Mitochondrial Dysfunction in Parkinson’s Disease: Focus on Mitochondrial DNA


Abstract
1. Introduction
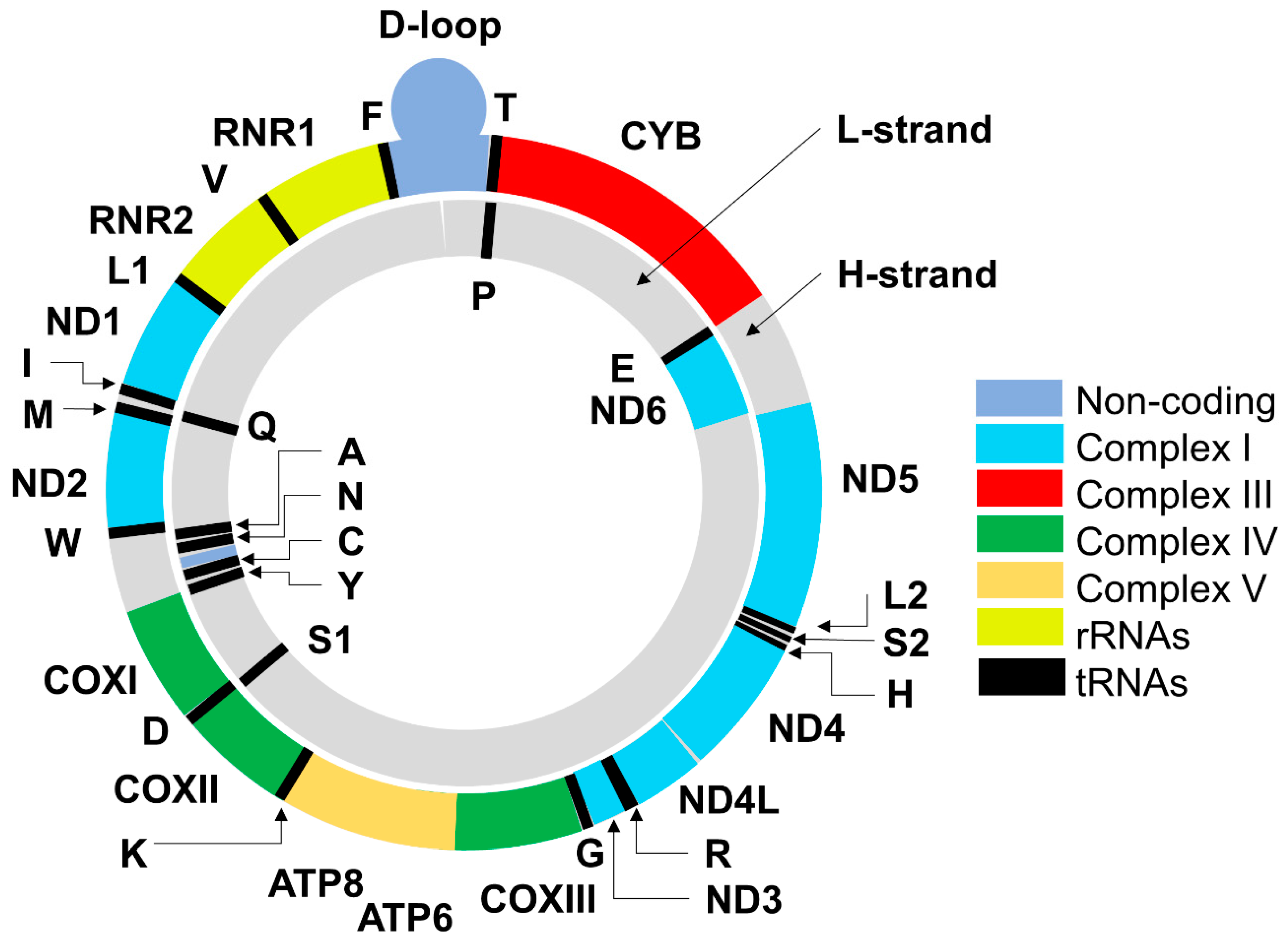
2. Mitochondrial DNA: Structure, Functions, Mode of Replication and Transcription of Mitochondrial Genes
3. MtDNA Oxidation and Repair Mechanisms
4. Age-Related Changes in mtDNA
5. MtDNA Changes in Parkinson’s Disease
5.1. MtDNA Mutations
5.2. Substantia Nigra Samples from PD Patients
5.3. Other Cells from PD Patients
5.4. Studies Using Cytoplasmic Hybrid (Cybrid) Cell Lines
6. Studies Using Animal Models of Parkinson’s Disease
6.1. MPTP-Induced Parkinsonism
6.2. Rotenone-Induced Parkinsonism
6.3. Polg Mutator Mice
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Goetz, C.G. The history of Parkinson’s disease: Early clinical descriptions and neurological therapies. Cold Spring Harb. Perspect. Med. 2011, 1, a008862. [Google Scholar] [CrossRef] [PubMed]
- Obeso, J.A.; Stamelou, M.; Goetz, C.G.; Poewe, W.; Lang, A.E.; Weintraub, D.; Burn, D.; Halliday, G.M.; Bezard, E.; Przedborski, S.; et al. Past, present, and future of Parkinson’s disease: A special essay on the 200th anniversary of the Shaking Palsy. Mov. Disord. 2017, 32, 1264–1310. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Langston, E.B.; Irwin, I. MPTP-induced parkinsonism in human and non-human primates--clinical and experimental aspects. Acta Neurologica Scandinavica 1984, 100, 49–54. [Google Scholar] [PubMed]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 1, 1269. [Google Scholar] [CrossRef]
- Schapira, A.H.; Mann, V.M.; Cooper, J.M.; Dexter, D.; Daniel, S.E.; Jenner, P.; Clark, J.B.; Marsden, C.D. Anatomic and disease specificity of NADH CoQ1 reductase (complex I) deficiency in Parkinson’s disease. J. Neurochem. 1990, 55, 2142–2145. [Google Scholar] [CrossRef]
- Blin, O.; Desnuelle, C.; Rascol, O.; Borg, M.; Peyro Saint Paul, H.; Azulay, J.P.; Billé, F.; Figarella, D.; Coulom, F.; Pellissier, J.F.; et al. Mitochondrial respiratory failure in skeletal muscle from patients with Parkinson’s disease and multiple system atrophy. J. Neurol. Sci. 1994, 125, 95–101. [Google Scholar] [CrossRef]
- Cardellach, F.; Marti, M.J.; Fernandez-Sola, J.; Marin, C.; Hoek, J.B.; Tolosa, E.; Urbano-Marquez, A. Mitochondrial respiratory chain activity in skeletal muscle from patients with Parkinson’s disease. Neurology 1993, 43, 2258–2262. [Google Scholar] [CrossRef]
- Haas, R.H.; Nasirian, F.; Nakano, K.; Ward, D.; Pay, M.; Hill, R.; Shults, C.W. Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson’s disease. Ann. Neurol. 1995, 37, 714–722. [Google Scholar] [CrossRef]
- Yoshino, H.; Nakagawa-Hattori, Y.; Kondo, T.; Mizuno, Y. Mitochondrial complex I and II activities of lymphocytes and platelets in Parkinson’s disease. J. Neural. Transm. Parkinson’s Dis. Dement. Sect. 1992, 4, 27–34. [Google Scholar] [CrossRef]
