

Lipid-Metabolism-Related Gene Signature Predicts Prognosis and Immune Microenvironment Alterations in Endometrial Cancer
 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection and Preprocessing
2.2. Construction and Validation of a Lipid Metabolism Risk Model
2.3. Prognostic Significance and Nomogram Development
2.4. Functional and Pathway Enrichment Analysis
2.5. Tumor Mutational Burden and Immune Infiltration Analysis
- Immune and stromal infiltration levels were inferred using the ESTIMATE algorithm via the “estimate” R package, providing immune and stromal scores for each tumor sample. These scores reflect both the content and spatial composition of tumor-infiltrating immune cells, offering an overview of immune infiltration.
- The “GSVA” R package was utilized to execute ssGSEA, 24 distinct immune cell populations were profiled within individual tumor samples. Enrichment scores from ssGSEA were compared across risk groups to elucidate immune cell composition patterns related to prognostic stratification.
- We explored the correlations between the risk score and the levels of immunological checkpoint molecules. These analyses offer insights into the potential role of the risk model in guiding immune checkpoint blockade (ICB) therapies.
- The TIDE algorithm was applied to estimate ICB therapy responsiveness by modeling immune evasion. It integrated two key mechanisms: T-cell dysfunction in tumors with high cytotoxic T lymphocyte (CTL) infiltration and T-cell exclusion in tumors with low CTL presence. The resulting TIDE scores provide individualized predictions of immunotherapeutic efficacy.
2.6. Validation of LMRG Protein Expression Using HPA
2.7. Cell Culture and Patient Sample Collection
2.8. Real-Time PCR (RT-PCR) Analysis
2.9. Gene Silencing via siRNA
2.10. Western Blot Analysis
2.11. Cell Viability and Migration Assays
- CCK-8 Viability Assay: Transfected Ishikawa and HEC-1-B cells were seeded in 96-well plates (2 × 103 cells/well). After incubation with 10% CCK-8 solution (Applygen, Beijing, China) for 1 h, the absorbance at 462 nm was measured using a microplate reader (BioTek Instruments, Winooski, VT, USA).
- Wound Healing Assay: Confluent monolayers of transfected cells were scratched with a sterile pipette tip, and wound closure was monitored and photographed at 0 and 24 h.
