food Components. Int. J. Mol. Sci. 2020, 21, 3688. [Google Scholar] [CrossRef]
- Rossi, T.; Vergara, D.; Fanini, F.; Maffia, M.; Bravaccini, S.; Pirini, F. Microbiota-Derived Metabolites in Tumor Progression and Metastasis. Int. J. Mol. Sci. 2020, 21, 5786. [Google Scholar] [CrossRef]
- Pappas-Gogos, G.; Tepelenis, K.; Fousekis, F.; Katsanos, K.; Pitiakoudis, M.; Vlachos, K. The Implication of Gastric Microbiome in the Treatment of Gastric Cancer. Cancers 2022, 14, 2039. [Google Scholar] [CrossRef]
- Engevik, M.A.; Morra, C.N.; Röth, D.; Engevik, K.; Spinler, J.K.; Devaraj, S.; Crawford, S.E.; Estes, M.K.; Kalkum, M.; Versalovic, J. Microbial Metabolic Capacity for Intestinal Folate Production and Modulation of Host Folate Receptors. Front. Microbiol. 2019, 10, 2305. [Google Scholar] [CrossRef]
- Reva, K.; Laranjinha, J.; Rocha, B.S. Epigenetic Modifications Induced by the Gut Microbiota May Result from What We Eat: Should We Talk about Precision Diet in Health and Disease? Metabolites 2023, 13, 375. [Google Scholar] [CrossRef]
- Lei, C.; Gong, D.; Zhuang, B.; Zhang, Z. Alterations in the gastric microbiota and metabolites in gastric cancer: An update review. Front. Oncol. 2022, 12, 960281. [Google Scholar] [CrossRef]
- Dai, D.; Yang, Y.; Yu, J.; Dang, T.; Qin, W.; Teng, L.; Ye, J.; Jiang, H. Interactions between gastric microbiota and metabolites in gastric cancer. Cell Death Dis. 2021, 12, 1104. [Google Scholar] [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef]
- Liatsos, C.; Papaefthymiou, A.; Kyriakos, N.; Galanopoulos, M.; Doulberis, M.; Giakoumis, M.; Petridou, E.; Mavrogiannis, C.; Rokkas, T.; Kountouras, J. Helicobacter pylori, gastric microbiota and gastric cancer relationship: Unrolling the tangle. World J. Gastrointest. Oncol. 2022, 14, 959–972. [Google Scholar] [CrossRef]
- Schiff, M.; Benoist, J.F.; Tilea, B.; Royer, N.; Giraudier, S.; Ogier de Baulny, H. Isolated remethylation disorders: Do our treatments benefit patients? J. Inherit. Metab. Dis. 2011, 34, 137–145. [Google Scholar] [CrossRef]
- Froese, D.S.; Fowler, B.; Baumgartner, M.R. Vitamin B(12), folate, and the methionine remethylation cycle-biochemistry, pathways, and regulation. J. Inherit. Metab. Dis. 2019, 42, 673–685. [Google Scholar] [CrossRef]
- Franceschi, F.; Annalisa, T.; Teresa, D.R.; Giovanna, D.; Ianiro, G.; Franco, S.; Viviana, G.; Valentina, T.; Riccardo, L.L.; Antonio, G. Role of Helicobacter pylori infection on nutrition and metabolism. World J. Gastroenterol. 2014, 20, 12809–12817. [Google Scholar] [CrossRef]
- Fox, J.G.; Wang, T.C. Inflammation, atrophy, and gastric cancer. J. Clin. Investig. 2007, 117, 60–69. [Google Scholar] [CrossRef]
- Yao, X.; Smolka, A.J. Gastric Parietal Cell Physiology and Helicobacter pylori-Induced Disease. Gastroenterology 2019, 156, 2158–2173. [Google Scholar] [CrossRef]
- McColl, K.E.; el-Omar, E.; Gillen, D. Interactions between H. pylori infection, gastric acid secretion and anti-secretory therapy. Br. Med. Bull. 1998, 54, 121–138. [Google Scholar] [CrossRef]
- Jaskiewicz, K.; Van Helden, P.D.; Wiid, I.J.; Steenkamp, H.J.; Van Wyk, M.J. Chronic atrophic gastritis, gastric pH, nitrites and micronutrient levels in a population at risk for gastric carcinoma. Anticancer Res. 1990, 10, 833–836. [Google Scholar]
- King, C.E.; Leibach, J.; Toskes, P.P. Clinically significant vitamin B12 deficiency secondary to malabsorption of protein-bound vitamin B12. Dig. Dis. Sci. 1979, 24, 397–402. [Google Scholar] [CrossRef]