- Mytilineou, C.; Werner, P.; Molinari, S.; Di Rocco, A.; Cohen, G.; Yahr, M.D. Impaired oxidative decarboxylation of pyruvate in fibroblasts from patients with Parkinson’s disease. J. Neural Transm. Park. Dis. Dement. Sect. 1994, 8, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Does mitochondrial DNA play a role in Parkinson’s disease? A review of cybrid and other supportive evidence. Antioxid. Redox Signal. 2012, 16, 950–964. [Google Scholar] [CrossRef] [PubMed]
- Nass, M.M.K.; Nass, S. Intramitochondrial fibers with DNA characteristics: I. Fixation and electron staining reactions. J. Cell Biol. 1963, 9, 593–611. [Google Scholar] [CrossRef] [PubMed]
- Nass, S.; Nass, M.M.K. Intramitochondrial fibers with DNA characteristics: II. Enzymatic and other hydrolytic treatments. J. Cell Biol. 1963, 9, 613–629. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K. Unique features of animal mitochondrial translation systems—The non-universal genetic code, unusual features of the translational apparatus and their relevance to human mitochondrial diseases. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 11–39. [Google Scholar] [CrossRef]
- Greber, B.; Ban, N. Structure and function of the mitochondrial ribosome. Ann. Rev. Biochem. 2016, 85, 103–132. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Gray, M.W.; Lang, B.F.; Cedergren, R.; Golding, G.B.; Lemieux, C.; Sankoff, D.; Turmel, M.; Brossard, N.; Delage, E.; Littlejohn, T.G.; et al. Genome structure and gene content in protist mitochondrial DNAs. Nucleic Acids Res. 1998, 26, 865–878. [Google Scholar] [CrossRef]
- Kogelnik, A.M.; Lott, M.T.; Brown, M.D.; Navathe, S.B.; Wallace, D.C. MITOMAP: A human mitochondrial genome database. Nucleic Acids Res. 1996, 24, 177–179. [Google Scholar] [CrossRef]
- Greaves, L.C.; Reeve, A.K.; Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA and disease. J. Pathol. 2012, 226, 274–286. [Google Scholar] [CrossRef]
- Vinothkumar, K.R.; Zhu, J.; Hirst, J. Architecture of mammalian respiratory complex I. Nature 2014, 515, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Esser, L.; Tang, W.-K.; Zhou, F.; Zhou, Y.; Yu, L.; Yu, C.-A. Structural analysis of cytochrome bc1 complexes: Implications to the mechanism of function. Biochim. Biophys. Acta (BBA) Bioenergetics 2013, 1827, 1278–1294. [Google Scholar] [CrossRef] [PubMed]
- Kadenbach, B.; Hüttemann, M. The subunit composition and function of mammalian cytochrome c oxidase. Mitochondrion 2015, 24, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Sousa, J.S.; D’Imprima, E.; Vonck, J. Mitochondrial respiratory chain complexes. Subcell. Biochem. 2018, 87, 167–227. [Google Scholar] [PubMed]
- Jonckheere, A.I.; Smeitink, J.A.; Rodenburg, R.J. Mitochondrial ATP synthase: Architecture, function and pathology. J. Inherit. Metab. Dis. 2012, 35, 211–225. [Google Scholar] [CrossRef]