- Transwell Migration Assay: Transfected cells (3 × 104) were seeded in the upper chambers of transwell inserts (8.0-μm, Corning, Shanghai, China) with serum-free medium, while the lower chambers contained DMEM with 10% fetal bovine serum. After 16 h of incubation, migrated cells were fixed, stained with crystal violet, and visualized under a microscope.
2.12. Immune-Related Analysis of LIPG Expression
2.13. Statistical Analysis
3. Results
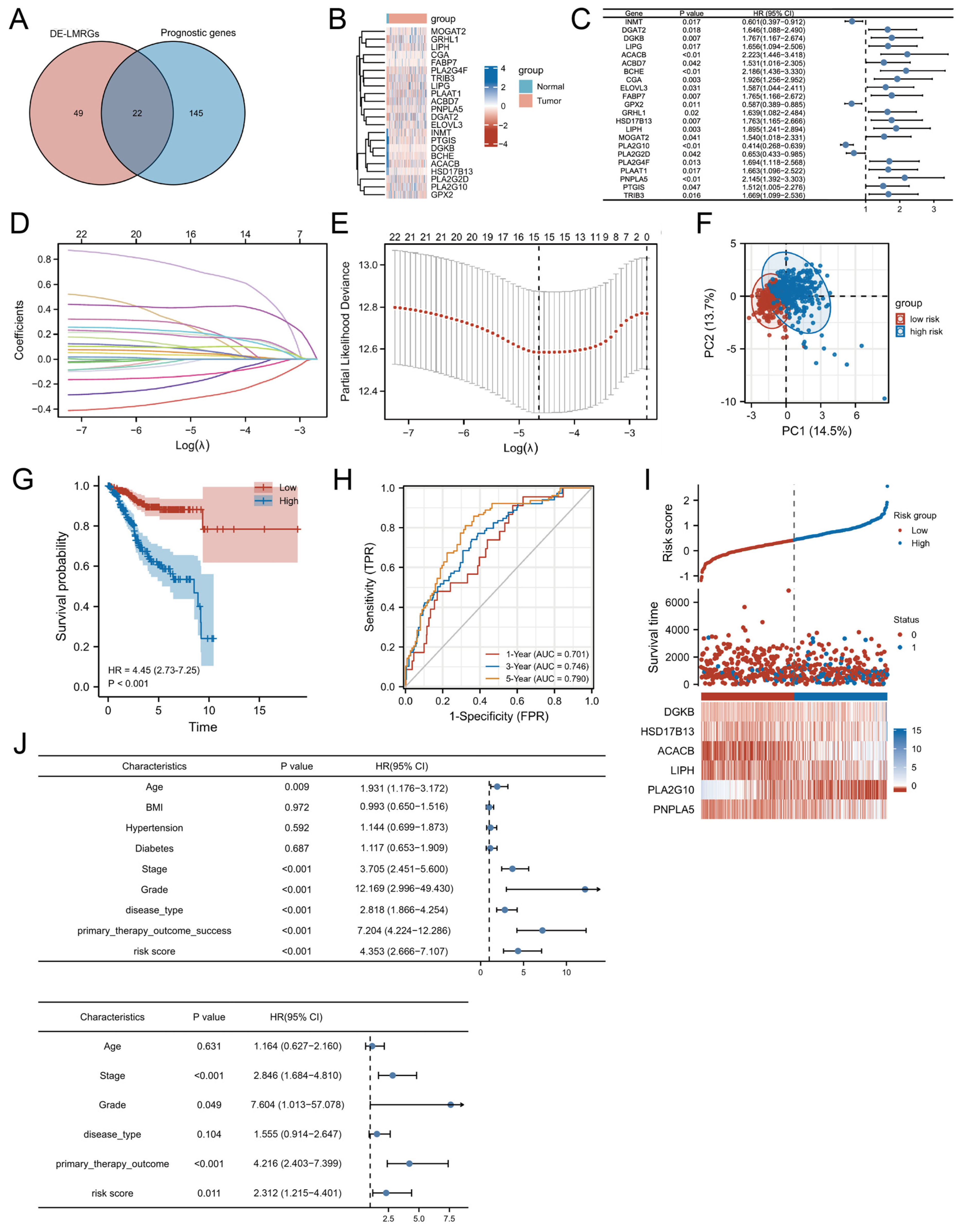
3.1. Identification of Prognostic LMRGs in UCEC
3.2. Construction of a Prognostic LMRG Signature
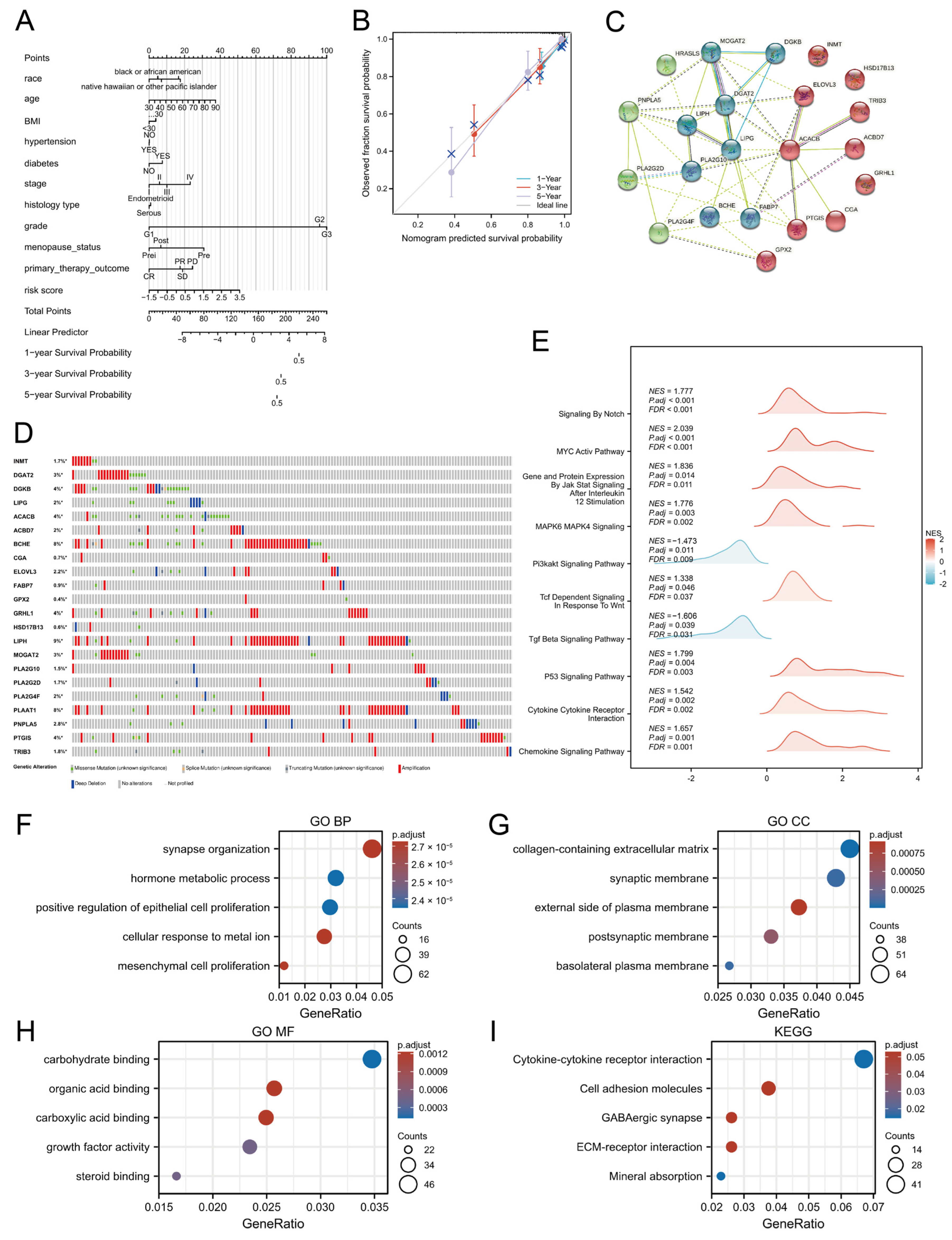
3.3. Development and Validation of an OS Prediction Nomogram
3.4. Functional Enrichment Analyses of LMRGs
3.5. TMB and Lipid-Metabolism-Related Risk Score
3.6. Connections Between Distinct Immune Cell Infiltration, Immune Checkpoint Genes, and Lipid-Metabolism-Related Risk Score
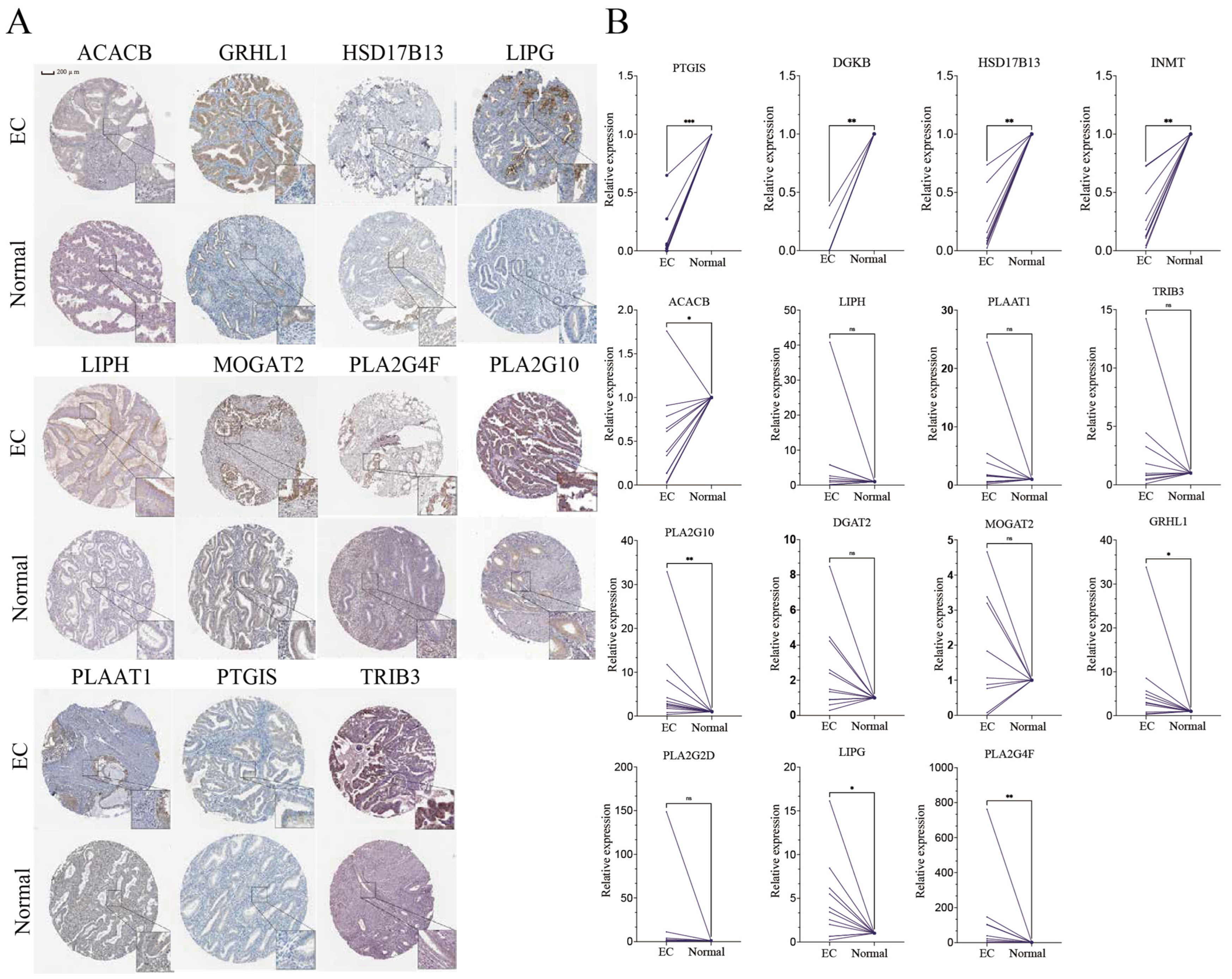
3.7. Expression of Key Genes in Clinical Samples
3.8. The Role of LIPG in EC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BMI | body mass index |
| CTLA4 | cytotoxic T-lymphocyte-associated protein 4 |
| CYT | cytolytic activity |
| DC | dendritic cells |
| DEGs | differentially expressed genes |
| DE-LMRGs | differentially expressed lipid-metabolism-associated genes |
| EaSIeR | Estimate Systems Immune Response |
| EC | endometrial cancer |
| FDR | false discovery Rate |
| GDC | Genomic Data Commons |
| GO | Gene Ontology |
| GSEA | Gene Set Enrichment Analysis |
| HAVCR2 | human activating vascular cell receptor 2 |
| HPA | Human Protein Atlas |
| HRD | homologous recombination defects |
| iDC | immature dendritic cells |
| IPS | Immune Prognostic Signature |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LASSO | least absolute shrinkage and selection operator |
| LIPG | endothelial lipase |
| LMRGs | Lipid-metabolism-related genes |
| MSigDB | Molecular Signatures Database |
| OS | overall survival |
| PCA | principal component analysis |
| PD1 | programmed cell death protein 1 |
| pDC | plasmacytoid dendritic cells |
| PPI | Protein-protein interaction |
| SREBP | sterol regulatory-element binding protein |
| ssGSEA | single-sample gene set enrichment analysis |
| TCGA | the Cancer Genome Atlas |
| TFH | follicular helper T |
| TIDE | Tumor Immune Dysfunction and Exclusion |