- Sugahara, H.; Odamaki, T.; Hashikura, N.; Abe, F.; Xiao, J.Z. Differences in folate production by bifidobacteria of different origins. Biosci. Microbiota Food Health 2015, 34, 87–93. [Google Scholar] [CrossRef]
- Rossi, M.; Amaretti, A.; Raimondi, S. Folate production by probiotic bacteria. Nutrients 2011, 3, 118–134. [Google Scholar] [CrossRef]
- Iino, C.; Shimoyama, T. Impact of Helicobacter pylori infection on gut microbiota. World J. Gastroenterol. 2021, 27, 6224–6230. [Google Scholar] [CrossRef]
- Forges, T.; Monnier-Barbarino, P.; Alberto, J.M.; Guéant-Rodriguez, R.M.; Daval, J.L.; Guéant, J.L. Impact of folate and homocysteine metabolism on human reproductive health. Hum. Reprod. Update 2007, 13, 225–238. [Google Scholar] [CrossRef]
- Zhao, W.X.; Liu, Z.F.; Li, X.L.; Li, Z. Correlations of serum homocysteine, VEGF and gastrin 17 with gastric cancer and precancerous lesions. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 4192–4198. [Google Scholar] [CrossRef]
- Yang, Z.; Yang, Y.; Yang, J.; Wan, X.; Yang, H.; Wang, Z. Hyperhomocysteinemia Induced by Methionine Excess Is Effectively Suppressed by Betaine in Geese. Animals 2020, 10, 1642. [Google Scholar] [CrossRef]
- Cravo, M.; Pinto, R.; Fidalgo, P.; Chaves, P.; Glória, L.; Nobre-Leitão, C.; Costa Mira, F. Global DNA hypomethylation occurs in the early stages of intestinal type gastric carcinoma. Gut 1996, 39, 434–438. [Google Scholar] [CrossRef]
- Ilver, D.; Arnqvist, A.; Ogren, J.; Frick, I.M.; Kersulyte, D.; Incecik, E.T.; Berg, D.E.; Covacci, A.; Engstrand, L.; Borén, T. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science 1998, 279, 373–377. [Google Scholar] [CrossRef]
- Mahdavi, J.; Sondén, B.; Hurtig, M.; Olfat, F.O.; Forsberg, L.; Roche, N.; Angstrom, J.; Larsson, T.; Teneberg, S.; Karlsson, K.A.; et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 2002, 297, 573–578. [Google Scholar] [CrossRef]
- Navabi, N.; Johansson, M.E.; Raghavan, S.; Lindén, S.K. Helicobacter pylori infection impairs the mucin production rate and turnover in the murine gastric mucosa. Infect. Immun. 2013, 81, 829–837. [Google Scholar] [CrossRef]
- Teixeira, A.; David, L.; Reis, C.A.; Costa, J.; Sobrinho-Simões, M. Expression of mucins (MUC1, MUC2, MUC5AC, and MUC6) and type 1 Lewis antigens in cases with and without Helicobacter pylori colonization in metaplastic glands of the human stomach. J. Pathol. 2002, 197, 37–43. [Google Scholar] [CrossRef]
- Watanabe, H. Intestinal metaplasia -the effect of Acid on the gastric mucosa and gastric carcinogenesis. J. Toxicol. Pathol. 2010, 23, 115–123. [Google Scholar] [CrossRef]
- Stockbruegger, R.W. Bacterial overgrowth as a consequence of reduced gastric acidity. Scand. J. Gastroenterol. 1985, 111, 7–16. [Google Scholar] [CrossRef]
- Aviles-Jimenez, F.; Vazquez-Jimenez, F.; Medrano-Guzman, R.; Mantilla, A.; Torres, J. Stomach microbiota composition varies between patients with non-atrophic gastritis and patients with intestinal type of gastric cancer. Sci. Rep. 2014, 4, 4202. [Google Scholar] [CrossRef]
- Wang, Z.; Gao, X.; Zeng, R.; Wu, Q.; Sun, H.; Wu, W.; Zhang, X.; Sun, G.; Yan, B.; Wu, L.; et al. Changes of the Gastric Mucosal Microbiome Associated With Histological Stages of Gastric Carcinogenesis. Front. Microbiol. 2020, 11, 997. [Google Scholar] [CrossRef]