- Marchington, D.R.; Scott Brown, M.S.G.; Lamb, V.K.; van Golde, R.J.T.; Kremer, J.A.M.; Tuerlings, J.H.A.M.; Mariman, E.C.M.; Balen, A.H.; Poulton, J. No evidence for paternal mtDNA transmission to offspring or extra-embryonic tissues after ICSI. Mol. Hum. Reprod. 2002, 8, 1046–1049. [Google Scholar] [CrossRef][Green Version]
- Danan, C.; Sternberg, D.; Van Steirteghem, A.; Cazeneuve, C.; Duquesnoy, P.; Besmond, C.; Goosens, M.; Lissens, W.; Amselem, S. Evaluation of parental mitochondrial inheritance in neonates born after intracytoplasmic sperm injection. Am. J. Hum. Genet. 1999, 65, 463–473. [Google Scholar] [CrossRef]
- Houshmand, M.; Holme, E.; Hanson, C.; Wennerholm, U.B.; Hamberger, L. Is paternal mitochondrial DNA transferred to the offspring following intracytoplasmic sperm injection? J. Assist. Reprod. Genet. 1997, 14, 223–227. [Google Scholar] [CrossRef][Green Version]
- Sutovsky, P.; Moreno, R.D.; Ramalho-Santos, J.; Dominko, T.; Simerly, C.; Schatten, G. Ubiquitin tag for sperm mitochondria. Nature 1999, 402, 371–372. [Google Scholar] [CrossRef]
- Song, W.-H.; Ballard, J.W.O.; Yi, Y.-J.; Sutovsky, P. Regulation of mitochondrial genome inheritance by autophagy and ubiquitin-proteasome system: Implications for health, fitness, and fertility. Biomed. Res. Int. 2014, 2014, 981867. [Google Scholar] [CrossRef]
- Buneeva, O.A.; Medvedev, A.E. Mitochondrial dysfunction in Parkinson’s disease. Biomed. Khim. 2011, 57, 246–281. [Google Scholar] [CrossRef]
- Wallace, D.C.; Chalkia, D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a021220. [Google Scholar] [CrossRef] [PubMed]
- Alexeyev, M. Mitochondrial DNA: The common confusions. Mitochondrial DNA Part A 2020, 31, 45–47. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, M. Mitochondrial DNA replication in mammalian cells: Overview of the pathway. Essays Biochem. 2018, 62, 287–296. [Google Scholar] [PubMed]
- Nicholls, T.J.; Minczuk, M. In D-loop: 40 years of mitochondrial 7S DNA. Exp. Gerontol. 2014, 56, 175–181. [Google Scholar] [CrossRef]
- Yasukawa, T.; Kang, D. An overview of mammalian mitochondrial DNA replication mechanisms. J. Biochem. 2018, 164, 183–193. [Google Scholar] [CrossRef]
- King, M.P.; Attardi, G. Isolation of human cell lines lacking mitochondrial DNA. Methods Enzymol. 1996, 264, 304–313. [Google Scholar]
- Gilkerson, R.; Bravo, L.; Garcia, I.; Gaytan, N.; Herrera, A.; Maldonado, A.; Quintanilla, B. The mitochondrial nucleoid: Integrating mitochondrial DNA into cellular homeostasis. Cold Spring Harb. Perspect. Biol. 2013, 5, a011080. [Google Scholar] [CrossRef]
- Farge, G.; Falkenberg, M. Organization of DNA in mammalian mitochondria. Int. J. Mol. Sci. 2019, 20, 2770. [Google Scholar] [CrossRef]
- Kaufman, B.A.; Newman, S.M.; Hallberg, R.L.; Slaughter, C.A.; Perlman, P.S.; Butow, R.A. In organello formaldehyde crosslinking of proteins to mtDNA: Identification of bifunctional proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 7772–7777. [Google Scholar] [CrossRef]
