

Ferroptosis: An Energetic Villain of Age-Related Macular Degeneration
 ,
,
Abstract
1. Introduction
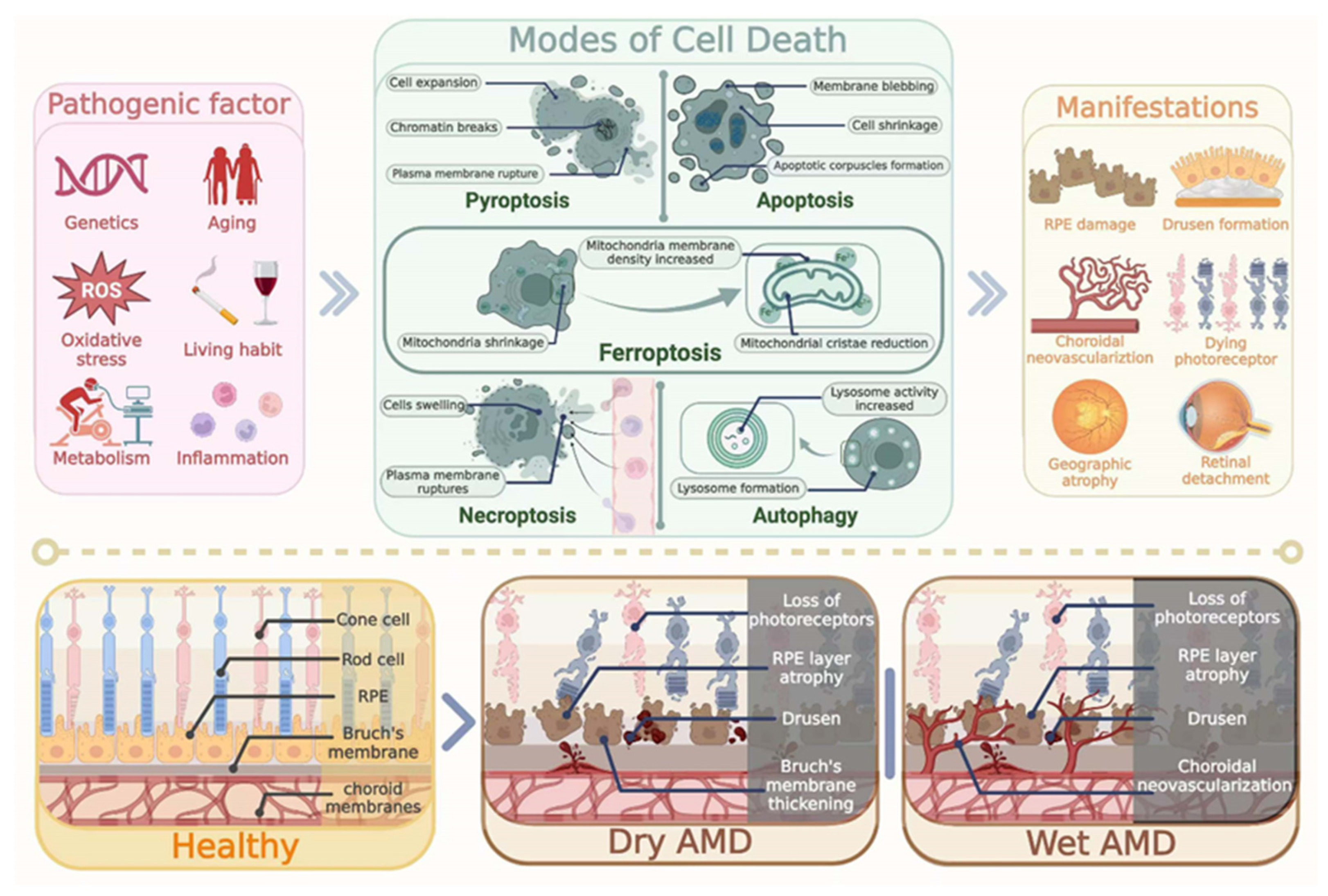
2. Pathological Features of AMD
3. Regulation of Iron in Retina
3.1. Iron Homeostasis in Normal Retina
3.2. Activity of Transferrin Receptor
3.3. Regulation of Ferritin
3.4. Regulation of Ferroportin
4. Lipid Peroxidation and Ferroptosis in AMD
5. Mitochondria Dysfunction of Ferroptosis in AMD
5.1. Mitochondrial Fission and Fusion in AMD
5.2. Ferritinophagy in AMD
6. The Regulatory Role of Ferroptosis in AMD
6.1. GSH-GPX4 Regulating Axis
6.2. GCH1–BH4-Regulating Axis
6.3. FSP1- Regulating Pathway
6.4. DOHDH–CoQH2 Pathway
6.5. Nrf2-Regulating Pathway
7. Potential Therapeutic Effects Against AMD
7.1. Iron-Chelating Agent
7.2. Lipophilic Antioxidant
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pennington, K.L.; DeAngelis, M.M. Epidemiology of age-related macular degeneration (AMD): Associations with cardiovascular disease phenotypes and lipid factors. Eye Vis. 2016, 3, 34. [Google Scholar] [CrossRef] [PubMed]
- Ach, T.; Huisingh, C.; McGwin, G., Jr.; Messinger, J.D.; Zhang, T.; Bentley, M.J.; Gutierrez, D.B.; Ablonczy, Z.; Smith, R.T.; Sloan, K.R.; et al. Quantitative autofluorescence and cell density maps of the human retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4832–4841. [Google Scholar] [CrossRef] [PubMed]
- Strauss, O. The Retinal Pigment Epithelium in Visual Function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [PubMed]
- Hanus, J.; Anderson, C.; Wang, S. RPE necroptosis in response to oxidative stress and in AMD. Ageing Res. Rev. 2015, 24, 286–298. [Google Scholar] [CrossRef]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef]
- Ding, J.D.; Johnson, L.V.; Herrmann, R.; Farsiu, S.; Smith, S.G.; Groelle, M.; Mace, B.E.; Sullivan, P.; Jamison, J.A.; Kelly, U.; et al. Anti-amyloid therapy protects against retinal pigmented epithelium damage and vision loss in a model of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, E279–E287. [Google Scholar] [CrossRef]
- Ramkumar, H.L.; Zhang, J.; Chan, C.C. Retinal ultrastructure of murine models of dry age-related macular degeneration (AMD). Prog. Retin. Eye Res. 2010, 29, 169–190. [Google Scholar] [CrossRef]
- Hobbs, S.D.; Pierce, K. Wet Age-Related Macular Degeneration (Wet AMD); StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Ueda-Consolvo, T.; Ozaki, H.; Nakamura, T.; Oiwake, T.; Hayashi, A. The association between cone density and visual function in the macula of patients with retinitis pigmentosa. Graefe’s Arch. Clin. Exp. Ophthalmol. 2019, 257, 1841–1846. [Google Scholar] [CrossRef]
- Nashine, S. Potential Therapeutic Candidates for Age-Related Macular Degeneration (AMD). Cells 2021, 10, 2483. [Google Scholar] [CrossRef]
- Bowes Rickman, C.; Farsiu, S.; Toth, C.A.; Klingeborn, M. Dry age-related macular degeneration: Mechanisms, therapeutic targets, and imaging. Investig. Ophthalmol. Vis. Sci. 2013, 54, ORSF68–ORSF80. [Google Scholar] [CrossRef]
- Jonasson, F.; Arnarsson, A.; Eiríksdottir, G.; Harris, T.B.; Launer, L.J.; Meuer, S.M.; Klein, B.E.; Klein, R.; Gudnason, V.; Cotch, M.F. Prevalence of age-related macular degeneration in old persons: Age, Gene/environment Susceptibility Reykjavik Study. Ophthalmology 2011, 118, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Gao, M.; Liang, J.; Chen, Y.; Wang, Y.; Wang, Y.; Xiao, Y.; Zhao, Z.; Wan, X.; Jiang, M.; et al. SLC7A11 Reduces Laser-Induced Choroidal Neovascularization by Inhibiting RPE Ferroptosis and VEGF Production. Front. Cell Dev. Biol. 2021, 9, 639851. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Jenkins, N.L.; James, S.A.; Salim, A.; Sumardy, F.; Speed, T.P.; Conrad, M.; Richardson, D.R.; Bush, A.I.; McColl, G. Changes in ferrous iron and glutathione promote ferroptosis and frailty in aging Caenorhabditis elegans. eLife 2020, 9, e56580. [Google Scholar] [CrossRef]
- Brock, J.H. B Iron-binding proteins. Acta Paediatr. Scand. Suppl. 1989, 361, 31–43. [Google Scholar] [CrossRef]
- Liu, K.; Li, H.; Wang, F.; Su, Y. Ferroptosis: Mechanisms and advances in ocular diseases. Mol. Cell Biochem. 2023, 478, 2081–2095. [Google Scholar] [CrossRef]
- Yant, L.J.; Ran, Q.; Rao, L.; Van Remmen, H.; Shibatani, T.; Belter, J.G.; Motta, L.; Richardson, A.; Prolla, T.A. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med. 2003, 34, 496–502. [Google Scholar] [CrossRef]
- Rochette, L.; Dogon, G.; Rigal, E.; Zeller, M.; Cottin, Y.; Vergely, C. Lipid Peroxidation and Iron Metabolism: Two Corner Stones in the Homeostasis Control of Ferroptosis. Int. J. Mol. Sci. 2023, 24, 449. [Google Scholar] [CrossRef]
- Koppula, P.; Zhang, Y.; Zhuang, L.; Gan, B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. 2018, 38, 12. [Google Scholar] [CrossRef]
- Tu, H.; Tang, L.-J.; Luo, X.-J.; Ai, K.-L.; Peng, J. Insights into the novel function of system Xc- in regulated cell death. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 1650–1662. [Google Scholar] [PubMed]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018, 9, 753. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, P.G.; Ferrington, D.A.; Kannan, R. Glutathione Metabolism and the Novel Role of Mitochondrial GSH in Retinal Degeneration. Antioxidants 2021, 10, 661. [Google Scholar] [CrossRef] [PubMed]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2013, 1830, 3217–3266. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Freitas, F.P.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422. [Google Scholar] [CrossRef]