- Liu, D.; Chen, S.; Gou, Y.; Yu, W.; Zhou, H.; Zhang, R.; Wang, J.; Ye, F.; Liu, Y.; Sun, B.; et al. Gastrointestinal Microbiota Changes in Patients with Gastric Precancerous Lesions. Front. Cell. Infect. Microbiol. 2021, 11, 749207. [Google Scholar] [CrossRef]
- Park, J.Y.; Seo, H.; Kang, C.S.; Shin, T.S.; Kim, J.W.; Park, J.M.; Kim, J.G.; Kim, Y.K. Dysbiotic change in gastric microbiome and its functional implication in gastric carcinogenesis. Sci. Rep. 2022, 12, 4285. [Google Scholar] [CrossRef]
- Gao, J.J.; Zhang, Y.; Gerhard, M.; Mejias-Luque, R.; Zhang, L.; Vieth, M.; Ma, J.L.; Bajbouj, M.; Suchanek, S.; Liu, W.D.; et al. Association Between Gut Microbiota and Helicobacter pylori-Related Gastric Lesions in a High-Risk Population of Gastric Cancer. Front. Cell. Infect. Microbiol. 2018, 8, 202. [Google Scholar] [CrossRef]
- Lofgren, J.L.; Whary, M.T.; Ge, Z.; Muthupalani, S.; Taylor, N.S.; Mobley, M.; Potter, A.; Varro, A.; Eibach, D.; Suerbaum, S.; et al. Lack of commensal flora in Helicobacter pylori-infected INS-GAS mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterology 2011, 140, 210–220. [Google Scholar] [CrossRef]
- Lertpiriyapong, K.; Whary, M.T.; Muthupalani, S.; Lofgren, J.L.; Gamazon, E.R.; Feng, Y.; Ge, Z.; Wang, T.C.; Fox, J.G. Gastric colonisation with a restricted commensal microbiota replicates the promotion of neoplastic lesions by diverse intestinal microbiota in the Helicobacter pylori INS-GAS mouse model of gastric carcinogenesis. Gut 2014, 63, 54–63. [Google Scholar] [CrossRef]
- Ma, T.; Shen, X.; Shi, X.; Sakandar, H.; Quan, K.; Li, Y.; Jin, H.; Kwok, L.-Y.; Zhang, H.; Sun, Z. Targeting gut microbiota and metabolism as the major probiotic mechanism—An evidence-based review. Trends Food Sci. Technol. 2023, 138, 178–198. [Google Scholar] [CrossRef]
- Qin, D.; Ma, Y.; Wang, Y.; Hou, X.; Yu, L. Contribution of Lactobacilli on Intestinal Mucosal Barrier and Diseases: Perspectives and Challenges of Lactobacillus casei. Life 2022, 12, 1910. [Google Scholar] [CrossRef]
- Mattar, A.F.; Teitelbaum, D.H.; Drongowski, R.A.; Yongyi, F.; Harmon, C.M.; Coran, A.G. Probiotics up-regulate MUC-2 mucin gene expression in a Caco-2 cell-culture model. Pediatr. Surg. Int. 2002, 18, 586–590. [Google Scholar] [CrossRef]
- Serpa, J. The putative role of gut microbiota in cancer: Cysteine is a pivotal coin. Front. Gastroenterol. 2022, 1, 966957. [Google Scholar] [CrossRef]
- Mihara, M.; Yoshida, Y.; Tsukamoto, T.; Inada, K.; Nakanishi, Y.; Yagi, Y.; Imai, K.; Sugimura, T.; Tatematsu, M.; Ushijima, T. Methylation of multiple genes in gastric glands with intestinal metaplasia: A disorder with polyclonal origins. Am. J. Pathol. 2006, 169, 1643–1651. [Google Scholar] [CrossRef]
- Kang, G.H.; Shim, Y.H.; Jung, H.Y.; Kim, W.H.; Ro, J.Y.; Rhyu, M.G. CpG island methylation in premalignant stages of gastric carcinoma. Cancer Res. 2001, 61, 2847–2851. [Google Scholar]
- Roessler, K.; Mönig, S.P.; Schneider, P.M.; Hanisch, F.G.; Landsberg, S.; Thiele, J.; Hölscher, A.H.; Dienes, H.P.; Baldus, S.E. Co-expression of CDX2 and MUC2 in gastric carcinomas: Correlations with clinico-pathological parameters and prognosis. World J. Gastroenterol. 2005, 11, 3182–3188. [Google Scholar] [CrossRef]
- Reis, C.A.; David, L.; Correa, P.; Carneiro, F.; de Bolós, C.; Garcia, E.; Mandel, U.; Clausen, H.; Sobrinho-Simões, M. Intestinal metaplasia of human stomach displays distinct patterns of mucin (MUC1, MUC2, MUC5AC, and MUC6) expression. Cancer Res. 1999, 59, 1003–1007. [Google Scholar]
