CDK5: Key Regulator of Apoptosis and Cell Survival



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
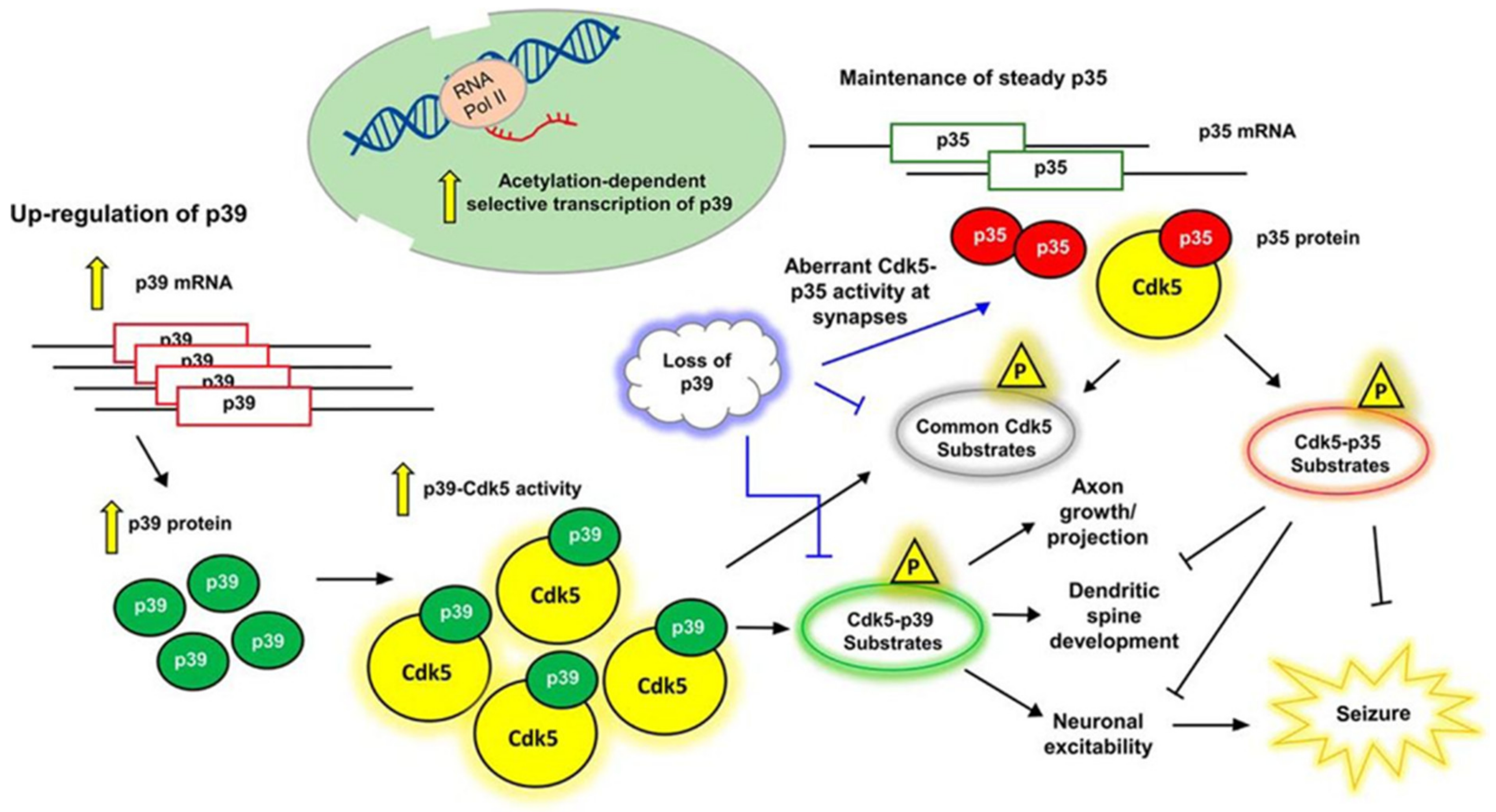
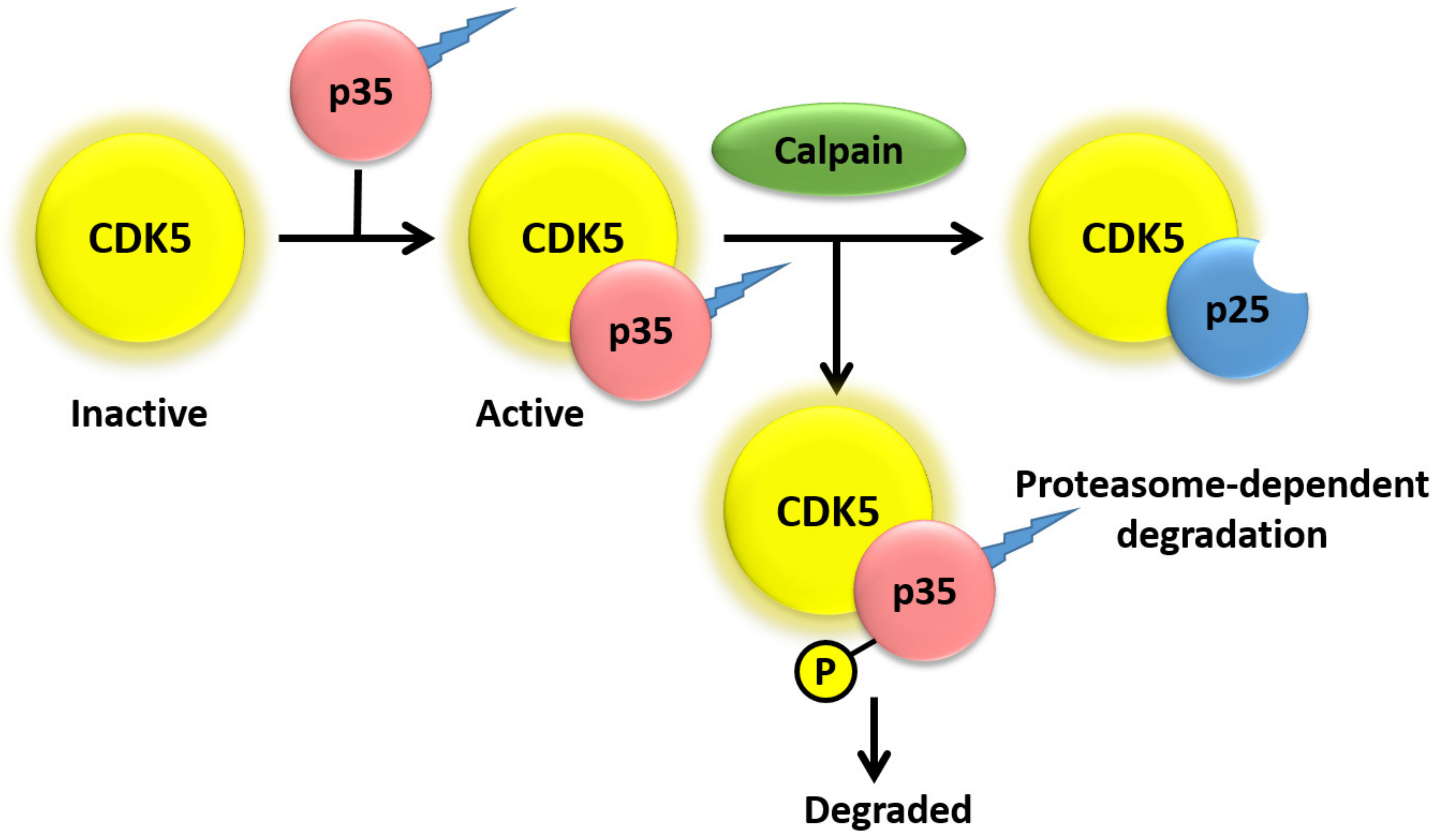
2. Activators of CDK5
3. Regulation of CDK5 Activity
4. Function of CDK5 in Neuronal Development
5. Function of CDK5 in Non-Neuronal Cells
6. Function of CDK5 in Autophagy
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dhariwala, F.A.; Rajadhyaksha, M.S. The unusual member of the Cdk family: Cdk5. Cell Mol. Neurobiol. 2008, 28, 351–369. [Google Scholar] [CrossRef]
- Hellmich, M.R.; Pant, H.C.; Wada, E.; Battey, J.F. Neuronal cdc2-like kinase: A cdc2-related protein kinase with predominantly neuronal expression. Proc. Natl. Acad. Sci. USA 1992, 88, 10867–10871. [Google Scholar] [CrossRef]
- Ishiguro, K.; Takamatsu, M.; Tomizawa, K.; Omori, A.; Takahashi, M.; Arioka, M.; Uchida, T.; Imahori, K. Tau protein kinase I converts normal tau protein into A68-like component of paired helical filaments. J. Biol. Chem. 1992, 267, 10897–10901. [Google Scholar]
- Cheung, Z.H.; Ip, N.Y. Cdk5: A multifaceted kinase in neurodegenerative diseases. Trends Cell Biol. 2012, 22, 169–175. [Google Scholar] [CrossRef]
- Shukla, V.; Skuntz, S.; Pant, H.C. Deregulated Cdk5 activity is involved in inducing Alzheimer’s disease. Arch. Med. Res. 2012, 43, 655–662. [Google Scholar] [CrossRef]
- Kimura, T.; Ishiguro, K.; Hisanaga, S.I. Physiological and pathological phosphorylation of tau by Cdk5. Front. Mol. Neurosci. 2014, 7, 1–10. [Google Scholar] [CrossRef]
- Imahori, K.; Uchida, T. Physiology and pathology of tau protein kinases in relation to Alzheimer’s disease. J. Biochem. 1997, 121, 179–188. [Google Scholar]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Involvement of aberrant glycosylation in phosphorylation of tau by cdk5 and GSK-3β. FEBS Lett. 2002, 530, 209–214. [Google Scholar] [CrossRef]
- Lew, J.; Beaudette, K.; Litwin, C.M.E.; Wang, J.H. Purification and characterization of a novel proline-directed protein kinase from bovine brain. J. Biol. Chem. 1992, 267, 13383–13390. [Google Scholar]
- Kobayashi, S.; Ishiguro, K.; Omori, A.; Takamatsu, M.; Arioka, M.; Imahori, K.; Uchida, T. A cdc2-related kinase PSSALRE/cdk5 is homologous with the 30 kDa subunit of tau protein kinase II, a proline-directed protein kinase associated with microtubule. FEBS Lett. 1993, 335, 171–175. [Google Scholar] [CrossRef] [Green Version]
- Shetty, K.T.; Link, W.T.; Pant, H.C. Cdc2-like kinase from rat spinal cord specifically phosphorylates KSPXK motifs in neurofilament proteins: Isolation and characterization. Proc. Natl. Acad. Sci. USA 1993, 90, 6844–6848. [Google Scholar] [CrossRef]
- Ko, J.; Humbert, S.; Bronson, R.T.; Takahashi, S.; Kulkarni, A.B.; Li, E.; Tsai, L.H. P35 and p39 are essential for cyclin-dependant kinase 5 function during neurodevelopment. J. Neurosci. 2001, 21, 6758–6771. [Google Scholar] [CrossRef]
- Paglini, G.; Cáceres, A. The role of the Cdk5-p35 kinase in neuronal development. Eur. J. Biochem. 2001, 268, 1528–1533. [Google Scholar] [CrossRef]
- Lee, K.Y.; Rosales, J.L.; Tang, D.; Wang, J.H. Interaction of cyclin-dependent kinase 5 (Cdk5) and neuronal Cdk5 activator in bovine brain. J. Biol. Chem. 1996, 271, 1538–1543. [Google Scholar] [CrossRef]
- Mapelli, M.; Musacchino, A. The structural perspective on CDK5. Neurosignals 2003, 12, 164–172. [Google Scholar] [CrossRef]