- Li, F.-J.; Long, H.-Z.; Zhou, Z.-W.; Luo, H.-Y.; Xu, S.-G.; Gao, L.-C. System Xc−/GSH/GPX4 axis: An important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Front. Pharmacol. 2022, 13, 910292. [Google Scholar] [CrossRef]
- Tonnus, W.; Gembardt, F.; Latk, M.; Parmentier, S.; Hugo, C.; Bornstein, S.R.; Linkermann, A. The clinical relevance of necroinflammation—Highlighting the importance of acute kidney injury and the adrenal glands. Cell Death Differ. 2018, 26, 68–82. [Google Scholar] [CrossRef]
- Liu, M.; Kong, X.Y.; Yao, Y.; Wang, X.A.; Yang, W.; Wu, H.; Li, S.; Ding, J.W.; Yang, J. The critical role and molecular mechanisms of ferroptosis in antioxidant systems: A narrative review. Ann. Transl. Med. 2022, 10, 368. [Google Scholar] [CrossRef]
- Wooff, Y.; Fernando, N.; Wong, J.H.C.; Dietrich, C.; Aggio-Bruce, R.; Chu-Tan, J.A.; Robertson, A.A.B.; Doyle, S.L.; Man, S.M.; Natoli, R. Caspase-1-dependent inflammasomes mediate photoreceptor cell death in photo-oxidative damage-induced retinal degeneration. Sci. Rep. 2020, 10, 2263. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Tokarz, P.; Koskela, A.; Paterno, J.; Blasiak, J. Autophagy regulates death of retinal pigment epithelium cells in age-related macular degeneration. Cell Biol. Toxicol. 2017, 33, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Biesemeier, A.; Yoeruek, E.; Eibl, O.; Schraermeyer, U. Iron accumulation in Bruch’s membrane and melanosomes of donor eyes with age-related macular degeneration. Exp. Eye Res. 2015, 137, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Ugarte, M.; Geraki, K.; Jeffery, G. Aging results in iron accumulations in the non-human primate choroid of the eye without an associated increase in zinc, copper or sulphur. BioMetals 2018, 31, 1061–1073. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Lüscher, T.F. Ageing, inflammation, and oxidative stress: Final common pathways of cardiovascular disease. Eur. Heart J. 2015, 36, 3381–3383. [Google Scholar] [CrossRef]
- Zhang, S.-M.; Fan, B.; Li, Y.L.; Zuo, Z.-Y.; Li, G.-Y. Oxidative Stress-Involved Mitophagy of Retinal Pigment Epithelium and Retinal Degenerative Diseases. Cell. Mol. Neurobiol. 2023, 43, 3265–3276. [Google Scholar] [CrossRef]
- Zou, M.; Ke, Q.; Nie, Q.; Qi, R.; Zhu, X.; Liu, W.; Hu, X.; Sun, Q.; Fu, J.-L.; Tang, X.; et al. Inhibition of cGAS-STING by JQ1 alleviates oxidative stress-induced retina inflammation and degeneration. Cell Death Differ. 2022, 29, 1816–1833. [Google Scholar] [CrossRef]
- Fleckenstein, M.; Keenan, T.D.; Guymer, R.H.; Chakravarthy, U.; Schmitz-Valckenberg, S.; Klaver, C.C.; Wong, W.T.; Chew, E.Y. Age-related macular degeneration. Nat. Rev. Dis. Primers 2021, 7, 31. [Google Scholar] [CrossRef]
- Hyttinen, J.M.; Blasiak, J.; Kaarniranta, K. Non-Coding RNAs Regulating Mitochondrial Functions and the Oxidative Stress Response as Putative Targets against Age-Related Macular Degeneration (AMD). Int. J. Mol. Sci. 2023, 24, 2636. [Google Scholar] [CrossRef]
- Yu, Y.; Yan, Y.; Niu, F.; Wang, Y.; Chen, X.; Su, G.; Liu, Y.; Zhao, X.; Qian, L.; Liu, P.; et al. Ferroptosis: A cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discov. 2021, 7, 193. [Google Scholar] [CrossRef]
- Lim, L.S.; Mitchell, P.; Seddon, J.M.; Holz, F.G.; Wong, T.Y. Age-related macular degeneration. Lancet 2012, 379, 1728–1738. [Google Scholar] [CrossRef] [PubMed]
- Bhutto, I.; Lutty, G. Understanding age-related macular degeneration (AMD): Relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol. Asp. Med. 2012, 33, 295–317. [Google Scholar] [CrossRef] [PubMed]
- Kokkinopoulos, I.; Shahabi, G.; Colman, A.; Jeffery, G. Mature peripheral RPE cells have an intrinsic capacity to proliferate; a potential regulatory mechanism for age-related cell loss. PLoS ONE 2011, 6, e18921. [Google Scholar] [CrossRef] [PubMed]
- Rashid, A.; Bhatia, S.K.; Mazzitello, K.I.; Chrenek, M.A.; Zhang, Q.; Boatright, J.H.; Grossniklaus, H.E.; Jiang, Y.; Nickerson, J.M. RPE Cell and Sheet Properties in Normal and Diseased Eyes. Adv. Exp. Med. Biol. 2016, 854, 757–763. [Google Scholar]
- Massé, A.; Buhannic, L. Comprendre la dégénérescence maculaire liée à l’âge. Actual. Pharm. 2017, 56, 18–21. [Google Scholar] [CrossRef]
- Bonilha, V.L.; Bell, B.A.; Hu, J.; Milliner, C.; Pauer, G.J.; Hagstrom, S.A.; Radu, R.A.; Hollyfield, J.G. Geographic Atrophy: Confocal Scanning Laser Ophthalmoscopy, Histology, and Inflammation in the Region of Expanding Lesions. Investig. Ophthalmol. Vis. Sci. 2020, 61, 15. [Google Scholar] [CrossRef]
- Yang, M.; So, K.F.; Lam, W.C.; Lo, A.C.Y. Novel Programmed Cell Death as Therapeutic Targets in Age-Related Macular Degeneration? Int. J. Mol. Sci. 2020, 21, 7279. [Google Scholar] [CrossRef]
