
Viroimmunotherapy for Colorectal Cancer: Clinical Studies


Abstract
:1. Introduction
2. Cancer Vaccines
3. Virus Infected Autologous Tumor Cell Vaccines
4. Viral Vector-Based Vaccines
5. Oncolytic Virus
6. Combination of Oncolytic Viruses with Immune Checkpoint Inhibitors
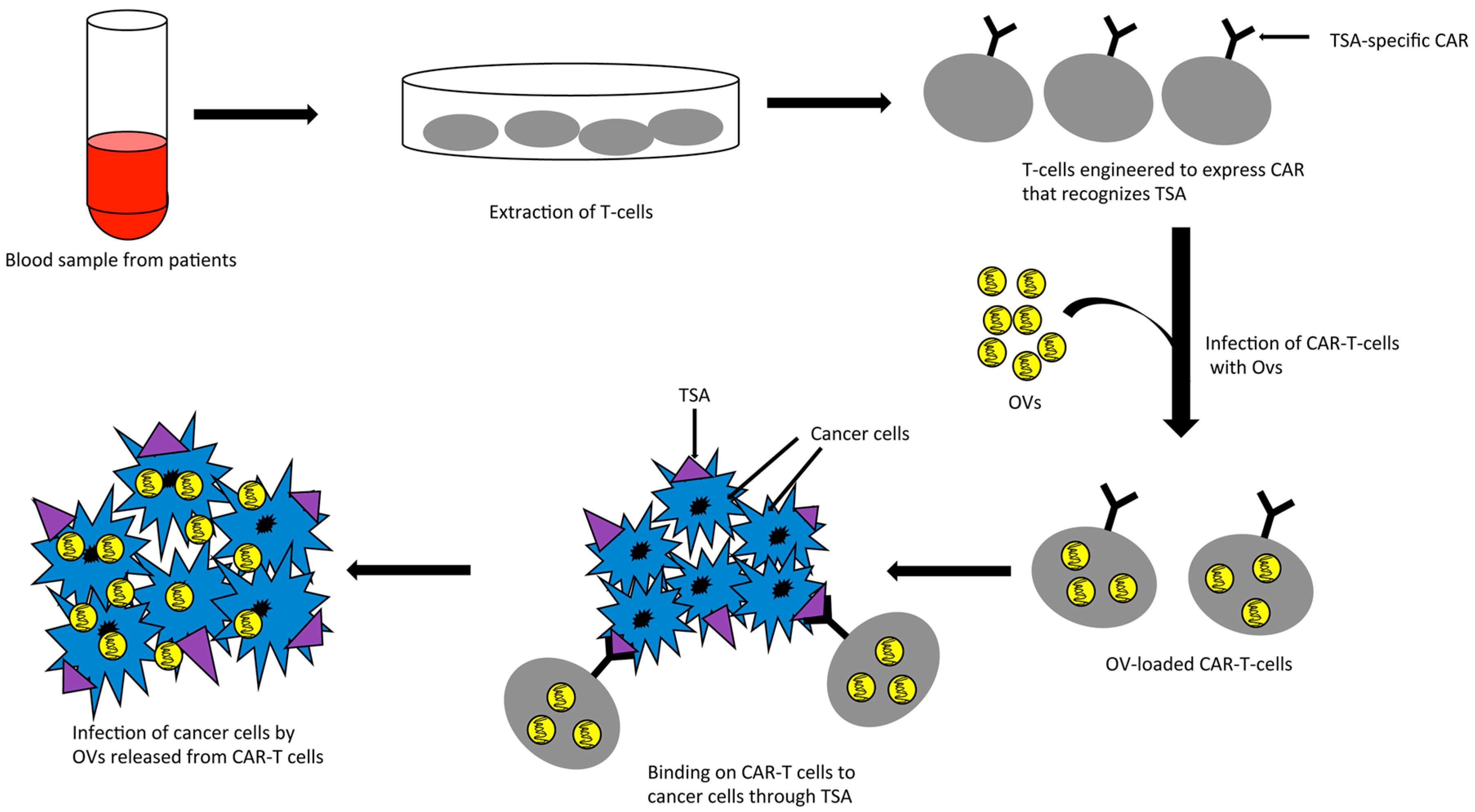
7. Combination of Oncolytic Viruses with T Cells Expressing Chimeric Antigen Receptor
8. Immune Analysis in Clinical Trials Examining Oncolytic Virus Versus Colorectal Cancer
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- Siegel, R.; Desantis, C.; Jemal, A. Colorectal cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Melcher, A.; Parato, K.; Rooney, C.M.; Bell, J.C. Thunder and lightning: Immunotherapy and oncolytic viruses collide. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Tsun, A.; Miao, X.N.; Wang, C.M.; Yu, D.C. Oncolytic Immunotherapy for Treatment of Cancer; Zhang, S., Ed.; Springer: Dordrecht, The Netherlands, 2016. [Google Scholar]
- Fulci, G.; Breymann, L.; Gianni, D.; Kurozomi, K.; Rhee, S.S.; Yu, J.; Kaur, B.; Louis, D.N.; Weissleder, R.; Caligiuri, M.A.; et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12873–12878. [Google Scholar] [CrossRef] [PubMed]
- Lun, X.Q.; Jang, J.H.; Tang, N.; Deng, H.; Head, R.; Bell, J.C.; Stojdl, D.F.; Nutt, C.L.; Senger, D.L.; Forsyth, P.A.; et al. Efficacy of systemically administered oncolytic vaccinia virotherapy for malignant gliomas is enhanced by combination therapy with rapamycin or cyclophosphamide. Clin. Cancer Res. 2009, 15, 2777–2788. [Google Scholar] [CrossRef]
- Parato, K.A.; Senger, D.; Forsyth, P.A.; Bell, J.C. Recent progress in the battle between oncolytic viruses and tumours. Nat. Rev. Cancer 2005, 5, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.J.; Errington, F.; Diaz, R.M.; Pandha, H.S.; Harrington, K.J.; Melcher, A.A.; Vile, R.G. The case of oncolytic viruses versus the immune system: Waiting on the judgment of Solomon. Hum. Gene Ther. 2009, 20, 1119–1132. [Google Scholar] [CrossRef] [PubMed]
- Blankenstein, T.; Coulie, P.G.; Gilboa, E.; Jaffee, E.M. The determinants of tumour immunogenicity. Nat. Rev. Cancer 2012, 12, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Komenaka, I.; Hoerig, H.; Kaufman, H.L. Immunotherapy for melanoma. Clin. Dermatol. 2004, 22, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, A.D.; Poletajew, S.; Wasiutynski, A. Spontaneous regression of renal cell carcinoma. Contemp. Oncol. (Pozn) 2013, 17, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Day, C.L., Jr.; Sober, A.J.; Kopf, A.W.; Lew, R.A.; Mihm, M.C., Jr.; Hennessey, P.; Golomb, F.M.; Harris, M.N.; Gumport, S.L.; Raker, J.W.; et al. A prognostic model for clinical stage I melanoma of the upper extremity. The importance of anatomic subsites in predicting recurrent disease. Ann. Surg. 1981, 193, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Belldegrun, A.; Muul, L.M.; Rosenberg, S.A. Interleukin 2 expanded tumor-infiltrating lymphocytes in human renal cell cancer: Isolation, characterization, and antitumor activity. Cancer Res. 1988, 48, 206–214. [Google Scholar] [PubMed]