- Yan, L.H.; Wang, X.T.; Yang, J.; Lian, C.; Kong, F.B.; Wei, W.Y.; Luo, W.; Xiao, Q.; Xie, Y.B. Reversal of multidrug resistance in gastric cancer cells by CDX2 downregulation. World J. Gastroenterol. 2013, 19, 4155–4165. [Google Scholar] [CrossRef]
- Wei, Z.; Liu, X.; Cheng, C.; Yu, W.; Yi, P. Metabolism of Amino Acids in Cancer. Front. Cell Dev. Biol. 2020, 8, 603837. [Google Scholar] [CrossRef]
- Kaiser, P. Methionine Dependence of Cancer. Biomolecules 2020, 10, 568. [Google Scholar] [CrossRef]
- Hoffman, R.M.; Stern, P.H.; Coalson, D.W.; Douglas Wallace, C.; Erbe, R.W. Altered Methionine Metabolism in Cancer Cells. Methionine Depend. Cancer Aging Methods Protoc. 2019, 1866, 13–26. [Google Scholar] [CrossRef]
- Módis, K.; Coletta, C.; Asimakopoulou, A.; Szczesny, B.; Chao, C.; Papapetropoulos, A.; Hellmich, M.R.; Szabo, C. Effect of S-adenosyl-L-methionine (SAM), an allosteric activator of cystathionine-β-synthase (CBS) on colorectal cancer cell proliferation and bioenergetics in vitro. Nitric Oxide 2014, 41, 146–156. [Google Scholar] [CrossRef]
- Lin, D.W.; Chung, B.P.; Kaiser, P. S-adenosylmethionine limitation induces p38 mitogen-activated protein kinase and triggers cell cycle arrest in G1. J. Cell Sci. 2014, 127, 50–59. [Google Scholar] [CrossRef]
- Orlov, E.N. Comparative S-adenosyl-L-methionine and S-adenosyl-L-homocysteine content in the tissues of experimental tumors in the process of their growth. Vopr. Med. Khimii 1980, 26, 699–704. [Google Scholar]
- German, D.C.; Bloch, C.A.; Kredich, N.M. Measurements of S-adenosylmethionine and L-homocysteine metabolism in cultured human lymphoid cells. J. Biol. Chem. 1983, 258, 10997–11003. [Google Scholar] [CrossRef]
- Li, K.; Dan, Z.; Nie, Y.Q. Gastric cancer stem cells in gastric carcinogenesis, progression, prevention and treatment. World J. Gastroenterol. 2014, 20, 5420–5426. [Google Scholar] [CrossRef]
- Sim, E.Z.; Enomoto, T.; Shiraki, N.; Furuta, N.; Kashio, S.; Kambe, T.; Tsuyama, T.; Arakawa, A.; Ozawa, H.; Yokoyama, M.; et al. Methionine metabolism regulates pluripotent stem cell pluripotency and differentiation through zinc mobilization. Cell Rep. 2022, 40, 111120. [Google Scholar] [CrossRef]
- Takuwa, N.; Fukui, Y.; Takuwa, Y. Cyclin D1 expression mediated by phosphatidylinositol 3-kinase through mTOR-p70(S6K)-independent signaling in growth factor-stimulated NIH 3T3 fibroblasts. Mol. Cell. Biol. 1999, 19, 1346–1358. [Google Scholar] [CrossRef]
- Fingar, D.C.; Richardson, C.J.; Tee, A.R.; Cheatham, L.; Tsou, C.; Blenis, J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol. Cell. Biol. 2004, 24, 200–216. [Google Scholar] [CrossRef]
- Tarn, W.Y.; Lai, M.C. Translational control of cyclins. Cell Div. 2011, 6, 5. [Google Scholar] [CrossRef]
- Kim, S.G.; Buel, G.R.; Blenis, J. Nutrient regulation of the mTOR complex 1 signaling pathway. Mol. Cells 2013, 35, 463–473. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef]
- Plugge, S.F.; Ma, H.; Vaart, J.Y.v.d.; Sprangers, J.; Morsink, F.H.M.; Xanthakis, D.; Jamieson, C.; Keijzer, A.R.; Margaritis, T.; Candelli, T.; et al. Intestinal LKB1 loss drives a pre-malignant program along the serrated cancer pathway. bioRxiv 2023. [Google Scholar] [CrossRef]
- Magaway, C.; Kim, E.; Jacinto, E. Targeting mTOR and Metabolism in Cancer: Lessons and Innovations. Cells 2019, 8, 1584. [Google Scholar] [CrossRef]