- Song, D.; Kanu, L.N.; Li, Y.; Kelly, K.L.; Bhuyan, R.K.; Aleman, T.; Morgan, J.I.W.; Dunaief, J.L. AMD-like retinopathy associated with intravenous iron. Exp. Eye Res. 2016, 151, 122–133. [Google Scholar] [CrossRef]
- Chua, S.Y.L.; Khawaja, A.P.; Dick, A.D.; Morgan, J.; Dhillon, B.; Lotery, A.J.; Strouthidis, N.G.; Reisman, C.; Peto, T.; Khaw, P.T.; et al. Ambient Air Pollution Associations with Retinal Morphology in the UK Biobank. Investig. Ophthalmol. Vis. Sci. 2020, 61, 32. [Google Scholar] [CrossRef]
- Moiseyev, G.; Takahashi, Y.; Chen, Y.; Gentleman, S.; Redmond, T.M.; Crouch, R.K.; Ma, J.X. RPE65 is an iron (II)-dependent isomerohydrolase in the retinoid visual cycle. J. Biol. Chem. 2006, 281, 2835–2840. [Google Scholar] [CrossRef]
- Gnana-Prakasam, J.P.; Martin, P.M.; Smith, S.B.; Ganapathy, V. Expression and function of iron-regulatory proteins in retina. IUBMB Life 2010, 62, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Gnana-Prakasam, J.P.; Ananth, S.; Prasad, P.D.; Zhang, M.; Atherton, S.S.; Martin, P.M.; Smith, S.B.; Ganapathy, V. Expression and iron-dependent regulation of succinate receptor GPR91 in retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3751–3758. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kang, R.; Tang, D. Signaling pathways and defense mechanisms of ferroptosis. FEBS J. 2022, 289, 7038–7050. [Google Scholar] [CrossRef]
- Kruszewski, M. Labile iron pool: The main determinant of cellular response to oxidative stress. Mutat. Res. 2003, 531, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Steere, A.N.; Byrne, S.L.; Chasteen, N.D.; Mason, A.B. Kinetics of iron release from transferrin bound to the transferrin receptor at endosomal pH. Biochim. Biophys. Acta 2012, 1820, 326–333. [Google Scholar] [CrossRef]
- Zhao, T.; Guo, X.; Sun, Y. Iron Accumulation and Lipid Peroxidation in the Aging Retina: Implication of Ferroptosis in Age-Related Macular Degeneration. Aging Dis. 2021, 12, 529–551. [Google Scholar] [CrossRef]
- Puri, C. Loss of Myosin VI No Insert Isoform (NoI) Induces a Defect in Clathrin-mediated Endocytosis and Leads to Caveolar Endocytosis of Transferrin Receptor. J. Biol. Chem. 2009, 284, 34998–35014. [Google Scholar] [CrossRef]
- Sterling, J.; Guttha, S.; Song, Y.; Song, D.; Hadziahmetovic, M.; Dunaief, J.L. Iron importers Zip8 and Zip14 are expressed in retina and regulated by retinal iron levels. Exp. Eye Res. 2017, 155, 15–23. [Google Scholar] [CrossRef]
- Shvartsman, M.; Ioav Cabantchik, Z. Intracellular iron trafficking: Role of cytosolic ligands. BioMetals 2012, 25, 711–723. [Google Scholar] [CrossRef]
- Rice, A.E.; Mendez, M.J.; Hokanson, C.A.; Rees, D.C.; Bjorkman, P.J. Investigation of the biophysical and cell biological properties of ferroportin, a multipass integral membrane protein iron exporter. J. Mol. Biol. 2009, 386, 717–732. [Google Scholar] [CrossRef]
- Ashok, A.; Chaudhary, S.; Wise, A.S.; Rana, N.A.; McDonald, D.; Kritikos, A.E.; Lindner, E.; Singh, N. Release of Iron-Loaded Ferritin in Sodium Iodate-Induced Model of Age Related Macular Degeneration: An In-Vitro and In-Vivo Study. Antioxidants 2021, 10, 1253. [Google Scholar] [CrossRef] [PubMed]
- Chowers, I.; Wong, R.; Dentchev, T.; Farkas, R.H.; Iacovelli, J.; Gunatilaka, T.L.; Medeiros, N.E.; Presley, J.B.; Campochiaro, P.A.; Curcio, C.A.; et al. The iron carrier transferrin is upregulated in retinas from patients with age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2135–2140. [Google Scholar] [CrossRef] [PubMed]
- Yeo, J.H.; Colonne, C.K.; Tasneem, N.; Cosgriff, M.P.; Fraser, S.T. The iron islands: Erythroblastic islands and iron metabolism. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 466–471. [Google Scholar] [CrossRef]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef]
- Kawabata, H.; Germain, R.S.; Vuong, P.T.; Nakamaki, T.; Said, J.W.; Koeffler, H.P. Transferrin receptor 2-alpha supports cell growth both in iron-chelated cultured cells and in vivo. J. Biol. Chem. 2000, 275, 16618–16625. [Google Scholar] [CrossRef]
- Lok, C.N.; Ponka, P. Identification of a hypoxia response element in the transferrin receptor gene. J. Biol. Chem. 1999, 274, 24147–24152. [Google Scholar] [CrossRef]
- Martin, P.M.; Gnana-Prakasam, J.P.; Roon, P.; Smith, R.G.; Smith, S.B.; Ganapathy, V. Expression and polarized localization of the hemochromatosis gene product HFE in retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4238–4244. [Google Scholar] [CrossRef]
- Testi, C.; Boffi, A.; Montemiglio, L.C. Structural analysis of the transferrin receptor multifaceted ligand(s) interface. Biophys. Chem. 2019, 254, 106242. [Google Scholar] [CrossRef]
- Wang, Z.; Ding, Y.; Wang, X.; Lu, S.; Wang, C.; He, C.; Wang, L.; Piao, M.; Chi, G.; Luo, Y.; et al. Pseudolaric acid B triggers ferroptosis in glioma cells via activation of Nox4 and inhibition of xCT. Cancer Lett. 2018, 428, 21–33. [Google Scholar] [CrossRef]
- Imamura, T.; Hirayama, T.; Tsuruma, K.; Shimazawa, M.; Nagasawa, H.; Hara, H. Hydroxyl radicals cause fluctuation in intracellular ferrous ion levels upon light exposure during photoreceptor cell death. Exp. Eye Res. 2014, 129, 24–30. [Google Scholar] [CrossRef]