- Tarricone, C.; Dhavan, R.; Peng, J.; Areces, L.B.; Tsai, L.; Musacchino, A. Structure and regulation of the CDK5-p25(nck5a) complex. Mol. Cell. 2001, 8, 657–669. [Google Scholar] [CrossRef]
- Lew, J.; Huang, Q.; Qi, Z.; Winkfein, R.J.; Aebersold, R.; Hunt, T.; Wang, J.H. A brain-specific activator of cyclin-dependant kinase 5. Nature 1994, 371, 423–426. [Google Scholar] [CrossRef]
- Tsai, L.; Delalle, I.; Caviness, J.R.; Chae, T.; Harlow, E. P35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 1994, 371, 419–423. [Google Scholar] [CrossRef]
- Zeb, A.; Son, M.; Yoon, S.; Kim, J.H.; Park, S.J.; Lee, K.W. Computational Simulations Identified Two Candidate Inhibitors of Cdk5/p25 to Abrogate Tau-associated Neurological Disorders. Comput. Struct. Biotechnol. J. 2019, 17, 579–590. [Google Scholar] [CrossRef]
- Tang, D.; Yeung, J.; Lee, K.; Matsushita, M.; Matsui, H.; Tomizawa, K.; Hatase, O.; Wang, J.H. An isoform of the neuronal cyclin-dependent kinase 5 (Cdk5) activator. J. Biol. Chem. 1995, 270, 26897–26903. [Google Scholar] [CrossRef]
- Patrick, G.N.; Zukerberg, L.; Nikolic, M.; de La Monte, S.; Dikkes, P.; Tsai, L.H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 1999, 402, 615–622. [Google Scholar] [CrossRef]
- Kusakawa, G.; Saito, T.; Onuki, R.; Ishiguro, K.; Kishimoto, T.; Hisanaga, S. Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J. Biol. Chem. 2000, 275, 17166–17172. [Google Scholar] [CrossRef]
- Gupta, K.K.; Singh, S.K. Cdk5: A main culprit in neurodegeneration. Int. J. Neurosci. 2019, 129, 1192–1197. [Google Scholar] [CrossRef]
- Kamei, H.; Saito, T.; Ozawa, M.; Fujita, Y.; Asada, A.; Bibb, J.A.; Saido, T.C.; Sorimachi, H.; Hisanaga, S. Suppression of calpain-dependent cleavage of the CDK5 activator p35 to p25 by site-specific phosphorylation. J. Biol. Chem. 2007, 282, 1687–1694. [Google Scholar] [CrossRef]
- Patzke, H.; Maddineni, U.; Ayala, R.; Morabito, M.; Janet, V.; Dikkes, P.; Ahlijanian, M.K.; Tsai, L.H. Partial rescue of the P35-/- brain phenotype by low expression of neuronal-specific enolase p25 transgene. J. Neurosci. 2003, 23, 2769–2778. [Google Scholar] [CrossRef]
- Morgan, D.O. Principles of CDK regulation. Nature 1995, 374, 131–134. [Google Scholar] [CrossRef]
- Brown, N.R.; Noble, M.E.; Endicott, J.A.; Garman, E.F.; Wakatsuki, S.; Mitchell, E.; Rasmussen, B.; Hunt, T.; Johnson, L.N. The crystal structure of cyclin A. Structure 1995, 3, 1235–1247. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Chun, A.C.S.; Zhang, M.; Wang, J.H. Cyclin-dependent kinase 5 (Cdk5) activation domain of neuronal Cdk5 activator. J. Biol. Chem. 1997, 272, 12318–12327. [Google Scholar] [CrossRef]
- Li, W.; Allen, M.E.; Rui, Y.; Ku, L.; Liu, G.; Bankston, A.N.; Zheng, J.Q.; Feng, Y. P39 Is Responsible for Increasing Cdk5 Activity during Postnatal Neuron Differentiation and Governs Neuronal Network Formation and Epileptic Responses. J. Neurosci. 2016, 36, 11283–11294. [Google Scholar] [CrossRef]
- Wu, D.C.; Yu, Y.P.; Lee, N.T.; Yu, A.C.; Wang, J.H.; Han, Y.F. The expression of Cdk5, p35, p39, and Cdk5 kinase activity in developing, adult, and aged rat brains. Neurochem. Res. 2000, 25, 923–929. [Google Scholar] [CrossRef]
- Asada, A.; Yamamoto, N.; Gohda, M.; Saito, T.; Hayashi, N.; Hisanaga, S. Myristoylation of p39 and p35 is a determinant of cytoplasmic or nuclear localization of active cyclin-dependent kinase 5 complexes. J. Neurochem. 2008, 106, 1325–1336. [Google Scholar] [CrossRef]