- Itsumi, M.; Tatsugami, K. Immunotherapy for renal cell carcinoma. Clin. Dev. Immunol. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L. Vaccines for melanoma and renal cell carcinoma. Semin. Oncol. 2012, 39, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.A.; Page, D.L.; Holt, J.T. Identification of genes expressed in premalignant breast disease by microscopy-directed cloning. Proc. Natl. Acad. Sci. USA 1994, 91, 9257–9261. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, H.; Maeurer, M.J.; Jager, E.; Wolfel, T.; Schneider, J.; Karbach, J.; Seliger, B.; Huber, C.; Storkus, W.S.; Lotze, M.T.; et al. Recognition of human renal cell carcinoma and melanoma by HLA-A2-restricted cytotoxic T lymphocytes is mediated by shared peptide epitopes and up-regulated by interferon-γ. Scand. J. Immunol. 1996, 44, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A. Development of cancer immunotherapies based on identification of the genes encoding cancer regression antigens. J. Natl. Cancer Inst. 1996, 88, 1635–1644. [Google Scholar] [CrossRef] [PubMed]
- De Vries, N.L.; Swets, M.; Vahrmeijer, A.L.; Hokland, M.; Kuppen, P.J. The Immunogenicity of Colorectal Cancer in Relation to Tumor Development and Treatment. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Kloor, M.; Michel, S.; von Knebel Doeberitz, M. Immune evasion of microsatellite unstable colorectal cancers. Int. J. Cancer 2010, 127, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Dalerba, P.; Maccalli, C.; Casati, C.; Castelli, C.; Parmiani, G. Immunology and immunotherapy of colorectal cancer. Crit. Rev. Oncol. Hematol. 2003, 46, 33–57. [Google Scholar] [CrossRef]
- Jochems, C.; Schlom, J. Tumor-infiltrating immune cells and prognosis: The potential link between conventional cancer therapy and immunity. Exp. Biol. Med. 2011, 236, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Van der Burg, S.H.; Arens, R.; Ossendorp, F.; van Hall, T.; Melief, C.J. Vaccines for established cancer: Overcoming the challenges posed by immune evasion. Nat. Rev. Cancer 2016, 16, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.Y. Therapeutic cancer vaccines: Past, present, and future. Adv. Cancer Res. 2013, 119, 421–475. [Google Scholar] [PubMed]