- Kawabata, H. Transferrin and transferrin receptors update. Free Radic. Biol. Med. 2019, 133, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Knovich, M.A.; Storey, J.A.; Coffman, L.G.; Torti, S.V.; Torti, F.M. Ferritin for the clinician. Blood Rev. 2009, 23, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Ropert, M.; Le Lan, C.; Loreal, O. Non-transferrin bound iron: A key role in iron overload and iron toxicity. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2012, 1820, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Ghosh, M.C.; Zhang, D.L.; Rouault, T.A. Iron misregulation and neurodegenerative disease in mouse models that lack iron regulatory proteins. Neurobiol. Dis. 2015, 81, 66–75. [Google Scholar] [CrossRef]
- Zhang, D.L.; Ghosh, M.C.; Rouault, T.A. The physiological functions of iron regulatory proteins in iron homeostasis—An update. Front. Pharmacol. 2014, 5, 124. [Google Scholar] [CrossRef]
- Hirota, K. An intimate crosstalk between iron homeostasis and oxygen metabolism regulated by the hypoxia-inducible factors (HIFs). Free Radic. Biol. Med. 2019, 133, 118–129. [Google Scholar] [CrossRef]
- Bourdon, E.; Kang, D.K.; Ghosh, M.C.; Drake, S.K.; Wey, J.; Levine, R.L.; Rouault, T.A. The role of endogenous heme synthesis and degradation domain cysteines in cellular iron-dependent degradation of IRP2. Blood Cells Mol. Dis. 2003, 31, 247–255. [Google Scholar] [CrossRef]
- Fuhrmann, D.C.; Mondorf, A.; Beifuß, J.; Jung, M.; Brüne, B. Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol. 2020, 36, 101670. [Google Scholar] [CrossRef]
- Cronin, S.J.F.; Woolf, C.J.; Weiss, G.; Penninger, J.M. The Role of Iron Regulation in Immunometabolism and Immune-Related Disease. Front. Mol. Biosci. 2019, 6, 116. [Google Scholar] [CrossRef]
- Zhang, D.L.; Wu, J.; Shah, B.N.; Greutélaers, K.C.; Ghosh, M.C.; Ollivierre, H.; Su, X.Z.; Thuma, P.E.; Bedu-Addo, G.; Mockenhaupt, F.P. Erythrocytic ferroportin reduces intracellular iron accumulation, hemolysis, and malaria risk. Science 2018, 359, 1520–1523. [Google Scholar] [CrossRef] [PubMed]
- Gnana-Prakasam, J.P.; Martin, P.M.; Mysona, B.A.; Roon, P.; Smith, S.B.; Ganapathy, V. Hepcidin expression in mouse retina and its regulation via lipopolysaccharide/Toll-like receptor-4 pathway independent of Hfe. Biochem. J. 2008, 411, 79–88. [Google Scholar] [CrossRef]
- Darshan, D.; Anderson, G.J. Interacting signals in the control of hepcidin expression. BioMetals 2009, 22, 77–87. [Google Scholar] [CrossRef]
- Aschemeyer, S.; Qiao, B.; Stefanova, D.; Valore, E.V.; Sek, A.C.; Ruwe, T.A.; Vieth, K.R.; Jung, G.; Casu, C.; Rivella, S.; et al. Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin. Blood 2018, 131, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.; Park, S.; Lee, Y.M.; Hong, S.; Cho, K.B.; Nam, W. Demonstration of the heterolytic O-O bond cleavage of putative nonheme iron(II)-OOH(R) complexes for Fenton and enzymatic reactions. Angew. Chem. Int. Ed. Engl. 2014, 53, 7843–7847. [Google Scholar] [CrossRef]
- Michalski, M.C.; Calzada, C.; Makino, A.; Michaud, S.; Guichardant, M. Oxidation products of polyunsaturated fatty acids in infant formulas compared to human milk–A preliminary study. Mol. Nutr. Food Res. 2008, 52, 1478–1485. [Google Scholar] [CrossRef]
- Masaldan, S.; Bush, A.I.; Devos, D.; Rolland, A.S.; Moreau, C. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radic. Biol. Med. 2019, 133, 221–233. [Google Scholar] [CrossRef]
- Dixon, S.J.; Winter, G.E.; Musavi, L.S.; Lee, E.D.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol. 2015, 10, 1604–1609. [Google Scholar] [CrossRef]
- Wong-Ekkabut, J.; Xu, Z.; Triampo, W.; Tang, I.M.; Tieleman, D.P.; Monticelli, L. Effect of lipid peroxidation on the properties of lipid bilayers: A molecular dynamics study. Biophys. J. 2007, 93, 4225–4236. [Google Scholar] [CrossRef]
- SanGiovanni, J.P.; Chew, E.Y.; Clemons, T.E.; Ferris, F.L., 3rd; Gensler, G.; Lindblad, A.S.; Milton, R.C.; Seddon, J.M.; Klein, R.; Sperduto, R.D. Age-Related Eye Disease Study Research Group (2008 Sep). The relationship of dietary omega-3 long-chain polyunsaturated fatty acid intake with incident age-related macular degeneration: AREDS report no. 23. Arch. Ophthalmol. 2007, 126, 1274–1279. [Google Scholar] [CrossRef] [PubMed]
- Hirschhorn, T.; Stockwell, B.R. The development of the concept of ferroptosis. Free Radic. Biol. Med. 2019, 133, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, J.; Wang, Y.; Liu, Z.; Wu, Y. Ferroptosis drives photoreceptor degeneration in mice with defects in all-trans-retinal clearance. J. Biol. Chem. 2021, 296, 100187. [Google Scholar] [CrossRef]
- Ng, M.Y.W.; Wai, T.; Simonsen, A. Quality control of the mitochondrion. Dev. Cell 2021, 56, 881–905. [Google Scholar] [CrossRef]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Romanello, V.; Sandri, M. The connection between the dynamic remodeling of the mitochondrial network and the regulation of muscle mass. Cell. Mol. Life Sci. 2021, 78, 1305–1328. [Google Scholar] [CrossRef]
- Mattie, S.; Riemer, J.; Wideman, J.G.; McBride, H.M. A new mitofusin topology places the redox-regulated C terminus in the mitochondrial intermembrane space. J. Cell Biol. 2017, 217, 507–515. [Google Scholar] [CrossRef]