- Hallows, J.L.; Chen, K.; DePinho, R.A.; Vincent, I. Decreased cyclin-dependent kinase 5 (Cdk5) activity is accompanied by redistribution of cdk5 and cytoskeletal proteins and increased cytoskeletal protein phosphorylation in p35 null mice. J. Neurosci. 2003, 23, 10633–10644. [Google Scholar] [CrossRef]
- Hisanaga, S.I.; Endo, R. Regulation and role of cylin-dependent kinase activity in neuronal survival and death. J. Neurochem. 2010, 115, 1309–1321. [Google Scholar] [CrossRef]
- Xiong, Y.; Zhang, H.; Beach, D. D type cyclins associate with multiple protein kinases and the DNA replication and repair factor PCNA. Cell 1992, 71, 505–514. [Google Scholar] [CrossRef]
- Miyajima, M.; Nornes, H.O.; Neuman, T. Cyclin E is expressed in neurons and forms complexes with cdk5. Neuroreport 1995, 6, 130–132. [Google Scholar] [CrossRef]
- Lalioti, V.; Pulido, D.; Sandoval, I.V. Cdk5, the multifunctional surveyor. Cell Cycle 2010, 9, 284–311. [Google Scholar] [CrossRef] [Green Version]
- Brinkkoetter, P.T.; Olivier, P.; Wu, J.S.; Henderson, S.; Krofft, R.D.; Pippin, J.W.; Hockenbery, D.; Roberts, J.M.; Shankland, S.J. Cyclin I activates Cdk5 and regulates expression of Bcl-2 and Bcl-xL in postmitotic mouse cells. J. Clin. Inv. 2009, 119, 3089–3101. [Google Scholar] [CrossRef]
- Brinkkoetter, P.T.; Pippin, J.W.; Shankland, S.J. Cyclin I-Cdk5 governs survival in post-mitotic cells. Cell Cycle 2010, 9, 1729–1731. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yang, X.; Zhang, H.; Li, X.; Zhang, Z.; Hou, L.; Wang, Z.; Niu, Q.; Wang, T. Neurotrophins and cholinergic enzyme regulated by calpain-2: New insights into neuronal apoptosis induced by polybrominated diphenyl ether-153. Toxicol. Lett. 2018, 291, 29–38. [Google Scholar] [CrossRef]
- Zhou, T.; Wang, H.; Shen, J.; Li, W.; Cao, M.; Hong, Y.; Cao, M. The p35/CDK5 signaling is regulated by p75NTR in neuronal apoptosis after intracerebral hemorrhage. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef]
- Tokuoka, H.; Saito, T.; Yorifuji, H.; Wei, F.Y.; Kishimoto, T.; Hisanaga, S.I. Brain-derived neurotrophic factor-induced phosphorylation of neurofilament-H subunit in primary cultures of embryo rat cortical neurons. J. Cell Sci. 2000, 116, 1059–1068. [Google Scholar]
- Bogush, A.; Pedrini, S.; Pelta-Heller, J.; Chan, T.; Yang, Q.; Mao, Z.; Sluzas, E.; Gieringer, T.; Ehrlich, M.E. AKT and CDK5/p35 mediate brain-derived neurotrophic factor induction of DARPP-32 in medium sized spiny neurons in vitro. J. Biol. Chem. 2007, 282, 7352–7359. [Google Scholar] [CrossRef]
- Harada, T.; Morooka, T.; Ogawa, S.; Nishida, E. ERK induces p35, a neuron-specific activator of Cdk5, through induction of Egr1. Nat. Cell Biol. 2001, 3, 453–459. [Google Scholar] [CrossRef]
- Li, T.; Chalifour, L.E.; Paudel, H.F. Phosphorylation of protein phosphatase 1 by cylin-dependent protein kinase 5 during nerve growth factor-induced PC12 cell differentiation. J. Biol. Chem. 2007, 228, 6619–6628. [Google Scholar] [CrossRef]
- Chang, Y.; Östling, P.; Åkerfelt, M.; Trouillet, D.; Rallu, M.; Gitton, Y.; El Fatimy, R.; Fardeau, V.; Le Crom, S.; Morange, M.; et al. Role of heat-shock factor 2 in cerebral cortex formation and as a regulator of p35 expression. Genes Dev. 2006, 20, 836–847. [Google Scholar] [CrossRef]