- Peng, Y.; Wang, Y.; Zhou, C.; Mei, W.; Zeng, C. PI3K/Akt/mTOR pathway and its role in cancer therapeutics: Are we making headway? Front. Oncol. 2022, 12, 819128. [Google Scholar] [CrossRef]
- Lu, W.; Ni, Z.; Jiang, S.; Tong, M.; Zhang, J.; Zhao, J.; Feng, C.; Jia, Q.; Wang, J.; Yao, T.; et al. Resveratrol inhibits bile acid-induced gastric intestinal metaplasia via the PI3K/AKT/p-FoxO4 signalling pathway. Phytother. Res. 2021, 35, 1495–1507. [Google Scholar] [CrossRef]
- Xu, W.; Huang, Y.; Yang, Z.; Hu, Y.; Shu, X.; Xie, C.; He, C.; Zhu, Y.; Lu, N. Helicobacter pylori promotes gastric epithelial cell survival through the PLK1/PI3K/Akt pathway. Onco Targets Ther. 2018, 11, 5703–5713. [Google Scholar] [CrossRef]
- Reines, M.; Soerensen, M.; Berger, H.; Schlaermann, P.; Patel, M.; Meyer, T.F. Helicobacter pylori cancer associated CagA protein drives intestinal metaplastic transition in human gastric organoids. bioRxiv 2023. [Google Scholar] [CrossRef]
- Kim, I.J.; Lee, J.; Oh, S.J.; Yoon, M.S.; Jang, S.S.; Holland, R.L.; Reno, M.L.; Hamad, M.N.; Maeda, T.; Chung, H.J.; et al. Helicobacter pylori Infection Modulates Host Cell Metabolism through VacA-Dependent Inhibition of mTORC1. Cell Host Microbe 2018, 23, 583–593.e8. [Google Scholar] [CrossRef]
- Fu, Z.; Kim, J.; Vidrich, A.; Sturgill, T.W.; Cohn, S.M. Intestinal cell kinase, a MAP kinase-related kinase, regulates proliferation and G1 cell cycle progression of intestinal epithelial cells. Am. J. Physiol.-Gastrointest. Liver Physiol. 2009, 297, G632–G640. [Google Scholar] [CrossRef]
- Bai, Z.G.; Ye, Y.J.; Shen, D.H.; Lu, Y.Y.; Zhang, Z.T.; Wang, S. PTEN expression and suppression of proliferation are associated with Cdx2 overexpression in gastric cancer cells. Int. J. Oncol. 2013, 42, 1682–1691. [Google Scholar] [CrossRef]
- Kim, B.; Kang, S.Y.; Kim, D.; Heo, Y.J.; Kim, K.M. PTEN Protein Loss and Loss-of-Function Mutations in Gastric Cancers: The Relationship with Microsatellite Instability, EBV, HER2, and PD-L1 Expression. Cancers 2020, 12, 1724. [Google Scholar] [CrossRef]
- Liu, P.; Ge, M.; Hu, J.; Li, X.; Che, L.; Sun, K.; Cheng, L.; Huang, Y.; Pilo, M.G.; Cigliano, A.; et al. A functional mammalian target of rapamycin complex 1 signaling is indispensable for c-Myc-driven hepatocarcinogenesis. Hepatology 2017, 66, 167–181. [Google Scholar] [CrossRef]
- Felsher, D.W.; Bishop, J.M. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol. Cell 1999, 4, 199–207. [Google Scholar] [CrossRef]
- Dong, Y.; Tu, R.; Liu, H.; Qing, G. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal Transduct. Target. Ther. 2020, 5, 124. [Google Scholar] [CrossRef]
- Melnik, S.; Werth, N.; Boeuf, S.; Hahn, E.M.; Gotterbarm, T.; Anton, M.; Richter, W. Impact of c-MYC expression on proliferation, differentiation, and risk of neoplastic transformation of human mesenchymal stromal cells. Stem Cell Res. Ther. 2019, 10, 73. [Google Scholar] [CrossRef]
- Hayashi, K.; Jutabha, P.; Endou, H.; Anzai, N. c-Myc is crucial for the expression of LAT1 in MIA Paca-2 human pancreatic cancer cells. Oncol. Rep. 2012, 28, 862–866. [Google Scholar] [CrossRef]
- Lu, J.J.; Li, P.; Yang, Y.; Wang, L.; Zhang, Y.; Zhu, J.Y.; Zhu, X.R.; Chen, M.B. Prognostic value of LAT-1 status in solid cancer: A systematic review and meta-analysis. PLoS ONE 2020, 15, e0233629. [Google Scholar] [CrossRef]