| TIGIT | T cell immunoglobulin and mucin domain containing |
| TIICs | tumor-infiltrating immune cells |
| TILs | tumor infiltrated lymphocytes |
| TIP | Tracking Tumor Immunophenotype |
| TLS | tertiary lymphoid structures |
| TMB | tumor mutational burden |
| Treg | regulatory T cells |
| UCEC | uterine corpus endometrial carcinoma |
| WHO | World Health Organization |
References
- Marti, C.; Deluche, E.; Jochum, F.; Bendifallah, S.; Azais, H.; Deidier, J.; Cockenpot, V.; Menoux, I.; Balaya, V.; Betrian, S.; et al. Management of Endometrial Cancer: French Society of Onco-Gynecology’s Evaluation through a Delphi Survey. J. Clin. Med. 2022, 11, 6765. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Kawaler, E.A.; Cui Zhou, D.; Gritsenko, M.A.; Huang, C.; Blumenberg, L.; Karpova, A.; Petyuk, V.A.; Savage, S.R.; Satpathy, S.; et al. Proteogenomic Characterization of Endometrial Carcinoma. Cell 2020, 180, 729–748.e26. [Google Scholar] [CrossRef] [PubMed]
- Makker, V.; Colombo, N.; Casado Herráez, A.; Santin, A.D.; Colomba, E.; Miller, D.S.; Fujiwara, K.; Pignata, S.; Baron-Hay, S.; Ray-Coquard, I.; et al. Lenvatinib plus Pembrolizumab for Advanced Endometrial Cancer. N. Engl. J. Med. 2022, 386, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Crosbie, E.J.; Kitson, S.J.; McAlpine, J.N.; Mukhopadhyay, A.; Powell, M.E.; Singh, N. Endometrial Cancer. Lancet Lond. Engl. 2022, 399, 1412–1428. [Google Scholar] [CrossRef]
- Lorentzen, G.M.; Łaniewski, P.; Cui, H.; Mahnert, N.D.; Mourad, J.; Borst, M.P.; Willmott, L.; Chase, D.M.; Roe, D.J.; Herbst-Kralovetz, M.M. Cervicovaginal Metabolome and Tumor Characteristics for Endometrial Cancer Detection and Risk Stratification. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2024, 30, 3073–3087. [Google Scholar] [CrossRef]
- Li, J.; Yang, H.; Zhang, L.; Zhang, S.; Dai, Y. Metabolic Reprogramming and Interventions in Endometrial Carcinoma. Biomed. Pharmacother. Biomed. Pharmacother. 2023, 161, 114526. [Google Scholar] [CrossRef]
- Yao, L.; Chen, S.; Li, W. Fatostatin Inhibits the Development of Endometrial Carcinoma in Endometrial Carcinoma Cells and a Xenograft Model by Targeting Lipid Metabolism. Arch. Biochem. Biophys. 2020, 684, 108327. [Google Scholar] [CrossRef]