- Takahashi, T.; Saito, T.; Hisanaga, S.; Pant, H.C.; Kulkarni, A.B. Tau phosphorylation by cyclin-dependent kinase 5/p39 during brain development reduces its affinity for microtubules. J. Biol. Chem. 2003, 278, 10506–10515. [Google Scholar] [CrossRef]
- Zheng, M.; Leung, C.L.; Liem, R.K. Region-specific expression of cyclin-dependent kinase 5 (cdk5) and its activators, p35 and p39, in the developing and adult rat central nervous system. J. Neruobiol. 1998, 35, 141–159. [Google Scholar] [CrossRef]
- Patrick, G.N.; Zhou, P.; Kwon, Y.T.; Howley, P.M.; Tsai, L.H. p35, the neuronal-specific activator of cyclin-dependent kinase 5 (Cdk5) is degraded by the ubiquitin-proteasome pathway. J. Biol. Chem. 1998, 273, 24057–24064. [Google Scholar] [CrossRef]
- Endo, R.; Saito, T.; Asada, A.; Kawahara, H.; Ohshima, T.; Hisanaga, S. Commitment of 1-methyl-4-phenylpyrinidinium ion-induced neuronal cell death by proteasome-mediated degradation of p35 cyclin-dependant kinase 5 activator. J. Biol. Chem. 2009, 284, 26029–26039. [Google Scholar] [CrossRef]
- Zhao, C.T.; Li, K.; Zheng, W.; Liang, X.J.; Geng, A.Q.; Li, N.; Yuan, X.B. PKCdelta regulates cortical radial migration by stabilizing the Cdk5 activator p35. Proc. Natl. Acad. Sci. USA 2009, 106, 21353–21358. [Google Scholar] [CrossRef]
- Saito, T.; Oba, T.; Shimizu, S.; Asada, A.; Iijima, K.M.; Ando, K. Cdk5 increases MARK4 activity and augments pathological tau accumulation and toxicity through tau phosphorylation at Ser262. Hum. Mol. Genet. 2019. [Google Scholar] [CrossRef]
- Minegishi, S.; Asada, A.; Miyauchi, S.; Fuchigami, T.; Saito, T.; Hisanaga, S. Membrane association facilitates degradation and cleavage of the cyclin-dependent kinase 5 activators p35 and p39. Biochemistry 2010, 49, 5482–5493. [Google Scholar] [CrossRef]
- Liebl, J.; Fürst, R.; Vollmar, A.M.; Zahler, S. Twice switched at birth: Cell cycle-independent roles of the “neuron-specific” cyclin-dependent kinase 5 (Cdk5) in non-neuronal cells. Cell Signal 2011, 23, 1698–1707. [Google Scholar] [CrossRef]
- Lapresa, R.; Agulla, J.; Sánchez-Morán, I.; Zamarreño, R.; Prieto, E.; Bolaños, J.P.; Almeida, A. Amyloid-ß promotes neurotoxicity by Cdk5-induced p53 stabilization. Neuropharmacology 2018, 146, 19–27. [Google Scholar] [CrossRef]
- Qi, Z.; Huang, Q.Q.; Lee, K.Y.; Lew, J.; Wang, J.H. Reconstitution of neuronal Cdc2-like kinase from bacterial-expressed Cdk5 and an active fragment of the brain-specific activator. Kinase activation in the absence of Cdk5 phosphorylation. J. Biol. Chem. 1995, 270, 10847–10854. [Google Scholar] [CrossRef]
- Poon, R.Y.C.; Lew, J.; Hunter, T. Identification of functional domains in the neuronal Cdk5 activator protein. J. Biol. Chem. 1997, 272, 5703–5708. [Google Scholar] [CrossRef]
- Matsuura, I.; Wang, J.H. Demonstration of cyclin-dependent kinase inhibitory serine/threonine kinase in bovine thymus. J. Biol. Chem. 1996, 271, 5443–5450. [Google Scholar] [CrossRef]
- Zukerberg, L.R.; Patrick, G.N.; Nikolic, M.; Humbert, S.; Wu, C.L.; Lanier, L.M.; Gertler, F.B.; Vidal, M.; Van Etten, R.A.; Tsai, L.H. Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth. Neuron 2000, 26, 633–646. [Google Scholar] [CrossRef]
- Sasaki, Y.; Cheng, C.; Uchida, Y.; Nakajima, O.; Ohshima, T.; Yagi, T.; Taniguchi, M.; Nakayama, T.; Kishida, R.; Kudo, Y.; et al. Fyn and Cdk5 mediate semaphorin-3A signaling, which is involved in regulation of dendrite orientation in cerebral cortex. Neuron 2002, 35, 907–920. [Google Scholar] [CrossRef]