- Schlag, P.; Manasterski, M.; Gerneth, T.; Hohenberger, P.; Dueck, M.; Herfarth, C.; Liebrich, W.; Schirrmacher, V. Active specific immunotherapy with Newcastle-disease-virus-modified autologous tumor cells following resection of liver metastases in colorectal cancer. First evaluation of clinical response of a phase II-trial. Cancer Immunol. Immunother. CII 1992, 35, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Ockert, D.; Schirrmacher, V.; Beck, N.; Stoelben, E.; Ahlert, T.; Flechtenmacher, J.; Hagmüller, E.; Buchcik, R.; Nagel, M.; Saeger, H.D. Newcastle disease virus-infected intact autologous tumor cell vaccine for adjuvant active specific immunotherapy of resected colorectal carcinoma. Clin. Cancer Res. 1996, 2, 21–28. [Google Scholar] [PubMed]
- Schulze, T.; Kemmner, W.; Weitz, J.; Wernecke, K.D.; Schirrmacher, V.; Schlag, P.M. Efficiency of adjuvant active specific immunization with Newcastle disease virus modified tumor cells in colorectal cancer patients following resection of liver metastases: Results of a prospective randomized trial. Cancer Immunol. Immunother. CII 2009, 58, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Ura, T.; Okuda, K.; Shimada, M. Developments in Viral Vector-Based Vaccines. Vaccines (Basel) 2014, 2, 624–641. [Google Scholar] [CrossRef] [PubMed]
- Larocca, C.; Schlom, J. Viral vector-based therapeutic cancer vaccines. Cancer J. 2011, 17, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Moss, B. Genetically engineered poxviruses for recombinant gene expression, vaccination, and safety. Proc. Natl. Acad. Sci. USA 1996, 93, 11341–11348. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kay, M.A.; Glorioso, J.C.; Naldini, L. Viral vectors for gene therapy: The art of turning infectious agents into vehicles of therapeutics. Nat. Med. 2001, 7, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Xiang, B.; Snook, A.E.; Magee, M.S.; Waldman, S.A. Colorectal cancer immunotherapy. Discov. Med. 2013, 15, 301–308. [Google Scholar] [PubMed]
- Rahma, O.E.; Khleif, S.N. Therapeutic vaccines for gastrointestinal cancers. Gastroenterol. Hepatol. (N Y) 2011, 7, 517–564. [Google Scholar]
- Lynch, D.; Murphy, A. The emerging role of immunotherapy in colorectal cancer. Ann. Transl. Med. 2016, 4, 305. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.L.; Gulley, J.L.; Arlen, P.M.; Beetham, P.K.; Tsang, K.Y.; Slack, R.; Hodge, J.W.; Doren, S.; Grosenbach, D.W.; Hwang, J.; et al. Phase I study of sequential vaccinations with fowlpox-CEA(6D)-TRICOM alone and sequentially with vaccinia-CEA(6D)-TRICOM, with and without granulocyte-macrophage colony-stimulating factor, in patients with carcinoembryonic antigen-expressing carcinomas. J. Clin. Oncol. 2005, 23, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Mullen, J.T.; Tanabe, K.K. Viral oncolysis. Oncologist 2002, 7, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Andreansky, S.; He, B.; van Cott, J.; McGhee, J.; Markert, J.M.; Gillespie, G.Y.; Roizman, B.; Whitley, R.J. Treatment of intracranial gliomas in immunocompetent mice using herpes simplex viruses that express murine interleukins. Gene Ther. 1998, 5, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.N.; Gillespie, G.Y.; Love, C.E.; Randall, S.; Whitley, R.J.; Markert, J.M. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc. Natl. Acad. Sci. USA 2000, 97, 2208–2213. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Oh, J.Y.; Park, B.H.; Lee, D.E.; Kim, J.S.; Park, H.E.; Roh, M.S.; Je, J.E.; Yoon, J.H.; Thorne, S.H.; et al. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol. Ther. 2006, 14, 361–370. [Google Scholar] [CrossRef]