- Martinez, R.S.; Salji, M.J.; Rushworth, L.; Ntala, C.; Rodriguez Blanco, G.; Hedley, A.; Clark, W.; Peixoto, P.; Hervouet, E.; Renaude, E.; et al. SLFN5 Regulates LAT1-Mediated mTOR Activation in Castration-Resistant Prostate Cancer. Cancer Res. 2021, 81, 3664–3678. [Google Scholar] [CrossRef]
- Kanai, Y. Amino acid transporter LAT1 (SLC7A5) as a molecular target for cancer diagnosis and therapeutics. Pharmacol. Ther. 2022, 230, 107964. [Google Scholar] [CrossRef]
- Salisbury, T.B.; Arthur, S. The Regulation and Function of the L-Type Amino Acid Transporter 1 (LAT1) in Cancer. Int. J. Mol. Sci. 2018, 19, 2373. [Google Scholar] [CrossRef]
- Zhang, S.; Lin, X.; Hou, Q.; Hu, Z.; Wang, Y.; Wang, Z. Regulation of mTORC1 by amino acids in mammalian cells: A general picture of recent advances. Anim. Nutr. 2021, 7, 1009–1023. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell Sci. 2013, 126, 1713–1719. [Google Scholar] [CrossRef]
- Zhou, Y.; Ren, J.; Song, T.; Peng, J.; Wei, H. Methionine Regulates mTORC1 via the T1R1/T1R3-PLCβ-Ca(2+)-ERK1/2 Signal Transduction Process in C2C12 Cells. Int. J. Mol. Sci. 2016, 17, 1684. [Google Scholar] [CrossRef]
- Kitada, M.; Xu, J.; Ogura, Y.; Monno, I.; Koya, D. Mechanism of Activation of Mechanistic Target of Rapamycin Complex 1 by Methionine. Front. Cell Dev. Biol. 2020, 8, 715. [Google Scholar] [CrossRef]
- Murphy, D.J.; Junttila, M.R.; Pouyet, L.; Karnezis, A.; Shchors, K.; Bui, D.A.; Brown-Swigart, L.; Johnson, L.; Evan, G.I. Distinct thresholds govern Myc’s biological output in vivo. Cancer Cell 2008, 14, 447–457. [Google Scholar] [CrossRef]
- Bazarov, A.V.; Adachi, S.; Li, S.-F.; Mateyak, M.K.; Wei, S.; Sedivy, J.M. A modest reduction in c-myc expression has minimal effects on cell growth and apoptosis but dramatically reduces susceptibility to Ras and Raf transformation. Cancer Res. 2001, 61, 1178–1186. [Google Scholar]
- Dai, J.; Yu, S.-X.; Qi, X.-L.; Bo, A.-H.; Xu, Y.-L.; Guo, Z.-Y. Expression of bcl-2 and c-myc protein in gastric carcinoma and precancerous lesions. World J. Gastroenterol. 1998, 4, 97. [Google Scholar] [CrossRef]
- Villa, E.; Sahu, U.; O’Hara, B.P.; Ali, E.S.; Helmin, K.A.; Asara, J.M.; Gao, P.; Singer, B.D.; Ben-Sahra, I. mTORC1 stimulates cell growth through SAM synthesis and m(6)A mRNA-dependent control of protein synthesis. Mol. Cell 2021, 81, 2076–2093.e9. [Google Scholar] [CrossRef]
- Maldonado, L.Y.; Arsene, D.; Mato, J.M.; Lu, S.C. Methionine adenosyltransferases in cancers: Mechanisms of dysregulation and implications for therapy. Exp. Biol. Med. 2018, 243, 107–117. [Google Scholar] [CrossRef]
- Frau, M.; Feo, F.; Pascale, R.M. Pleiotropic effects of methionine adenosyltransferases deregulation as determinants of liver cancer progression and prognosis. J. Hepatol. 2013, 59, 830–841. [Google Scholar] [CrossRef]
- Martínez-Chantar, M.L.; Latasa, M.U.; Varela-Rey, M.; Lu, S.C.; García-Trevijano, E.R.; Mato, J.M.; Avila, M.A. L-methionine availability regulates expression of the methionine adenosyltransferase 2A gene in human hepatocarcinoma cells: Role of S-adenosylmethionine. J. Biol. Chem. 2003, 278, 19885–19890. [Google Scholar] [CrossRef]
- Guo, J.; Yang, Y.; Buettner, R.; Rosen, S.T. Targeting the methionine-methionine adenosyl transferase 2A- S -adenosyl methionine axis for cancer therapy. Curr. Opin. Oncol. 2022, 34, 546–551. [Google Scholar] [CrossRef]