- Feher, J.; Kovacs, I.; Artico, M.; Cavallotti, C.; Papale, A.; Gabrieli, C.B. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol. Aging 2006, 27, 983–993. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 79, 100858. [Google Scholar] [CrossRef]
- Wang, L.; Yu, X.; Zhang, D.; Wen, Y.; Zhang, L.; Xia, Y.; Chen, J.; Xie, C.; Zhu, H.; Tong, J.; et al. Long-term blue light exposure impairs mitochondrial dynamics in the retina in light-induced retinal degeneration in vivo and in vitro. J. Photochem. Photobiol. B Biol. 2023, 240, 112654. [Google Scholar] [CrossRef]
- Li, J.; Feng, Y.; Li, Y.; He, P.; Zhou, Q.; Tian, Y.; Yao, R.; Yao, Y. Ferritinophagy: A novel insight into the double-edged sword in ferritinophagy–ferroptosis axis and human diseases. Cell Prolif. 2024, 57, e13621. [Google Scholar] [CrossRef] [PubMed]
- Moroishi, T.; Yamauchi, T.; Nishiyama, M.; Nakayama, K.I. HERC2 Targets the Iron Regulator FBXL5 for Degradation and Modulates Iron Metabolism. J. Biol. Chem. 2014, 289, 16430–16441. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Vaites, L.P.; Nissim, S.; Biancur, E.D.; Kim, A.J.; Wang, X.; Liu, Y.; Goessling, W.; Kimmelman, A.C.; Harper, J.W. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. eLife 2015, 4, e10308. [Google Scholar] [CrossRef]
- Yao, F.; Peng, J.; Zhang, E.; Ji, D.; Gao, Z.; Tang, Y.; Yao, X.; Xia, X. Pathologically high intraocular pressure disturbs normal iron homeostasis and leads to retinal ganglion cell ferroptosis in glaucoma. Cell Death Differ. 2023, 30, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Y.; Wei, T.T.; Zhuang, M.; Tan, C.Y.; Xie, T.H.; Cai, J.; Yao, Y.; Zhu, L. Iron derived from NCOA4-mediated ferritinophagy causes cellular senescence via the cGAS-STING pathway. Cell Death Discov. 2023, 9, 419. [Google Scholar] [CrossRef] [PubMed]
- Stoyanovsky, D.A.; Tyurina, Y.Y.; Shrivastava, I.; Bahar, I.; Tyurin, V.A.; Protchenko, O.; Jadhav, S.; Bolevich, S.B.; Kozlov, A.V.; Vladimirov, Y.A.; et al. Iron catalysis of lipid peroxidation in ferroptosis: Regulated enzymatic or random free radical reaction? Free Radic. Biol. Med. 2019, 133, 153–161. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to Tango: Regulation of Mammalian Iron Metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef]
- Xiang, Y.; Song, X.; Long, D. Ferroptosis regulation through Nrf2 and implications for neurodegenerative diseases. Arch. Toxicol. 2024, 98, 579–615. [Google Scholar] [CrossRef]
- Imai, H.; Matsuoka, M.; Kumagai, T.; Sakamoto, T.; Koumura, T. Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. In Apoptotic and Non-Apoptotic Cell Death; Springer: Cham, Switzerland, 2016; pp. 143–170. [Google Scholar]
- Conrad, M.; Proneth, B. Selenium: Tracing Another Essential Element of Ferroptotic Cell Death. Cell Chem. Biol. 2020, 27, 409–419. [Google Scholar] [CrossRef]
- Shimada, K.; Hayano, M.; Pagano, N.C.; Stockwell, B.R. Cell-Line Selectivity Improves the Predictive Power of Pharmacogenomic Analyses and Helps Identify NADPH as Biomarker for Ferroptosis Sensitivity. Cell Chem. Biol. 2016, 23, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, e1800311. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Chen, D.; Tavana, O.; Chu, B.; Erber, L.; Chen, Y.; Baer, R.; Gu, W. NRF2 Is a Major Target of ARF in p53-Independent Tumor Suppression. Mol. Cell 2017, 68, 224–232.e224. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, J.; Liu, X.; Feng, L.; Gong, Z.; Koppula, P.; Sirohi, K.; Li, X.; Wei, Y.; Lee, H.; et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat. Cell Biol. 2018, 20, 1181–1192. [Google Scholar] [CrossRef]
- Tang, W.; Guo, J.; Liu, W.; Ma, J.; Xu, G. Ferrostatin-1 attenuates ferroptosis and protects the retina against light-induced retinal degeneration. Biochem. Biophys. Res. Commun. 2021, 548, 27–34. [Google Scholar] [CrossRef]
- Yang, M.; Tsui, M.G.; Tsang, J.K.W.; Goit, R.K.; Yao, K.M.; So, K.F.; Lam, W.C.; Lo, A.C.Y. Involvement of FSP1-CoQ (10)-NADH and GSH-GPx-4 pathways in retinal pigment epithelium ferroptosis. Cell Death Dis. 2022, 13, 468. [Google Scholar] [CrossRef]
- Xiang, W.; Li, L.; Zhao, Q.; Zeng, Y.; Shi, J.; Chen, Z.; Gao, G.; Lai, K. PEDF protects retinal pigment epithelium from ferroptosis and ameliorates dry AMD-like pathology in a murine model. GeroScience 2023, 46, 2697–2714. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, Y.; Deng, Y.; Lu, J.; Xiao, L.; Li, J.; Zhou, Y.; Nie, F.; Chen, X.; Peng, J.; et al. Fructus Lycii and Salvia miltiorrhiza Bunge extract attenuate oxidative stress-induced photoreceptor ferroptosis in retinitis pigmentosa. Biomed. Pharmacother. 2023, 167, 115547. [Google Scholar] [CrossRef]
- Sakai, O.; Uchida, T.; Roggia, M.F.; Imai, H.; Ueta, T.; Amano, S. Role of Glutathione Peroxidase 4 in Glutamate-Induced Oxytosis in the Retina. PLoS ONE 2015, 10, e0130467. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wu, Y.; Song, P.; Zhang, M.; Wang, S.; Zou, M.-H. Proteasome-Dependent Degradation of Guanosine 5′-Triphosphate Cyclohydrolase I Causes Tetrahydrobiopterin Deficiency in Diabetes Mellitus. Circulation 2007, 116, 944–953. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Wei, W.; Wu, D.; Huang, F.; Li, M.; Li, W.; Yin, J.; Peng, Y.; Lu, Y.; Zhao, Q.; et al. Blockade of GCH1/BH4 Axis Activates Ferritinophagy to Mitigate the Resistance of Colorectal Cancer to Erastin-Induced Ferroptosis. Front. Cell Dev. Biol. 2022, 10, 810327. [Google Scholar] [CrossRef] [PubMed]