- Han, Z.Q.; Assenberg, M.; Liu, B.L.; Wang, Y.B.; Simpson, G.; Thomas, S.; Coffin, R.S. Development of a second-generation oncolytic Herpes simplex virus expressing TNFα for cancer therapy. J. Gene Med. 2007, 9, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Heiber, J.F.; Barber, G.N. Vesicular stomatitis virus expressing tumor suppressor p53 is a highly attenuated, potent oncolytic agent. J. Virol. 2011, 85, 10440–10450. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.L.; Yu, Y.H.; Tian, H.; Ren, G.P.; Wang, H.; Zhou, B.; Han, X.H.; Yu, Q.Z.; Li, D.S. Genetically engineered Newcastle disease virus expressing interleukin-2 and TNF-related apoptosis-inducing ligand for cancer therapy. Cancer Biol. Ther. 2014, 15, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Takasu, A.; Masui, A.; Hamada, M.; Imai, T.; Iwai, S.; Yura, Y. Immunogenic cell death by oncolytic herpes simplex virus type 1 in squamous cell carcinoma cells. Cancer Gene Ther. 2016, 23, 107–113. [Google Scholar] [CrossRef]
- Angelova, A.L.; Grekova, S.P.; Heller, A.; Kuhlmann, O.; Soyka, E.; Giese, T.; Aprahamian, M.; Bour, G.; Rüffer, S.; Cziepluch, C.; et al. Complementary induction of immunogenic cell death by oncolytic parvovirus H-1PV and gemcitabine in pancreatic cancer. J. Virol. 2014, 88, 5263–5276. [Google Scholar] [CrossRef] [PubMed]
- Workenhe, S.T.; Mossman, K.L. Oncolytic virotherapy and immunogenic cancer cell death: Sharpening the sword for improved cancer treatment strategies. Mol. Ther. 2014, 22, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Yamano, T.; Kubo, S.; Fukumoto, M.; Yano, A.; Mawatari-Furukawa, Y.; Okamura, H.; Tomita, N. Whole cell vaccination using immunogenic cell death by an oncolytic adenovirus is effective against a colorectal cancer model. Mol. Ther. Oncol. 2016, 3, 16031. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; Thompson, C.B. At the bench: Preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy. J. Leukoc. Biol. 2013, 94, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Boutros, C.; Tarhini, A.; Routier, E.; Lambotte, O.; Ladurie, F.L.; Carbonnel, F.; Izzeddine, H.; Marabelle, A.; Champiat, S.; Berdelou, A.; et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat. Rev. Clin. Oncol. 2016, 13, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Marincola, F.M. 2015: The Year of Anti-PD-1/PD-L1s Against Melanoma and Beyond. EBioMedicine 2015, 2, 92–93. [Google Scholar] [CrossRef] [PubMed]