- Wang, Y.; Bogenhagen, D.F. Human mitochondrial DNA nucleoids are linked to protein folding machinery and metabolic enzymes at the mitochondrial inner membrane. J. Biol. Chem. 2006, 281, 25791–25802. [Google Scholar] [CrossRef] [PubMed]
- Bogenhagen, D.F.; Rousseau, D.; Burke, S. The layered structure of human mitochondrial DNA nucleoids. J. Biol. Chem. 2008, 283, 3665–3675. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.-S.; Jeong, J.H.; Min, H.-K.; Jung, H.-J.; Hwang, D.; Lee, S.-W.; Pak, Y.K. Shot-gun proteomic analysis of mitochondrial D-loop DNA binding proteins: Identification of mitochondrial histones. Mol. Biosyst. 2011, 7, 1523–1536. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Cooper, H.M.; Reyes, A.; Di Re, M.; Sembongi, H.; Litwin, T.R.; Gao, J.; Neuman, K.C.; Fearnley, I.M.; Spinazzola, A.; et al. Mitochondrial nucleoid interacting proteins support mitochondrial protein synthesis. Nucleic Acids Res. 2012, 40, 6109–6121. [Google Scholar] [CrossRef] [PubMed]
- Hensen, F.; Cansiz, S.; Gerhold, J.M.; Spelbrink, J.N. To be or not to be a nucleoid protein: A comparison of mass-spectrometry based approaches in the identification of potential mtDNA-nucleoid associated proteins. Biochimie 2014, 100, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Udeshi, N.D.; Deerinck, T.J.; Svinkina, T.; Ellisman, M.H.; Carr, S.A.; Ting, A.Y. Proximity Biotinylation as a method for mapping proteins associated with mtDNA in living cells. Cell Chem. Biol. 2017, 24, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Trinkle-Mulcahy, L. Recent advances in proximity-based labeling methods for interactome mapping. Version 1. F1000Ressearch 2019, 8. [Google Scholar] [CrossRef]
- Kopek, B.G.; Shtengel, G.; Xu, C.S.; Clayton, D.A.; Hess, H.F. Correlative 3D superresolution fluorescence and electron microscopy reveal the relationship of mitochondrial nucleoids to membranes. Proc. Natl. Acad. Sci. USA 2012, 109, 6136–6141. [Google Scholar] [CrossRef]
- Qin, J.; Guo, Y.; Xue, B.; Shi, P.; Chen, Y.; Su, Q.P.; Hao, H.; Zhao, S.; Wu, C.; Yu, L.; et al. ER-mitochondria contacts promote mtDNA nucleoids active transportation via mitochondrial dynamic tubulation. Nat. Commun. 2020, 11, 4471. [Google Scholar] [CrossRef]
- Robberson, D.L.; Kasamatsu, H.; Vinograd, J. Replication of mitochondrial DNA. Circular replicative intermediates in mouse L cells. Proc. Natl. Acad. Sci. USA 1972, 69, 737–741. [Google Scholar] [CrossRef]