- Jin, H.-R.; Wang, J.; Wang, Z.-J.; Xi, M.-J.; Xia, B.-H.; Deng, K.; Yang, J.-L. Lipid Metabolic Reprogramming in Tumor Microenvironment: From Mechanisms to Therapeutics. J. Hematol. Oncol. 2023, 16, 103. [Google Scholar] [CrossRef]
- Dyck, L.; Prendeville, H.; Raverdeau, M.; Wilk, M.M.; Loftus, R.M.; Douglas, A.; McCormack, J.; Moran, B.; Wilkinson, M.; Mills, E.L.; et al. Suppressive Effects of the Obese Tumor Microenvironment on CD8 T Cell Infiltration and Effector Function. J. Exp. Med. 2022, 219, e20210042. [Google Scholar] [CrossRef]
- Mendiola, M.; Pellinen, T.; Ramon-Patino, J.L.; Berjon, A.; Bruck, O.; Heredia-Soto, V.; Turkki, R.; Escudero, J.; Hemmes, A.; Garcia de la Calle, L.E.; et al. Prognostic Implications of Tumor-Infiltrating T Cells in Early-Stage Endometrial Cancer. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc. 2022, 35, 256–265. [Google Scholar] [CrossRef]
- Mou, L.; Pu, Z.; Luo, Y.; Quan, R.; So, Y.; Jiang, H. Construction of a Lipid Metabolism-Related Risk Model for Hepatocellular Carcinoma by Single Cell and Machine Learning Analysis. Front. Immunol. 2023, 14, 1036562. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xiong, Z.; Han, J.; Nian, W.; Wang, Z.; Cai, K.; Gao, J.; Wang, G.; Tao, K.; Cai, M. Identification of a lipid homeostasis-related gene signature for predicting prognosis, immunity, and chemotherapeutic effect in patients with gastric cancer. Sci. Rep. 2024, 14, 2895. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Deng, R.; Wang, P.; Lu, X.; Zhao, X.; Wang, X.; Cai, R.; Lin, J. LIPG: An Inflammation and Cancer Modulator. Cancer Gene Ther. 2021, 28, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Minhajuddin, M.; Krug, A.; Pei, S.; Chou, C.-H.; Culp-Hill, R.; Ponder, J.; De Bloois, E.; Schniedewind, B.; Amaya, M.L.; et al. The Hepatic Microenvironment Uniquely Protects Leukemia Cells through Induction of Growth and Survival Pathways Mediated by LIPG. Cancer Discov. 2021, 11, 500–519. [Google Scholar] [CrossRef]
- Santos, C.R.; Schulze, A. Lipid Metabolism in Cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef]
- Welte, M.A.; Gould, A.P. Lipid Droplet Functions beyond Energy Storage. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1260–1272. [Google Scholar] [CrossRef]
- Long, J.; Zhang, C.-J.; Zhu, N.; Du, K.; Yin, Y.-F.; Tan, X.; Liao, D.-F.; Qin, L. Lipid Metabolism and Carcinogenesis, Cancer Development. Am. J. Cancer Res. 2018, 8, 778–791. [Google Scholar]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid Metabolic Reprogramming in Cancer Cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef]