- West, H.J. JAMA Oncology Patient Page. Immune Checkpoint Inhibitors. JAMA Oncol. 2015, 1, 115. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.Y.; Gore, I.; Fong, L.; Venook, A.; Beck, S.B.; Dorazio, P.; Criscitiello, P.J.; Healey, D.I.; Huang, B.; Gomez-Navarro, J.; et al. Phase II study of the anti-cytotoxic T-lymphocyte-associated antigen 4 monoclonal antibody, tremelimumab, in patients with refractory metastatic colorectal cancer. J. Clin. Oncol. 2010, 28, 3485–3490. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.P.; Sharma, P.K.; Krishnan, G.; Lockhart, A.C. Immune checkpoints and immunotherapy for colorectal cancer. Gastroenterol. Rep. (Oxf) 2015, 3, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [CrossRef] [PubMed]
- Banerjea, A.; Ahmed, S.; Hands, R.E.; Huang, F.; Han, X.; Shaw, P.M.; Feakins, R.; Bustin, S.A.; Dorudi, S. Colorectal cancers with microsatellite instability display mRNA expression signatures characteristic of increased immunogenicity. Mol. Cancer. 2004, 3, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer-the stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 2016, 14. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Whitaker-Dowling, P.; Griffin, J.A.; Barmada, M.A.; Bergman, I. Recombinant vesicular stomatitis virus targeted to Her2/neu combined with anti-CTLA4 antibody eliminates implanted mammary tumors. Cancer Gene Ther. 2009, 16, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.J.; Sampath, P.; Hou, W.; Thorne, S.H. Defining Effective Combinations of Immune Checkpoint Blockade and Oncolytic Virotherapy. Clin. Cancer Res. 2015, 21, 5543–5551. [Google Scholar] [CrossRef] [PubMed]
- Sobol, R.E.; Shawler, D.L.; Carson, C.; Van Beveren, C.; Mercola, D.; Fakhrai, H.; Garrett, M.A.; Barone, R.; Goldfarb, P.; Bartholomew, R.M.; et al. Interleukin 2 gene therapy of colorectal carcinoma with autologous irradiated tumor cells and genetically engineered fibroblasts: A Phase I study. Clin. Cancer Res. 1999, 5, 2359–2365. [Google Scholar] [PubMed]
- Conry, R.M.; Khazaeli, M.B.; Saleh, M.N.; Allen, K.O.; Barlow, D.L.; Moore, S.E.; Craig, D.; Arani, R.B.; Schlom, J.; LoBuglio, A.F. Phase I trial of a recombinant vaccinia virus encoding carcinoembryonic antigen in metastatic adenocarcinoma: Comparison of intradermal versus subcutaneous administration. Clin. Cancer Res. 1999, 5, 2330–2337. [Google Scholar] [PubMed]
- Morse, M.A.; Chaudhry, A.; Gabitzsch, E.S.; Hobeika, A.C.; Osada, T.; Clay, T.M.; Amalfitano, A.; Burnett, B.K.; Devi, G.R.; Hsu, D.S.; et al. Novel adenoviral vector induces T-cell responses despite anti-adenoviral neutralizing antibodies in colorectal cancer patients. Cancer Immunol. Immunother. CII 2013, 62, 1293–1301. [Google Scholar] [CrossRef] [PubMed]