- Miralles Fusté, J.M.; Wanrooij, S.; Jemt, E.; Granycome, C.E.; Cluett, T.J.; Shi, Y.; Atanassova, N.; Holt, I.J.; Gustafsson, C.M.; Falkenberg, M. Mitochondrial RNA polymerase is needed for activation of the origin of light-strand DNA replication. Mol. Cell 2010, 37, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Miralles Fusté, J.; Shi, Y.; Wanrooij, S.; Zhu, X.; Jemt, E.; Persson, Ö.; Sabouri, N.; Gustafsson, C.M.; Falkenberg, M. In vivo occupancy of mitochondrial single-stranded DNA binding protein supports the strand displacement mode of DNA replication. PLoS Genet. 2014, 10, e1004832. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-L. Novel lines of evidence for the asymmetric strand displacement model of mitochondrial DNA replication. Mol. Cell. Biol. 2019, 39, e00406-18. [Google Scholar] [CrossRef] [PubMed]
- Agaronyan, K.; Morozov, Y.I.; Anikin, M.; Temiakov, D. Mitochondrial biology. Replication-transcription switch in human mitochondria. Science 2015, 347, 548–551. [Google Scholar] [CrossRef]
- D’Souza, A.R.; Minczuk, M. Mitochondrial transcription and translation: Overview. Essays Biochem. 2018, 62, 309–320. [Google Scholar] [PubMed]
- Posse, V.; Gustafsson, C.M. Human mitochondrial transcription factor B2 is required for promoter melting during initiation of transcription. J. Biol. Chem. 2017, 292, 2637–2645. [Google Scholar] [CrossRef]
- Litonin, D.; Sologub, M.; Shi, Y.; Savkina, M.; Anikin, M.; Falkenberg, M.; Gustafsson, C.M.; Temiakov, D. Human mitochondrial transcription revisited: Only TFAM and TFB2M are required for transcription of the mitochondrial genes in vitro. J. Biol. Chem. 2010, 285, 18129–18133. [Google Scholar] [CrossRef]
- Jiang, S.; Koolmeister, C.; Misic, J.; Siira, S.; Kühl, I.; Silva Ramos, E.; Miranda, M.; Jiang, M.; Posse, V.; Lytovchenko, O.; et al. TEFM regulates both transcription elongation and RNA processing in mitochondria. EMBO Rep. 2019, 20, e48101. [Google Scholar] [CrossRef]
- Tomecki, R.; Dmochowska, A.; Gewartowski, K.; Dziembowski, A.; Stepien, P.P. Identification of a novel human nuclear-encoded mitochondrial poly(A) polymerase. Nucleic Acids Res. 2004, 32, 6001–6014. [Google Scholar] [CrossRef]
- Nagaike, T.; Suzuki, T.; Katoh, T.; Ueda, T. Human mitochondrial mRNAs are stabilized with polyadenylation regulated by mitochondria-specific poly(A) polymerase and polynucleotide phosphorylase. J. Biol. Chem. 2005, 280, 19721–19727. [Google Scholar] [CrossRef]
- Lapkouski, M.; Hällberg, B.M. Structure of mitochondrial poly(A) RNA polymerase reveals the structural basis for dimerization, ATP selectivity and the SPAX4 disease phenotype. Nucleic Acids Res. 2015, 43, 9065–9075. [Google Scholar] [CrossRef] [PubMed]
- Ruzzenente, B.; Metodiev, M.D.; Wredenberg, A.; Bratic, A.; Park, C.B.; Cámara, Y.; Milenkovic, D.; Zickermann, V.; Wibom, R.; Hultenby, K.; et al. LRPPRC is necessary for polyadenylation and coordination of translation of mitochondrial mRNAs. EMBO J. 2012, 31, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Wang, L.; Ren, X.; Zhang, Y.; Zhang, H. LRPPRC: A multifunctional protein involved in energy metabolism and human disease. Front. Physiol. 2019, 10, 595. [Google Scholar] [CrossRef] [PubMed]