- Kuhajda, F.P.; Pizer, E.S.; Li, J.N.; Mani, N.S.; Frehywot, G.L.; Townsend, C.A. Synthesis and Antitumor Activity of an Inhibitor of Fatty Acid Synthase. Proc. Natl. Acad. Sci. USA 2000, 97, 3450–3454. [Google Scholar] [CrossRef]
- Benjamin, D.I.; Li, D.S.; Lowe, W.; Heuer, T.; Kemble, G.; Nomura, D.K. Diacylglycerol Metabolism and Signaling Is a Driving Force Underlying FASN Inhibitor Sensitivity in Cancer Cells. ACS Chem. Biol. 2015, 10, 1616–1623. [Google Scholar] [CrossRef]
- Ligorio, F.; Pellegrini, I.; Castagnoli, L.; Vingiani, A.; Lobefaro, R.; Zattarin, E.; Santamaria, M.; Pupa, S.M.; Pruneri, G.; de Braud, F.; et al. Targeting Lipid Metabolism Is an Emerging Strategy to Enhance the Efficacy of Anti-HER2 Therapies in HER2-Positive Breast Cancer. Cancer Lett. 2021, 511, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, Y.; Koo, J.S. Differential Expression of Lipid Metabolism-Related Proteins in Different Breast Cancer Subtypes. PLoS ONE 2015, 10, e0119473. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.P.; Gómez de Cedrón, M.; Ramírez de Molina, A. Alterations of Lipid Metabolism in Cancer: Implications in Prognosis and Treatment. Front. Oncol. 2020, 10, 577420. [Google Scholar] [CrossRef] [PubMed]
- Sangineto, M.; Villani, R.; Cavallone, F.; Romano, A.; Loizzi, D.; Serviddio, G. Lipid Metabolism in Development and Progression of Hepatocellular Carcinoma. Cancers 2020, 12, 1419. [Google Scholar] [CrossRef]
- Qu, J.; Liu, B.; Li, B.; Du, G.; Li, Y.; Wang, J.; He, L.; Wan, X. TRIB3 Suppresses Proliferation and Invasion and Promotes Apoptosis of Endometrial Cancer Cells by Regulating the AKT Signaling Pathway. OncoTargets Ther. 2019, 12, 2235–2245. [Google Scholar] [CrossRef]
- Dai, D.; Chen, B.; Feng, Y.; Wang, W.; Jiang, Y.; Huang, H.; Liu, J. Prognostic Value of Prostaglandin I2 Synthase and Its Correlation with Tumor-Infiltrating Immune Cells in Lung Cancer, Ovarian Cancer, and Gastric Cancer. Aging 2020, 12, 9658–9685. [Google Scholar] [CrossRef]
- Lu, C.; Fang, S.; Weng, Q.; Lv, X.; Meng, M.; Zhu, J.; Zheng, L.; Hu, Y.; Gao, Y.; Wu, X.; et al. Integrated Analysis Reveals Critical Glycolytic Regulators in Hepatocellular Carcinoma. Cell Commun. Signal. CCS 2020, 18, 97. [Google Scholar] [CrossRef]