- Neidhart, J.; Allen, K.O.; Barlow, D.L.; Carpenter, M.; Shaw, D.R.; Triozzi, P.L.; Conry, R.M. Immunization of colorectal cancer patients with recombinant baculovirus-derived KSA (Ep-CAM) formulated with monophosphoryl lipid A in liposomal emulsion, with and without granulocyte-macrophage colony-stimulating factor. Vaccine 2004, 22, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Ullenhag, G.J.; Frodin, J.E.; Mosolits, S.; Kiaii, S.; Hassan, M.; Bonnet, M.C.; Moingeon, P.; Mellstedt, H.; Rabbani, H. Immunization of colorectal carcinoma patients with a recombinant canarypox virus expressing the tumor antigen Ep-CAM/KSA (ALVAC-KSA) and granulocyte macrophage colony—Stimulating factor induced a tumor-specific cellular immune response. Clin. Cancer Res. 2003, 9, 2447–2456. [Google Scholar] [PubMed]
- Von Mehren, M.; Arlen, P.; Tsang, K.Y.; Rogatko, A.; Meropol, N.; Cooper, H.S.; Davey, M.; McLaughlin, S.; Schlom, J.; Weiner, L.M. Pilot study of a dual gene recombinant avipox vaccine containing both carcinoembryonic antigen (CEA) and B7.1 transgenes in patients with recurrent CEA-expressing adenocarcinomas. Clin. Cancer Res. 2000, 6, 2219–2228. [Google Scholar] [PubMed]
- Marshall, J.L.; Hoyer, R.J.; Toomey, M.A.; Faraguna, K.; Chang, P.; Richmond, E.; Pedicano, J.E.; Gehan, E.; Peck, R.A.; Arlen, P.; et al. Phase I study in advanced cancer patients of a diversified prime-and-boost vaccination protocol using recombinant vaccinia virus and recombinant nonreplicating avipox virus to elicit anti-carcinoembryonic antigen immune responses. J. Clin. Oncol. 2000, 18, 3964–3973. [Google Scholar] [CrossRef] [PubMed]
- Hodge, J.W.; Grosenbach, D.W.; Aarts, W.M.; Poole, D.J.; Schlom, J. Vaccine therapy of established tumors in the absence of autoimmunity. Clin. Cancer Res. 2003, 9, 1837–1849. [Google Scholar] [PubMed]
- Gulley, J.L.; Arlen, P.M.; Tsang, K.Y.; Yokokawa, J.; Palena, C.; Poole, D.J.; Remondo, C.; Cereda, V.; Jones, J.L.; Pazdur, M.P.; et al. Pilot study of vaccination with recombinant CEA-MUC-1-TRICOM poxviral-based vaccines in patients with metastatic carcinoma. Clin. Cancer Res. 2008, 14, 3060–3069. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Teachey, D.T.; Porter, D.L.; Grupp, S.A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 2015, 125, 4017–4023. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Wang, Y.; Lu, X.; Han, W. Chimeric Antigen Receptors Modified T-Cells for Cancer Therapy. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Kakarla, S.; Gottschalk, S. CAR T cells for solid tumors: Armed and ready to go? Cancer J. 2014, 20, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Abken, H. Adoptive therapy with CAR redirected T cells: The challenges in targeting solid tumors. Immunotherapy 2015, 7, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Yu, D.; Essand, M. Prospects to improve chimeric antigen receptor T-cell therapy for solid tumors. Immunotherapy 2016, 8, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.A.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Nishio, N.; Diaconu, I.; Liu, H.; Cerullo, V.; Caruana, I.; Hoyos, V.; Bouchier-Hayes, L.; Savoldo, B.; Dotti, G. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 2014, 74, 5195–5205. [Google Scholar] [CrossRef] [PubMed]