- Fu, W.Y.; Chen, Y.; Sahin, M.; Zhao, X.S.; Shi, L.; Bikoff, J.B.; Lai, K.O.; Yung, W.H.; Fu, A.K.; Greenberg, M.E.; et al. Cdk5 regulates EphA4-mediated dendritic spine retraction through an ephexin1-dependent mechanism. Nat. Neurosci. 2007, 10, 67–76. [Google Scholar] [CrossRef]
- Veselý, J.; Havlicek, L.; Strnad, M.; Blow, J.J.; Donella-Deana, A.; Pinna, L.; Letham, D.S.; Kato, J.; Detivaud, L.; Leclerc, S.; et al. Inhibition of cyclin-dependent kinases by purine analogues. Eur. J. Biochem. 1994, 224, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Meijer, L.; Borgne, A.; Mulner, O.; Chong, J.P.J.; Blow, J.; Inagaki, N.; Inagaki, M.; Delcros, J.G.; Moulinoux, J.P. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2, and cdk5. Eur. J. Biochem. 1997, 243, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Tomov, N.; Surchev, L.; Wiedenmann, C.; Döbrössy, M.; Nikkhah, G. Roscovitine, an experimental CDK5 inhibitor, causes delayed suppression of microglial, but not astroglial recruitment around intracerebral dopaminergic grafts. Exp. Neurol. 2019, 318, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Krystof, V.; Uldrijan, S. Cyclin-dependent kinase inhibitors as anticancer drugs. Curr. Drug Targets 2010, 11, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Knockaert, M.; Greengard, P.; Meijer, L. Pharmacological inhibitors of cyclin-dependent kinases. Trends Pharmacol. Sci. 2002, 23, 417–425. [Google Scholar] [CrossRef]
- Bach, S.; Knockaert, M.; Reinhardt, J.; Lozach, O.; Schmitt, S.; Baratte, B.; Koken, M.; Coburn, S.P.; Tang, L.; Jiang, T.; et al. Roscovitine targets, protein kinases and pyridoxal kinase. J. Biol. Chem. 2005, 280, 31208–31219. [Google Scholar] [CrossRef]
- Galimberti, F.; Thompson, S.L.; Liu, X.; Li, H.; Memoli, V.; Green, S.R.; DiRenzo, J.; Greninger, P.; Sharma, S.V.; Settleman, J.; et al. Targeting the cyclin-E-Cdk-2 complex represses lung cancer growth by triggering anaphase catastrophe. Clin. Cancer Res. 2010, 16, 109–120. [Google Scholar] [CrossRef]
- Smith, D.S.; Tsai, L.H. Cdk5 behind the wheel: A role in trafficking and transport? Trends Cell Biol. 2002, 12, 28–36. [Google Scholar] [CrossRef]
- Xiao, N.; Zhang, F.; Zhu, B.; Liu, C.; Lin, Z.; Wang, H.; Xie, W.B. CDK5-mediated tau accumulation triggers methamphetamine-induced neuronal apoptosis via endoplasmic reticulum-associated degradation pathway. Toxicol. Lett. 2018, 292, 97–107. [Google Scholar] [CrossRef]
- Hirasawa, M.; Ohshima, T.; Takahashi, S.; Longenecker, G.; Honjo, Y.; Veeranna; Pant, H.C.; Mikoshiba, K.; Brady, R.O.; Kulkarni, A.B. Perinatal abrogation of Cdk5 expression in brain results in neuronal migration defects. Proc. Natl. Acad. Sci. USA 2004, 101, 6249–6254. [Google Scholar] [CrossRef] [Green Version]
- Gilmore, E.C.; Ohshima, T.; Goffinet, A.M.; Kulkarni, A.B.; Herrup, K. Cyclin-dependent kinase 5-deficeint mice demonstrate novel developmental arrest in cerebral cortex. J. Neurosci. 1998, 18, 6370–6377. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Veeranna; Ohshima, T.; Amin, N.D.; Cho, A.; Sreenath, T.; Pant, H.C.; Brady, R.O.; Kulkarni, A.B.; Ashok, B.; et al. Neuronal cyclin-dependent kinase 5 activity is critical for survival. J. Neurosci. 2001, 21, 550–558. [Google Scholar] [CrossRef]
- Li, B.S.; Zhang, L.; Takahashi, S.; Ma, W.; Jaffe, H.; Kulkarni, A.B.; Pant, H.C. Cyclin-dependent kinase 5 prevents neuronal apoptosis by negative regulation of c-Jun N-terminal kinase 3. EMBO J. 2002, 21, 324–333. [Google Scholar] [CrossRef] [Green Version]