- Costigan, M.; Latremoliere, A.; Woolf, C.J. Analgesia by inhibiting tetrahydrobiopterin synthesis. Curr. Opin. Pharmacol. 2012, 12, 92–99. [Google Scholar] [CrossRef]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Müller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kössl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Central Sci. 2019, 6, 41–53. [Google Scholar] [CrossRef]
- Cronin, S.J.; Yu, W.; Hale, A.; Licht-Mayer, S.; Crabtree, M.J.; Korecka, J.A.; Tretiakov, E.O.; Sealey-Cardona, M.; Somlyay, M.; Onji, M.; et al. Crucial neuroprotective roles of the metabolite BH4 in dopaminergic neurons. bioRxiv 2023. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Yan, H.F.; Zou, T.; Tuo, Q.Z.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and links with diseases. Signal Transduct. Target. Ther. 2021, 6, 49. [Google Scholar] [CrossRef]
- Lee, J.; Roh, J.L. Unleashing Ferroptosis in Human Cancers: Targeting Ferroptosis Suppressor Protein 1 for Overcoming Therapy Resistance. Antioxidants 2023, 12, 1218. [Google Scholar] [CrossRef]
- Shukla, S.; Dubey, K.K. CoQ10 a super-vitamin: Review on application and biosynthesis. 3 Biotech 2018, 8, 249. [Google Scholar] [CrossRef]
- Song, Y.; Lei, H.; Yu, D.; Zhu, H.; Hao, M.; Cui, R.; Meng, X.; Sheng, X.; Zhang, L. Endogenous chemicals guard health through inhibiting ferroptotic cell death. BioFactors 2023, 50, 266–293. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Alehagen, U.; Johansson, P.; Aaseth, J.; Alexander, J.; Brismar, K. Increase in insulin-like growth factor 1 (IGF-1) and insulin-like growth factor binding protein 1 after supplementation with selenium and coenzyme Q10. A prospective randomized double-blind placebo-controlled trial among elderly Swedish citizens. PLoS ONE 2017, 12, e0178614. [Google Scholar] [CrossRef]
- Miriyala, S.; Thippakorn, C.; Chaiswing, L.; Xu, Y.; Noel, T.; Tovmasyan, A.; Batinic-Haberle, I.; Kooi, C.W.V.; Chi, W.; Latif, A.A.; et al. Novel role of 4-hydroxy-2-nonenal in AIFm2-mediated mitochondrial stress signaling. Free Radic. Biol. Med. 2016, 91, 68–80. [Google Scholar] [CrossRef]
- Yin, S.; Kabashima, T.; Zhu, Q.; Shibata, T.; Kai, M. Fluorescence assay of dihydroorotate dehydrogenase that may become a cancer biomarker. Sci. Rep. 2017, 7, 40670. [Google Scholar] [CrossRef]
- Zhou, Y.; Tao, L.; Zhou, X.; Zuo, Z.; Gong, J.; Liu, X.; Zhou, Y.; Liu, C.; Sang, N.; Liu, H.; et al. DHODH and cancer: Promising prospects to be explored. Cancer Metab. 2021, 9, 22. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, Q.; Peng, Z.; Li, X.J.; Ding, P.F.; Gao, S.; Sheng, B.; Liu, Y.; Lu, Y.; Zhuang, Z.; et al. Menaquinone-4 attenuates ferroptosis by upregulating DHODH through activation of SIRT1 after subarachnoid hemorrhage. Free Radic. Biol. Med. 2024, 210, 416–429. [Google Scholar] [CrossRef]
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575. [Google Scholar] [CrossRef]
- Hellyer, J.A.; Padda, S.K.; Diehn, M.; Wakelee, H.A. Clinical Implications of KEAP1-NFE2L2 Mutations in NSCLC. J. Thorac. Oncol. 2021, 16, 395–403. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; Gladwell, W.; Wang, X.; Chorley, B.; Bell, D.; Reddy, S.P.; Kleeberger, S.R. Nrf2-regulated PPARγ expression is critical to protection against acute lung injury in mice. Am. J. Respir. Crit. Care Med. 2010, 182, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Song, Q.; Jian, W.; Zhang, Y.; Li, Q.; Zhao, Y.; Liu, R.; Zeng, Y.; Zhang, F.; Duan, J. Puerarin Attenuates Iron Overload—Induced Ferroptosis in Retina through a Nrf2—Mediated Mechanism. Mol. Nutr. Food Res. 2024, 68, e2300123. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, X.; Wang, K.; Zhu, L.; Murray, M.; Zhou, F. Ginkgo biloba extracts (GBE) protect human RPE cells from t-BHP-induced oxidative stress and necrosis by activating the Nrf2-mediated antioxidant defence. J. Pharm. Pharmacol. 2022, 75, 105–116. [Google Scholar] [CrossRef]
- Hybertson, B.; Gao, B.; Doan, A. The clinical potential of influencing Nrf2 signaling in degenerative and immunological disorders. Clin. Pharmacol. Adv. Appl. 2014, 6, 19–34. [Google Scholar] [CrossRef]
- Wang, L.; Ebrahimi, K.B.; Chyn, M.; Cano, M.; Handa, J.T. Biology of p62/sequestosome-1 in Age-Related Macular Degeneration (AMD). Adv. Exp. Med. Biol. 2016, 854, 17–22. [Google Scholar]
- Zhao, Z.; Chen, Y.; Wang, J.; Sternberg, P.; Freeman, M.L.; Grossniklaus, H.E.; Cai, J. Age-Related Retinopathy in NRF2-Deficient Mice. PLoS ONE 2011, 6, e19456. [Google Scholar] [CrossRef]
- Totsuka, K.; Ueta, T.; Uchida, T.; Roggia, M.F.; Nakagawa, S.; Vavvas, D.G.; Honjo, M.; Aihara, M. Oxidative stress induces ferroptotic cell death in retinal pigment epithelial cells. Exp. Eye Res. 2019, 181, 316–324. [Google Scholar] [CrossRef]