- Thorne, S.H.; Negrin, R.S.; Contag, C.H. Synergistic antitumor effects of immune cell-viral biotherapy. Science 2006, 311, 1780–1784. [Google Scholar] [CrossRef] [PubMed]
- Edinger, M.; Cao, Y.A.; Verneris, M.R.; Bachmann, M.H.; Contag, C.H.; Negrin, R.S. Revealing lymphoma growth and the efficacy of immune cell therapies using in vivo bioluminescence imaging. Blood 2003, 101, 640–648. [Google Scholar] [CrossRef] [PubMed]
- VanSeggelen, H.; Tantalo, D.G.; Afsahi, A.; Hammill, J.A.; Bramson, J.L. Chimeric antigen receptor-engineered T cells as oncolytic virus carriers. Mol. Ther. Oncol. 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Kemeny, N.; Brown, K.; Covey, A.; Kim, T.; Bhargava, A.; Brody, L.; Guilfoyle, B.; Haag, N.P.; Karrasch, M.; Glasschroeder, B.; et al. Phase, I.; open-label, dose-escalating study of a genetically engineered herpes simplex virus, NV1020, in subjects with metastatic colorectal carcinoma to the liver. Hum. Gene Ther. 2006, 17, 1214–1224. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Breitbach, C.J.; Lee, J.; Park, J.O.; Lim, H.Y.; Kang, W.K.; Moon, A.; Mun, J.H.; Sommermann, E.M.; Avidal, L.M.; et al. Phase 1b Trial of Biweekly Intravenous Pexa-Vec (JX-594), an Oncolytic and Immunotherapeutic Vaccinia Virus in Colorectal Cancer. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 1532–1540. [Google Scholar] [CrossRef] [PubMed]
- Balint, J.P.; Gabitzsch, E.S.; Rice, A.; Latchman, Y.; Xu, Y.; Messerschmidt, G.L.; Chaudhry, A.; Morse, M.A.; Jones, F.R. Extended evaluation of a phase 1/2 trial on dosing, safety, immunogenicity, and overall survival after immunizations with an advanced-generation Ad5 [E1-, E2b-]-CEA(6D) vaccine in late-stage colorectal cancer. Cancer Immunol. Immunother. CII 2015, 64, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Pecora, A.L.; Rizvi, N.; Cohen, G.I.; Meropol, N.J.; Sterman, D.; Marshall, J.L.; Goldberg, S.; Gross, P.; O’neil, J.D.; Groene, W.S.; et al. Phase I trial of intravenous administration of PV701, an oncolytic virus, in patients with advanced solid cancers. J. Clin. Oncol. 2002, 20, 2251–2266. [Google Scholar] [CrossRef] [PubMed]
- Calvo, E.; Gil-Martin, M.; Machiels, J.-P.; Rottey, S.; Cubillo, A.; Salazar, R.; Mardjuadi, F.I.; Geboes, K.P.; Ellis, C.; Beadle, J.W.; et al. (Eds.) A first-in-class, first-in-human phase I study of enadenotucirev, an oncolytic Ad11/Ad3 chimeric group B adenovirus, administered intravenously in patients with metastatic epithelial tumors. In Proceedings of the 2014 ASCO Annual meeting, Chicago, IL, USA, 30 May–3 June 2014.