- Milgraum, L.Z.; Witters, L.A.; Pasternack, G.R.; Kuhajda, F.P. Enzymes of the Fatty Acid Synthesis Pathway Are Highly Expressed in in Situ Breast Carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1997, 3, 2115–2120. [Google Scholar]
- Swinnen, J.V.; Brusselmans, K.; Verhoeven, G. Increased Lipogenesis in Cancer Cells: New Players, Novel Targets. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 358–365. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhu, X.; Qiao, X.; Gu, X.; Xue, J.; Han, Y.; Sun, L.; Cui, M.; Liu, C. LIPH Promotes Metastasis by Enriching Stem-like Cells in Triple-Negative Breast Cancer. J. Cell. Mol. Med. 2020, 24, 9125–9134. [Google Scholar] [CrossRef]
- Li, S.; Wu, T.; Lu, Y.-X.; Wang, J.-X.; Yu, F.-H.; Yang, M.-Z.; Huang, Y.-J.; Li, Z.-J.; Wang, S.-L.; Huang, L.; et al. Obesity Promotes Gastric Cancer Metastasis via Diacylglycerol Acyltransferase 2-Dependent Lipid Droplets Accumulation and Redox Homeostasis. Redox Biol. 2020, 36, 101596. [Google Scholar] [CrossRef] [PubMed]
- Fabian, J.; Lodrini, M.; Oehme, I.; Schier, M.C.; Thole, T.M.; Hielscher, T.; Kopp-Schneider, A.; Opitz, L.; Capper, D.; von Deimling, A.; et al. GRHL1 Acts as Tumor Suppressor in Neuroblastoma and Is Negatively Regulated by MYCN and HDAC3. Cancer Res. 2014, 74, 2604–2616. [Google Scholar] [CrossRef] [PubMed]
- Lo, P.-K.; Yao, Y.; Lee, J.S.; Zhang, Y.; Huang, W.; Kane, M.A.; Zhou, Q. LIPG Signaling Promotes Tumor Initiation and Metastasis of Human Basal-like Triple-Negative Breast Cancer. eLife 2018, 7, e31334. [Google Scholar] [CrossRef] [PubMed]
- Jonusiene, V.; Sasnauskiene, A.; Lachej, N.; Kanopiene, D.; Dabkeviciene, D.; Sasnauskiene, S.; Kazbariene, B.; Didziapetriene, J. Down-Regulated Expression of Notch Signaling Molecules in Human Endometrial Cancer. Med. Oncol. Northwood Lond. Engl. 2013, 30, 438. [Google Scholar] [CrossRef]
- Fatima, I.; Barman, S.; Rai, R.; Thiel, K.W.W.; Chandra, V. Targeting Wnt Signaling in Endometrial Cancer. Cancers 2021, 13, 2351. [Google Scholar] [CrossRef]
- Jin, W. Role of JAK/STAT3 Signaling in the Regulation of Metastasis, the Transition of Cancer Stem Cells, and Chemoresistance of Cancer by Epithelial-Mesenchymal Transition. Cells 2020, 9, 217. [Google Scholar] [CrossRef]