- Song, D.; Zhao, L.; Li, Y.; Hadziahmetovic, M.; Song, Y.; Connelly, J.; Spino, M.; Dunaief, J.L. The Oral Iron Chelator Deferiprone Protects Against Systemic Iron Overload–Induced Retinal Degeneration in Hepcidin Knockout Mice. Investig. Opthalmology Vis. Sci. 2014, 55, 4525–4532. [Google Scholar] [CrossRef]
- Hadziahmetovic, M.; Pajic, M.; Grieco, S.; Song, Y.; Song, D.; Li, Y.; Cwanger, A.; Iacovelli, J.; Chu, S.; Ying, G.-S.; et al. The Oral Iron Chelator Deferiprone Protects Against Retinal Degeneration Induced through Diverse Mechanisms. Transl. Vis. Sci. Technol. 2012, 1, 2. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Suzuki, T.; Takahashi, K.; Koguchi, T.; Hirayama, T.; Mori, A.; Nakahara, T.; Nagasawa, H.; Ishii, K. Iron-chelating agents attenuate NMDA-Induced neuronal injury via reduction of oxidative stress in the rat retina. Exp. Eye Res. 2018, 171, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Obolensky, A.; Berenshtein, E.; Lederman, M.; Bulvik, B.; Alper-Pinus, R.; Yaul, R.; Deleon, E.; Chowers, I.; Chevion, M.; Banin, E. Zinc–desferrioxamine attenuates retinal degeneration in the rd10 mouse model of retinitis pigmentosa. Free Radic. Biol. Med. 2011, 51, 1482–1491. [Google Scholar] [CrossRef] [PubMed]
- Lukinova, N.; Iacovelli, J.; Dentchev, T.; Wolkow, N.; Hunter, A.; Amado, D.; Ying, G.-S.; Sparrow, J.R.; Dunaief, J.L. Iron chelation protects the retinal pigment epithelial cell line ARPE-19 against cell death triggered by diverse stimuli. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1440–1447. [Google Scholar] [CrossRef]
- Lin, B.; Youdim, M.B.H. The protective, rescue and therapeutic potential of multi-target iron-chelators for retinitis pigmentosa. Free Radic. Biol. Med. 2021, 174, 1–11. [Google Scholar] [CrossRef]
- Tang, Z.; Huo, M.; Ju, Y.; Dai, X.; Ni, N.; Liu, Y.; Gao, H.; Zhang, D.; Sun, H.; Fan, X.; et al. Nanoprotection Against Retinal Pigment Epithelium Degeneration via Ferroptosis Inhibition. Small Methods 2021, 5, 2100848. [Google Scholar] [CrossRef]
- Mandala, A.; Armstrong, A.; Girresch, B.; Zhu, J.; Chilakala, A.; Chavalmane, S.; Chaudhary, K.; Biswas, P.; Ogilvie, J.; Gnana-Prakasam, J.P. Fenofibrate prevents iron induced activation of canonical Wnt/β-catenin and oxidative stress signaling in the retina. npj Aging Mech. Dis. 2020, 6, 12. [Google Scholar] [CrossRef]
- Liu, B.; Wang, W.; Shah, A.; Yu, M.; Liu, Y.; He, L.; Dang, J.; Yang, L.; Yan, M.; Ying, Y.; et al. Sodium iodate induces ferroptosis in human retinal pigment epithelium ARPE-19 cells. Cell Death Dis. 2021, 12, 230. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Gao, S.; Wang, Y.; Li, N.; Yang, Z.; Yao, H.; Chen, Y.; Cheng, Y.; Zhong, Y.; Shen, X. Inhibition of Ferroptosis Ameliorates Photoreceptor Degeneration in Experimental Diabetic Mice. Int. J. Mol. Sci. 2023, 24, 16946. [Google Scholar] [CrossRef]
- Lee, J.J.; Chang-Chien, G.P.; Lin, S.; Hsiao, Y.T.; Ke, M.C.; Chen, A.; Lin, T.K. 5-Lipoxygenase Inhibition Protects Retinal Pigment Epithelium from Sodium Iodate-Induced Ferroptosis and Prevents Retinal Degeneration. Oxid. Med. Cell. Longev. 2022, 2022, 1792894. [Google Scholar] [CrossRef]
- Tang, X.; Li, X.; Zhang, D.; Han, W. Astragaloside-IV alleviates high glucose-induced ferroptosis in retinal pigment epithelial cells by disrupting the expression of miR-138-5p/Sirt1/Nrf2. Bioengineered 2022, 13, 8238–8253. [Google Scholar] [CrossRef] [PubMed]
- Shamsi, F.A.; Chaudhry, I.A.; Boulton, M.E.; Al-Rajhi, A.A. L-Carnitine Protects Human Retinal Pigment Epithelial Cells from Oxidative Damage. Curr. Eye Res. 2009, 32, 575–584. [Google Scholar] [CrossRef]
- Figon, F.; Casas, J. Ommochromes in invertebrates: Biochemistry and cell biology. Biol. Rev. Camb. Philos. Soc. 2019, 94, 156–183. [Google Scholar] [CrossRef] [PubMed]
- Brittenham, G.M. Iron-chelating therapy for transfusional iron overload. N. Engl. J. Med. 2011, 364, 146–156. [Google Scholar] [CrossRef]
- Li, X.; Zhu, S.; Qi, F. Blue light pollution causes retinal damage and degeneration by inducing ferroptosis. J. Photochem. Photobiol. B. 2023, 238, 112617. [Google Scholar] [CrossRef]
- Timoshnikov, V.A.; Kobzeva, T.V.; Polyakov, N.E.; Kontoghiorghes, G.J. Redox Interactions of Vitamin C and Iron: Inhibition of the Pro-Oxidant Activity by Deferiprone. Int. J. Mol. Sci. 2020, 21, 3967. [Google Scholar] [CrossRef] [PubMed]
- Hadziahmetovic, M.; Song, Y.; Wolkow, N.; Iacovelli, J.; Grieco, S.; Lee, J.; Lyubarsky, A.; Pratico, D.; Connelly, J.; Spino, M.; et al. The Oral Iron Chelator Deferiprone Protects against Iron Overload–Induced Retinal Degeneration. Investig. Opthalmology Vis. Sci. 2011, 52, 959–968. [Google Scholar] [CrossRef]
- Song, D.; Song, Y.; Hadziahmetovic, M.; Zhong, Y.; Dunaief, J.L. Systemic administration of the iron chelator deferiprone protects against light-induced photoreceptor degeneration in the mouse retina. Free Radic. Biol. Med. 2012, 53, 64–71. [Google Scholar] [CrossRef]
- Caro, A.A.; Commissariat, A.; Dunn, C.; Kim, H.; García, S.L.; Smith, A.; Strang, H.; Stuppy, J.; Desrochers, L.P.; Goodwin, T.E. Prooxidant and antioxidant properties of salicylaldehyde isonicotinoyl hydrazone iron chelators in HepG2 cells. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2015, 1850, 2256–2264. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, W.; Li, Y.; Xiao, Y.; Cheng, J.; Jia, J. The 5-Lipoxygenase Inhibitor Zileuton Confers Neuroprotection against Glutamate Oxidative Damage by Inhibiting Ferroptosis. Biol. Pharm. Bull. 2015, 38, 1234–1239. [Google Scholar] [CrossRef]