- Ikenoyama, Y.; Hirasawa, T.; Ishioka, M.; Namikawa, K.; Yoshimizu, S.; Horiuchi, Y.; Ishiyama, A.; Yoshio, T.; Tsuchida, T.; Takeuchi, Y.; et al. Detecting early gastric cancer: Comparison between the diagnostic ability of convolutional neural networks and endoscopists. Dig. Endosc. 2021, 33, 141–150. [Google Scholar] [CrossRef]
- Di Giulio, E.; Hassan, C.; Marmo, R.; Zullo, A.; Annibale, B. Appropriateness of the indication for upper endoscopy: A meta-analysis. Dig. Liver Dis. 2010, 42, 122–126. [Google Scholar] [CrossRef]
- Xue, H.; Yang, A.; Liu, F.; Sun, X.; Liu, X. Clinical significance of Serum Pepsinogen I/II and gastrin-17 determination in gastric cancer diagnosis and prognosis. Eur. J. Inflamm. 2018, 16, 2058739218781291. [Google Scholar] [CrossRef]
- Asaka, M.; Kimura, T.; Kudo, M.; Takeda, H.; Mitani, S.; Miyazaki, T.; Miki, K.; Graham, D.Y. Relationship of Helicobacter pylori to serum pepsinogens in an asymptomatic Japanese population. Gastroenterology 1992, 102, 760–766. [Google Scholar] [CrossRef]
- Haj–sheykholeslami, A.; Rakhshani, N.; Amirzargar, A.; Rafiee, R.; Shahidi, S.M.; Nikbin, B.; Khosravi, F.; Massarrat, S. Serum Pepsinogen I, Pepsinogen II, and Gastrin 17 in Relatives of Gastric Cancer Patients: Comparative Study With Type and Severity of Gastritis. Clin. Gastroenterol. Hepatol. 2008, 6, 174–179. [Google Scholar] [CrossRef]
- Qin, Y.; Geng, J.X.; Huang, B. Clinical value of serum pepsinogen in the diagnosis and treatment of gastric diseases. World J. Gastrointest. Oncol. 2023, 15, 1174–1181. [Google Scholar] [CrossRef]
- Chinda, D.; Shimoyama, T.; Sawaya, M.; Hanabata, N.; Mikami, T.; Fukuda, S. Administration of Gastric Acid Suppressive Agents Modulates the Level of Serum Pepsinogen in Patients with Early Gastric Cancer: 112. Off. J. Am. Coll. Gastroenterol. ACG 2012, 107, S48. [Google Scholar] [CrossRef]
- Abuduwaili, M.; Boda, T.; Ito, M.; Takigawa, H.; Kotachi, T.; Matsuo, T.; Oka, S.; Tanaka, S. Serum Gastrin and Pepsinogen Levels after Administration of Acid Secretion Inhibitors for Ulcers due to Endoscopic Submucosal Dissection in Patients with Early Gastric Cancer. Gastroenterol. Res. Pract. 2022, 2022, 2830227. [Google Scholar] [CrossRef]
- Drnovsek, J.; Homan, M.; Zidar, N.; Smid, L.M. Pathogenesis and potential reversibility of intestinal metaplasia—A milestone in gastric carcinogenesis. Radiol. Oncol. 2024, 58, 186–195. [Google Scholar] [CrossRef]
- Tahara, S.; Tahara, T.; Tuskamoto, T.; Horiguchi, N.; Kawamura, T.; Okubo, M.; Ishizuka, T.; Nagasaka, M.; Nakagawa, Y.; Shibata, T.; et al. Morphologic characterization of residual DNA methylation in the gastric mucosa after Helicobacter pylori eradication. Cancer Med. 2017, 6, 1730–1737. [Google Scholar] [CrossRef]
- Kim, M.J.; Kim, H.N.; Jacobs, J.P.; Yang, H.J. Combined DNA Methylation and Gastric Microbiome Marker Predicts Helicobacter pylori-Negative Gastric Cancer. Gut Liver 2024, 18, 611–620. [Google Scholar] [CrossRef]
- Muscaritoli, M.; Conversano, L.; Petti, M.C.; Torelli, G.F.; Cascino, A.; Mecarocci, S.; Annicchiarico, M.A.; Rossi Fanelli, F. Plasma amino acid concentrations in patients with acute myelogenous leukemia. Nutrition 1999, 15, 195–199. [Google Scholar] [CrossRef]
- Greenberg, A.K.; Rimal, B.; Felner, K.; Zafar, S.; Hung, J.; Eylers, E.; Phalan, B.; Zhang, M.; Goldberg, J.D.; Crawford, B.; et al. S-adenosylmethionine as a biomarker for the early detection of lung cancer. Chest 2007, 132, 1247–1252. [Google Scholar] [CrossRef]