- Slebe, F.; Rojo, F.; Vinaixa, M.; García-Rocha, M.; Testoni, G.; Guiu, M.; Planet, E.; Samino, S.; Arenas, E.J.; Beltran, A.; et al. FoxA and LIPG Endothelial Lipase Control the Uptake of Extracellular Lipids for Breast Cancer Growth. Nat. Commun. 2016, 7, 11199. [Google Scholar] [CrossRef]
- Cadenas, C.; Vosbeck, S.; Edlund, K.; Grgas, K.; Madjar, K.; Hellwig, B.; Adawy, A.; Glotzbach, A.; Stewart, J.D.; Lesjak, M.S.; et al. LIPG-Promoted Lipid Storage Mediates Adaptation to Oxidative Stress in Breast Cancer. Int. J. Cancer 2019, 145, 901–915. [Google Scholar] [CrossRef]
- Lo, P.-K.; Yao, Y.; Zhou, Q. Inhibition of LIPG Phospholipase Activity Suppresses Tumor Formation of Human Basal-like Triple-Negative Breast Cancer. Sci. Rep. 2020, 10, 8911. [Google Scholar] [CrossRef]
- Zhao, J.; Li, G.; Zhao, G.; Wang, W.; Shen, Z.; Yang, Y.; Huang, Y.; Ye, L. Prognostic Signature of Lipid Metabolism Associated LncRNAs Predict Prognosis and Treatment of Lung Adenocarcinoma. Front. Oncol. 2022, 12, 986367. [Google Scholar] [CrossRef]
- Siddiqui, S.; Glauben, R. Fatty Acid Metabolism in Myeloid-Derived Suppressor Cells and Tumor-Associated Macrophages: Key Factor in Cancer Immune Evasion. Cancers 2022, 14, 250. [Google Scholar] [CrossRef] [PubMed]
- Tu, B.; Gao, Y.; Sun, F.; Shi, M.; Huang, Y. Lipid Metabolism Regulation Based on Nanotechnology for Enhancement of Tumor Immunity. Front. Pharmacol. 2022, 13, 840440. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Ye, Q.; Gu, H.; Chen, Z. The Role of Lipid Metabolism in Tumor Immune Microenvironment and Potential Therapeutic Strategies. Front. Oncol. 2022, 12, 984560. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Wang, X.; Song, C.; He, Z.; Wang, R.; Xu, Y.; Jiang, G.; Wan, Y.; Mei, J.; Mao, W. The Role of Lipid Metabolic Reprogramming in Tumor Microenvironment. Theranostics 2023, 13, 1774–1808. [Google Scholar] [CrossRef]
- Tan, X.; Liu, S.; Yao, L.; Cui, G.; Liu, J.; Ding, J. Comprehensive Analysis of a Novel Lipid Metabolism-Related Gene Signature for Predicting the Prognosis and Immune Landscape in Uterine Corpus Endometrial Carcinoma. J. Oncol. 2022, 80, 28825. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathways | Database | Gene Count |
|---|---|---|
| HALLMARK_FATTY_ACID_METABOLISM | HALLMARK | 158 |
| KEGG_GLYCEROLIPID_METABOLISM | KEGG | 49 |