- Chae, T.; Kwon, Y.T.; Bronson, R.; Dikkes, P.; Li, E.; Tsai, L.H. Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron 1997, 18, 29–42. [Google Scholar] [CrossRef]
- Paglini, G.; Pigino, G.; Kunda, P.; Morfini, G.; Maccioni, R.; Quiroga, S.; Ferreira, A.; Cáceres, A. Evidence for the participation of the neuron-specific CDK5 activator P35 during laminin-enhanced axonal growth. J. Neurosci. 1998, 18, 9858–9869. [Google Scholar] [CrossRef]
- Nikolic, M.; Dudek, H.; Kwon, Y.T.; Ramos, Y.F.; Tsai, L.H. The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes. Dev. 1996, 10, 816–825. [Google Scholar] [CrossRef]
- Carter, J.M.; Waite, K.A.; Campenot, R.B.; Vance, J.E.; Vance, D.E. Enhanced expression and activation of CTP:phosphocholine cytidylyltransferase beta2 during neurite outgrowth. J. Biol. Chem. 2003, 278, 44988–44994. [Google Scholar] [CrossRef]
- Carter, J.M.; Demizieux, L.; Campenot, R.B.; Vance, D.E.; Vance, J.E. Phosphatidylcholine biosynthesis via CTP:phosphocholine cytidylyltransferase 2 facilitates neurite outgrowth and branching. J. Biol. Chem. 2008, 283, 202–212. [Google Scholar] [CrossRef]
- Lee, M.S.; Kwon, Y.T.; Li, M.; Peng, J.; Friedlander, R.M.; Tsai, L.H. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 2000, 405, 360–364. [Google Scholar] [CrossRef]
- Zhou, Y.; Deng, J.; Chu, X.; Zhao, Y.; Guo, Y. Role of Post-Transcriptional Control of Calpain by miR-124-3p in the Development of Alzheimer’s Disease. J. Alzheimers Dis. 2019, 67, 571–581. [Google Scholar] [CrossRef]
- Cheung, Z.H.; Chin, W.H.; Chen, Y.; Ng, Y.P.; Ip, N.Y. Cdk5 is involved in BDNF-stimulated dendritic growth in hippocampal neurons. PLoS Bio. 2007, 5, e63. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, C.; Ji, Y.; Teng, L.; Guo, Y. Neuregulin-1β Plays a Neuroprotective Role by Inhibiting the Cdk5 Signaling Pathway after Cerebral Ischemia-Reperfusion Injury in Rats. J. Mol. Neurosci. 2018. [Google Scholar] [CrossRef]
- Fu, A.K.; Fu, W.Y.; Cheung, J.; Tsim, K.W.; Ip, F.C.; Wang, J.H.; Ip, N.Y. Cdk5 is involved in neuregulin-induced AChR expression at the neuromuscular junction. Nat. Neurosci. 2001, 4, 374–381. [Google Scholar] [CrossRef]
- Humbert, S.; Lanier, L.M.; Tsai, L.H. Synaptic localization of p39, a neuronal activator of cdk5. Neuroreport 2000, 11, 2213–2216. [Google Scholar] [CrossRef]
- Niethammer, M.; Smith, D.S.; Ayala, R.; Peng, J.; Ko, J.; Lee, M.S.; Morabito, M.; Tsai, L.H. NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron 2000, 28, 697–711. [Google Scholar] [CrossRef]
- Bibb, J.A.; Snyder, G.L.; Nishi, A.; Yan, Z.; Meijer, L.; Fienberg, A.A.; Tsai, L.H.; Kwon, Y.T.; Girault, J.A.; Czernik, A.J.; et al. Phosphorylation of DARPP-32 by Cdk5 modulates dopamine signalling in neurons. Nature 1999, 402, 669–671. [Google Scholar] [CrossRef]
- Karunakaran, U.; Elumalai, S.; Moon, J.S.; Jeon, J.H.; Kim, N.D.; Park, K.G.; Won, K.C.; Leem, J.; Lee, I.K. Myricetin Protects Against High Glucose-Induced β-Cell Apoptosis by Attenuating Endoplasmic Reticulum Stress via Inactivation of Cyclin-Dependent Kinase 5. Diabetes Metab. J. 2019, 43, 192–205. [Google Scholar] [CrossRef]
- Guo, D.; Xie, W.; Xiong, P.; Li, H.; Wang, S.; Chen, G.; Gao, Y.; Zhou, J.; Zhang, Y.; Bu, G.; et al. Cyclin-dependent kinase 5-mediated phosphorylation of chloride intracellular channel 4 promotes oxidative stress-induced neuronal death. Cell Death Dis. 2018, 9, 951. [Google Scholar] [CrossRef]