- Li, T.; Yu, G.; Guo, T.; Qi, H.; Bing, Y.; Xiao, Y.; Li, C.; Liu, W.; Yuan, Y.; He, Y.; et al. The plasma S-adenosylmethionine level is associated with the severity of hepatitis B-related liver disease. Medicine 2015, 94, e489. [Google Scholar] [CrossRef]
- Xin, L.; Lu, H.; Liu, C.; Zeng, F.; Yuan, Y.W.; Wu, Y.; Wang, J.L.; Wu, D.Z.; Zhou, L.Q. Methionine deficiency promoted mitophagy via lncRNA PVT1-mediated promoter demethylation of BNIP3 in gastric cancer. Int. J. Biochem. Cell Biol. 2021, 141, 106100. [Google Scholar] [CrossRef]
- Xin, L.; Yuan, Y.-W.; Liu, C.-X.; Sheng, J.; Zhou, Q.; Liu, Z.-Y.; Yue, Z.-Q.; Zeng, F. Methionine restriction attenuates the migration and invasion of gastric cancer cells by inhibiting nuclear p65 translocation through TRIM47. Biol. Chem. 2023, 405, 257–265. [Google Scholar] [CrossRef]
- Gao, X.; Sanderson, S.M.; Dai, Z.; Reid, M.A.; Cooper, D.E.; Lu, M.; Richie, J.P., Jr.; Ciccarella, A.; Calcagnotto, A.; Mikhael, P.G.; et al. Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature 2019, 572, 397–401. [Google Scholar] [CrossRef]
- Prasad, M.; Banerjee, S.; Suman; Kumar, R.; Buragohain, L.; Ghosh, M. Proteomics and Metabolomics in Cancer Diagnosis and Therapy. In Handbook of Oxidative Stress in Cancer: Mechanistic Aspects; Springer: Berlin/Heidelberg, Germany, 2021; pp. 1–31. [Google Scholar]
- Danzi, F.; Pacchiana, R.; Mafficini, A.; Scupoli, M.T.; Scarpa, A.; Donadelli, M.; Fiore, A. To metabolomics and beyond: A technological portfolio to investigate cancer metabolism. Signal Transduct. Target. Ther. 2023, 8, 137. [Google Scholar] [CrossRef]
- Yu, L.; Aa, J.; Xu, J.; Sun, M.; Qian, S.; Cheng, L.; Yang, S.; Shi, R. Metabolomic phenotype of gastric cancer and precancerous stages based on gas chromatography time-of-flight mass spectrometry. J. Gastroenterol. Hepatol. 2011, 26, 1290–1297. [Google Scholar] [CrossRef]
- Aitmanaitė, L.; Širmonaitis, K.; Russo, G. Microbiomes, Their Function, and Cancer: How Metatranscriptomics Can Close the Knowledge Gap. Int. J. Mol. Sci. 2023, 24, 13786. [Google Scholar] [CrossRef]
- Zhang, X.; Li, L.; Butcher, J.; Stintzi, A.; Figeys, D. Advancing functional and translational microbiome research using meta-omics approaches. Microbiome 2019, 7, 154. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gebrehiwot, N.T.; Liu, Y.; Li, J.; Liu, H.-M. Molecular Alterations in Gastric Intestinal Metaplasia Shed Light on Alteration of Methionine Metabolism: Insight into New Diagnostic and Treatment Approaches. Biomedicines 2025, 13, 964. https://doi.org/10.3390/biomedicines13040964
Gebrehiwot NT, Liu Y, Li J, Liu H-M. Molecular Alterations in Gastric Intestinal Metaplasia Shed Light on Alteration of Methionine Metabolism: Insight into New Diagnostic and Treatment Approaches. Biomedicines. 2025; 13(4):964. https://doi.org/10.3390/biomedicines13040964
Chicago/Turabian StyleGebrehiwot, Nigatu Tadesse, Ying Liu, Juan Li, and Hong-Min Liu. 2025. "Molecular Alterations in Gastric Intestinal Metaplasia Shed Light on Alteration of Methionine Metabolism: Insight into New Diagnostic and Treatment Approaches" Biomedicines 13, no. 4: 964. https://doi.org/10.3390/biomedicines13040964
APA StyleGebrehiwot, N. T., Liu, Y., Li, J., & Liu, H.-M. (2025). Molecular Alterations in Gastric Intestinal Metaplasia Shed Light on Alteration of Methionine Metabolism: Insight into New Diagnostic and Treatment Approaches. Biomedicines, 13(4), 964. https://doi.org/10.3390/biomedicines13040964