| REACTOME_METABOLISM_OF_LIPIDS | Reactome | 741 |
| REACTOME_PHOSPHOLIPID_METABOLISM | Reactome | 211 |
| Sum | 1159 (unique: 865) | |
| Clinical Feature | Number | Univariate Analysis | Multivariate Analysis | ||||
|---|---|---|---|---|---|---|---|
| HR | 95% CI | p Value | HR | 95% CI | p Value | ||
| DE-LMG Risk Parameter (high-risk/low-risk) | 271/273 | 4.353 | 2.666–7.107 | <0.001 | 2.312 | 1.215–4.401 | 0.011 |
| Age (≥60/<60) | 364/178 | 1.931 | 1.176–3.172 | 0.009 | 1.164 | 0.627–2.160 | 0.631 |
| BMI (≥30/<30) | 304/208 | 0.993 | 0.650–1.516 | 0.972 | |||
| Hypertension (YES/NO) | 232/161 | 1.144 | 0.699–1.873 | 0.592 | |||
| Diabetes (YES/NO) | 100/267 | 1.117 | 0.653–1.909 | 0.687 | |||
| Stage (III–IV/I–II) | 155/389 | 3.705 | 2.451–5.600 | <0.001 | 2.846 | 1.684–4.810 | <0.001 |
| Grade (G2.G3/G1) | 446/98 | 12.169 | 2.996–49.4320 | <0.001 | 7.604 | 1.013–57.078 | 0.049 |
| Disease type (Serous/Endometrioid) | 141/401 | 2.818 | 1.866–4.254 | <0.001 | 1.555 | 0.914–2.647 | 0.104 |
| Primary therapy outcome (CR/not CR) | 386/33 | 7.204 | 4.224–12.286 | <0.001 | 4.216 | 2.403–7.399 | <0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Z.; Nie, Y.; Kong, D.; Xue, L.; He, T.; Zhang, K.; Zhang, J.; Shang, C.; Guo, H. Lipid-Metabolism-Related Gene Signature Predicts Prognosis and Immune Microenvironment Alterations in Endometrial Cancer. Biomedicines 2025, 13, 1050. https://doi.org/10.3390/biomedicines13051050
Wu Z, Nie Y, Kong D, Xue L, He T, Zhang K, Zhang J, Shang C, Guo H. Lipid-Metabolism-Related Gene Signature Predicts Prognosis and Immune Microenvironment Alterations in Endometrial Cancer. Biomedicines. 2025; 13(5):1050. https://doi.org/10.3390/biomedicines13051050
Chicago/Turabian StyleWu, Zhangxin, Yufei Nie, Deshui Kong, Lixiang Xue, Tianhui He, Kuaile Zhang, Jie Zhang, Chunliang Shang, and Hongyan Guo. 2025. "Lipid-Metabolism-Related Gene Signature Predicts Prognosis and Immune Microenvironment Alterations in Endometrial Cancer" Biomedicines 13, no. 5: 1050. https://doi.org/10.3390/biomedicines13051050
APA StyleWu, Z., Nie, Y., Kong, D., Xue, L., He, T., Zhang, K., Zhang, J., Shang, C., & Guo, H. (2025). Lipid-Metabolism-Related Gene Signature Predicts Prognosis and Immune Microenvironment Alterations in Endometrial Cancer. Biomedicines, 13(5), 1050. https://doi.org/10.3390/biomedicines13051050