- Kianpour, R.S.; Arya, A.; Karimian, H.; Madhavan, P.; Rizwan, F.; Koshy, S.; Prabhu, G. Mechanism involved in insulin resistance via accumulation of β-amyloid and neurofibrillary tangles: Link between type 2 diabetes and Alzheimer’s disease. Drug Des. Dev. Ther. 2018, 12, 3999–4021. [Google Scholar] [CrossRef]
- NavaneethaKrishnan, S.; Rosales, J.L.; Lee, K.Y. Loss of Cdk5 in breast cancer cells promotes ROS-mediated cell death through dysregulation of the mitochondrial permeability transition pore. Oncogene 2018, 37, 1788. [Google Scholar] [CrossRef]
- Contreras-Vallejos, E.; Utreras, E.; Gonzalez-Billault, C. Going out of the brain: Non-nervous system physiological and pathological functions of Cdk5. Cell Signal. 2012, 24, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Lenjisa, J.L.; Tadesse, S.; Khair, N.Z.; Kumarasiri, M.; Yu, M.; Albrecht, H.; Robert Milne, R.; Wang, S. CDK5 in oncology: Recent advances and future prospects. Future Med. Chem. 2017, 9, 1939–1962. [Google Scholar] [CrossRef] [PubMed]
- Ubeda, M.; Kemp, D.M.; Habener, J.F. Glucose-induced expression of the cyclin-dependent protein kinase 5 activator p35 involved in Alzheimer’s disease regulates insulin gene transcription in pancreatic beta-cells. Endocrinology 2004, 145, 3023–3031. [Google Scholar] [CrossRef]
- Lilja, L.; Johansson, J.U.; Gromada, J.; Mandic, S.A.; Fried, G.; Berggren, P.O.; Bark, C. Cyclin-dependent kinase 5 associated with p39 promotes Munc18-1 phosphorylation and Ca2+-dependent exocytosis. J. Biol. Chem. 2004, 279, 29534–29541. [Google Scholar] [CrossRef]
- Studzinski, G.P.; Harrison, J.S. The neuronal cyclin-dependent kinase 5 activator p35Nck5a and Cdk5 activity in monocytic cells. Leuk. Lymphoma. 2003, 44, 235–240. [Google Scholar] [CrossRef]
- Sandal, T.; Stapnes, C.; Kleivdal, H.; Hedin, L.; Døskeland, S.O. A novel, extraneuronal role for cyclin-dependent protein kinase 5 (CDK5): Modulation of cAMP-induced apoptosis in rat leukemia cells. J. Biol. Chem. 2002, 277, 20783–20793. [Google Scholar] [CrossRef]
- Alvi, A.J.; Austen, B.; Weston, V.J.; Fegan, C.; MacCallum, D.; Gianella-Borradori, A.; Lane, D.P.; Hubank, M.; Powell, J.E.; Wei, W.; et al. A novel CDK inhibitor, CYC202(R-roscovitine), overcomes the defect in p35-dependent apoptosis in B-CLL by down-regulation of genes involved in transcription regulation and survival. Blood 2005, 105, 4484–4491. [Google Scholar] [CrossRef]
- Raje, N.; Kumar, S.; Hideshima, T.; Roccaro, A.; Ishitsuka, K.; Yasui, H.; Shiraishi, N.; Chauhan, D.; Munschi, N.C.; Green, S.R.; et al. Seliciclib (CYC202 or R-roscovitine), a small-molecule cyclin-dependent kinase inhibitor, mediates activity via down-regulation of Mcl-1 in multiple myeloma. Blood 2005, 106, 1042–1047. [Google Scholar] [CrossRef] [Green Version]
- MacCallum, D.E.; Melville, J.; Frame, S.; Watt, K.; Anderson, S.; Gianella-Borradori, A.; Lane, D.P.; Green, S.R. Seliciclib (CY202, R-Roscovitine) induces cell death in multiple myeloma cells by inhibition of RNA polymerase II-dependent transcription and down-regulation of Mcl-1. Cancer Res. 2005, 65, 5399–5407. [Google Scholar] [CrossRef]