- Ribas, G.S.; Vargas, C.R.; Wajner, M. L-carnitine supplementation as a potential antioxidant therapy for inherited neurometabolic disorders. Gene 2014, 533, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.L.M.; Dörschmann, P.; Seeba, C.; Thalenhorst, T.; Roider, J.; Iloki Assanga, S.B.; Ruiz, J.C.G.; Del Castillo Castro, T.; Rosas-Burgos, E.C.; Plascencia-Jatomea, M.; et al. Properties of Cephalopod Skin Ommochromes to Inhibit Free Radicals, and the Maillard Reaction and Retino-Protective Mechanisms in Cellular Models Concerning Oxidative Stress, Angiogenesis, and Inflammation. Antioxidants 2022, 11, 1574. [Google Scholar] [CrossRef]
- Liu, Y.; Baumann, B.; Song, Y.; Zhang, K.; Sterling, J.K.; Lakhal-Littleton, S.; Kozmik, Z.; Su, G.; Dunaief, J.L. Minimal effect of conditional ferroportin KO in the neural retina implicates ferrous iron in retinal iron overload and degeneration. Exp. Eye Res. 2022, 218, 108988. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.-L.; Yang, Q.-L.; Wei, Y.-Z.; Qiu, X.; Zeng, H.-Y.; Yan, A.-M.; Peng, K.; Li, Y.-L.; Rao, F.-Q.; Chen, F.-H.; et al. Identification of a novel ferroptosis-related gene signature associated with retinal degeneration induced by light damage in mice. Heliyon 2023, 9, e23002. [Google Scholar] [CrossRef]
- Zheng, N.; Liao, T.; Zhang, C.; Zhang, Z.; Yan, S.; Xi, X.; Ruan, F.; Yang, C.; Zhao, Q.; Deng, W.; et al. Quantum Dots-caused Retinal Degeneration in Zebrafish Regulated by Ferroptosis and Mitophagy in Retinal Pigment Epithelial Cells through Inhibiting Spliceosome. Adv. Sci. 2024, 11, e2406343. [Google Scholar] [CrossRef]
- Huang, K.; Deng, H.; Wang, S.; Zhang, F.; Huang, G.; Wang, L.; Liu, J.; Zhao, X.; Ren, H.; Yang, G.; et al. Melanin-Like Nanomedicine Functions as a Novel RPE Ferroptosis Inhibitor to Ameliorate Retinal Degeneration and Visual Impairment in Dry Age-Related Macular Degeneration. Adv. Healthc. Mater. 2024, 13, e2401613. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Drugs | Experimental Objects | Mechanisms and Targets | Effect | References |
|---|---|---|---|---|---|
| Iron-chelating agent | Deferoxamine | ARPE-19 cells | Decreased iron level | Enhanced cell viability | [151] |
| Deferiprone | Hepc KO mice/rd6 mice | Decreased iron levels, alleviating oxidative stress | Preserved photoreceptor and RPE cell | [152,153] | |
| Deferasirox | Rats | Reduced iron content and oxidative stress | Protected retinal neurons | [154] | |
| Zinc deferriamine | rd10 mice | Reduced iron content and lipid peroxidation | Preserved photoreceptors | [155] | |
| Salicylaldehyde isonicotinoyl hydrazine | ARPE-19 cells | Reduced iron levels and ROS levels | Enhanced cell viability | [156] | |
| VK-28/VAR10303/M30 | rd10 mice | Downregulation of Tf, reduced the production of TNF-α and IL-1β | Preserved photoreceptors, improved visual behaviors | [157] | |
| KCa[FeIII(CN)6] | C57BL/6J mice | Reduced iron levels | Halted the degeneration of RPE cells and photoreceptors | [158] | |
| Fenofibrate | ARPE-19 cells | Reduced iron levels and inhibited ROS generation by downregulating the Wnt/β-catenin signaling pathway | Enhanced cell viability | [159] | |
| Ferrostatin-1 | HRPEpiC cells/661w cells | Reduced lipid peroxidation by mediating the GSH-GPX4 pathway | Enhanced viability of primary HRPEpiC cells | [120,160,161] | |
| Lipophilic antioxidant | liproxstatin-1 | ARPE-19 cells | Reduced lipid peroxidation | Rescued cell viability | [13] |
| Zileuton | ARPE-19 cells/mice | Inhibited 5-LOX | Increased cell viability, reduced photoreceptor death | [162] | |
| Astragaloside IV | ARPE-19 cells | Promoted the expression of Sirt1 and Nrf2 | Decreased cell death | [163] | |
| L-carnitine | HRPE cells | Alleviated oxidative damage | Protected RPE cells | [164] | |
| Ommochromes | ARPE-19 cells | Mitigated the levels of inflammatory cytokines and oxidative stress | Inhibited ferroptosis | [165] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, N.; Li, S.; Wu, H.; Wei, D.; Pu, N.; Wang, K.; Liu, Y.; Tao, Y.; Song, Z. Ferroptosis: An Energetic Villain of Age-Related Macular Degeneration. Biomedicines 2025, 13, 986. https://doi.org/10.3390/biomedicines13040986
Zhao N, Li S, Wu H, Wei D, Pu N, Wang K, Liu Y, Tao Y, Song Z. Ferroptosis: An Energetic Villain of Age-Related Macular Degeneration. Biomedicines. 2025; 13(4):986. https://doi.org/10.3390/biomedicines13040986
Chicago/Turabian StyleZhao, Na, Siyu Li, Hao Wu, Dong Wei, Ning Pu, Kexin Wang, Yashuang Liu, Ye Tao, and Zongming Song. 2025. "Ferroptosis: An Energetic Villain of Age-Related Macular Degeneration" Biomedicines 13, no. 4: 986. https://doi.org/10.3390/biomedicines13040986
APA StyleZhao, N., Li, S., Wu, H., Wei, D., Pu, N., Wang, K., Liu, Y., Tao, Y., & Song, Z. (2025). Ferroptosis: An Energetic Villain of Age-Related Macular Degeneration. Biomedicines, 13(4), 986. https://doi.org/10.3390/biomedicines13040986