{kind=link}
| Virus | Treatment Type | Transgene (Tumor Antigen or Cytokine) | Phase of Trial | Outcome | Immune Response | References |
|---|---|---|---|---|---|---|
| Retrovirus | Therapeutic vaccination | IL-2 | I | No objective response demonstrated | Tumor-specific CTL | [68] |
| Vaccinia virus | Therapeutic vaccination | CEA | I | No objective response | Not reported | [69] |
| Adenovirus | Therapeutic vaccination | CEA | I | Increased overall survival | CEA-specific immunity | [70] |
| Adenovirus | Therapeutic vaccination | GUCY2C | I | Not published | GUCY2C-specific antibody and T-cell responses | NCT01972737 |
| Baculovirus | Therapeutic vaccination | Ep-CAM | I | Not published | Ep-CAM-specific cellular immune response | [71] |
| Canarypox virus | Therapeutic vaccination | Ep-CAM | I | Not published | Ep-CAM-specific cellular immune response | [72] |
| Avipox virus | Therapeutic vaccination | CEA, B7-1 | Pilot | Stable disease in some patients | CEA-specific CTL | [73] |
| Vaccinia + Avipox virus | Therapeutic vaccination | CEA | I | No objective anti-tumor response | Antibody against CEA | [74] |
| Vaccinia + Fowlpox | Therapeutic vaccination | CEA, B7-1, ICAM-1, LFA-3 | I | Stable disease in some patients | CEA-specific CTL | [75,76] |
| Vaccinia virus | Oncolytic virotherapy | GM-CSF | I | Not published | Not published | NCT01394939 |
| Herpes simplex virus | Oncolytic virotherapy | None | I | Not published | Not published | NCT00149396 |
| Adenovirus | Oncolytic virotherapy | None | I | Not published | Not published | NCT02028442 |
| Authors & Year | Vector | Phase | N | Delivery | Results | Adverse Effects | Immune Investigations |
|---|---|---|---|---|---|---|---|
| Kemeny & Fong 2006 [88] NCT00149396 | NV1020 HSV+ GMCSF | I | 12 | 3 × 106 3 × 107 1 × 108 | GGT rise, diarrhea, elev WBC | TNF-α, IL-2, IL-1, IFN-γ, CD4+/CD8+ ratio | |
| Calvo 2014 [92] NCT02028442 | Ad11/ad3 Enadenotucirev | I/II | 161 | 1 × 1010–6 × 1012 | No survival data reported yet | Flu-like sx, elevated GGT | Elevated TNF, IFN, IL-6, and IL-12 on Day 1 after higher doses |
| Park SH 2015 [89] NCT01380600 | JX-594 tk attenuated Vaccinia | Ib | 15 | Up to 4 IV q14 days Dose 1 × 106 pfu/kg, 1 × 107, 3 × 107 | 67% stable disease | Pox skin lesions Flu like symptoms | IL-2, IL-6, IL-8, IL-10, IL-18, MIP-1α, MCP-1, MIP-1β, and TNF-α |
| Balint 2015 [90] NCT02028442 | A11/Ad3 group B adenovirus | I/II | 32 | 1 × 109 q3 weeks × 3 1 × 1010 q3 weeks × 3 1 × 1011 q3 weeks × 3 5 × 1011 q3 weeks × 3 | No objective ant-tumor responses; Median survival 13mos in optimal tx grp | Injection site rxn Fever, flu-like symptoms | Cytolytic T cell responses IFN-γ TNF-α |
| NCT01274624 | Reolysin + Folfiri + avastin in Folfiri naïve KRAS mutants | I | 12 | No data reported yet Due Fall 2017 | |||
| NCT02636036 | Ad11/Ad3 Enadenotucirev + Anti-PD-1 | I | Study completion June 2019 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaurasiya, S.; Warner, S. Viroimmunotherapy for Colorectal Cancer: Clinical Studies. Biomedicines 2017, 5, 11. https://doi.org/10.3390/biomedicines5010011
Chaurasiya S, Warner S. Viroimmunotherapy for Colorectal Cancer: Clinical Studies. Biomedicines. 2017; 5(1):11. https://doi.org/10.3390/biomedicines5010011
Chicago/Turabian StyleChaurasiya, Shyambabu, and Susanne Warner. 2017. "Viroimmunotherapy for Colorectal Cancer: Clinical Studies" Biomedicines 5, no. 1: 11. https://doi.org/10.3390/biomedicines5010011
APA StyleChaurasiya, S., & Warner, S. (2017). Viroimmunotherapy for Colorectal Cancer: Clinical Studies. Biomedicines, 5(1), 11. https://doi.org/10.3390/biomedicines5010011