- Hallaert, D.Y.; Spijker, R.; Jak, M.; Derks, I.A.; Alves, N.L.; Wensveen, F.M.; de Boer, J.P.; de Jong, D.; Green, S.R.; van Oers, M.H.; et al. Crosstalk among Bcl-2 family members in B-CLL: Seliciclib acts via the Mcl-1/Noxa axis and gradual exhaustion of Bcl-2 protection. Cell Death Differ. 2007, 14, 1958–1967. [Google Scholar] [CrossRef]
- Lowman, X.H.; McDonnel, M.A.; Kosloske, A.; Odumade, O.A.; Jenness, C.; Karim, C.B.; Jemmerson, R.; Kelekar, A. The proapoptotic function of Noxa in human leukemia cells is regulated by the kinase Cdk5 and by glucose. Mol. Cell. 2010, 40, 823–833. [Google Scholar] [CrossRef]
- Morey, T.M.; Roufayel, R.; Johnston, D.S.; Fletcher, A.S.; Mosser, D.D. Heat shock inhibition of CDK5 increases NOXA levels through miR-23a repression. J. Biol. Chem. 2015, 290, 11443–11454. [Google Scholar] [CrossRef]
- Stankiewicz, A.R.; Livingstone, A.M.; Mohseni, N.; Mosser, D.D. Regulation of heat-induced apoptosis by Mcl-1 degradation and its inhibition by Hsp70. Cell Death Differ. 2009, 16, 638–647. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.W.; Lee, S.H. The roles of autophagy in cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef]
- Mizumura, K.; Maruoka, S.; Shimizu, T.; Gon, Y. Autophagy, selective autophagy, and necroptosis in COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 2018, 13, 3165–3172. [Google Scholar] [CrossRef]
- Tan, Y.; Gong, Y.; Dong, M.; Pei, Z.; Ren, J. Role of autophagy in inherited metabolic and endocrine myopathies. BBA Mol. Basis Dis. 2019, 1865, 48–55. [Google Scholar] [CrossRef]
- Iachettini, S.; Trisciuoglio, D.; Rotili, D.; Lucidi, A.; Salvati, E.; Zizza, P.; Di Leo, L.; Del Bufalo, D.; Ciriolo, M.R.; Leonetti, C.; et al. Pharmacological activation of SIRT6 triggers lethal autophagy in human cancer cells. Cell Death Dis. 2018, 9, 996. [Google Scholar] [CrossRef]
- Serrano-Oviedo, L.; Ortega-Muelas, M.; García-Cano, J.; Valero, M.L.; Cimas, F.J.; Pascual-Serra, R.; Fernandez-Aroca, D.M.; Roche, O.; Ruiz-Hidalgo, M.J.; Belandia, B.; et al. Autophagic cell death associated to Sorafenib in renal cell carcinoma is mediated through Akt inhibition in an ERK1/2 independent fashion. PLoS ONE 2018, 13, e0200878. [Google Scholar] [CrossRef]
- Li, J.; Hu, X.; Su, M.; Shen, H.; Qiu, W.; Tian, Y. CDK5RAP3 Participates in Autophagy Regulation and Is Downregulated in Renal Cancer. Dis. Markers 2019. [Google Scholar] [CrossRef]
- Wong, A.S.; Lee, R.H.; Cheung, A.Y.; Yeung, P.K.; Chung, S.K.; Cheung, Z.H.; Ip, N.Y. Cdk5-mediated phosphorylation of endophilin B1 is required for induced autophagy in models of Parkinson’s disease. Nat. Cell Biol. 2011, 13, 568–579. [Google Scholar] [CrossRef]
- Nandi, N.; Krämer, H. Cdk5-mediated Acn/Acinus phosphorylation regulates basal autophagy independently of metabolic stress. Autophagy 2018, 14, 1271–1272. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roufayel, R.; Murshid, N. CDK5: Key Regulator of Apoptosis and Cell Survival. Biomedicines 2019, 7, 88. https://doi.org/10.3390/biomedicines7040088
Roufayel R, Murshid N. CDK5: Key Regulator of Apoptosis and Cell Survival. Biomedicines. 2019; 7(4):88. https://doi.org/10.3390/biomedicines7040088
Chicago/Turabian StyleRoufayel, Rabih, and Nimer Murshid. 2019. "CDK5: Key Regulator of Apoptosis and Cell Survival" Biomedicines 7, no. 4: 88. https://doi.org/10.3390/biomedicines7040088
APA StyleRoufayel, R., & Murshid, N. (2019). CDK5: Key Regulator of Apoptosis and Cell Survival. Biomedicines, 7(4), 88. https://doi.org/10.3390/biomedicines7040088