- Linnane, A.W.; Marzuki, S.; Ozawa, T.; Tanaka, M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet 1989, 1, 642–645. [Google Scholar] [CrossRef]
- Kruman, I.I.; Wersto, R.P.; Cardozo-Pelaez, F.; Smilenov, L.; Chan, S.L.; Chrest, F.J.; Emokpae, R., Jr.; Gorospe, M.; Mattson, M.P. Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron 2004, 41, 549–561. [Google Scholar] [CrossRef]
- Nakabeppu, Y.; Tsuchimoto, D.; Yamaguchi, H.; Sakumi, K. Oxidative damage in nucleic acids and Parkinson’s disease. J. Neurosci. Res. 2007, 85, 919–934. [Google Scholar] [CrossRef]
- Santos, R.X.; Correia, S.C.; Zhu, X.; Smith, M.A.; Moreira, P.I.; Castellani, R.J.; Nunomura, A.; Perry, G. Mitochondrial DNA oxidative damage and repair in aging and Alzheimer’s disease. Antioxid. Redox Signal. 2013, 18, 2444–2457. [Google Scholar] [CrossRef]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef]
- Horowitz, M.P.; Greenamyre, J.T. Mitochondrial iron metabolism and its role in neurodegeneration. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S551–S568. [Google Scholar] [CrossRef]
- Sies, H. Strategies of antioxidant defense. Eur. J. Biochem. 1993, 215, 213–219. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, D.; Sakumi, K.; Ohno, M.; Sakai, Y.; Furuichi, M.; Iwai, S.; Nakabeppu, Y. An oxidized purine nucleoside triphosphatase, MTH1, suppresses cell death caused by oxidative stress. J. Biol. Chem. 2003, 278, 37965–37973. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Bjørås, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, K.; Ohtsubo, T.; Oda, H.; Fujiwara, T.; Kang, D.; Sugimachi, K.; Nakabeppu, Y. Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol. Biol Cell. 1999, 10, 1637–1652. [Google Scholar] [CrossRef]
- De Souza-Pinto, N.C.; Eide, L.; Hogue, B.A.; Thybo, T.; Stevnsner, T.; Seeberg, E.; Klungland, A.; Bohr, V.A. Repair of 8-oxodeoxyguanosine lesions in mitochondrial dna depends on the oxoguanine dna glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial dna of OGG1-defective mice. Cancer Res. 2001, 61, 5378–5381. [Google Scholar]
- Yamaguchi, H.; Kajitani, K.; Dan, Y.; Furuichi, M.; Ohno, M.; Sakumi, K.; Kang, D.; Nakabeppu, Y. MTH1, an oxidized purine nucleoside triphosphatase, protects the dopamine neurons from oxidative damage in nucleic acids caused by 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine. Cell Death Differ. 2006, 13, 551–563. [Google Scholar] [CrossRef]
- Lauritzen, K.H.; Dalhus, B.; Storm, J.F.; Bjørås, M.; Klungland, A. Modeling the impact of mitochondrial DNA damage in forebrain neurons and beyond. Mech. Ageing Dev. 2011, 132, 424–428. [Google Scholar] [CrossRef]
- Lauritzen, K.H.; Moldestad, O.; Eide, L.; Carlsen, H.; Nesse, G.; Storm, J.F.; Mansuy, I.M.; Bergersen, L.H.; Klungland, A. Mitochondrial DNA toxicity in forebrain neurons causes apoptosis, neurodegeneration, and impaired behavior. Mol. Cell Biol. 2010, 30, 1357–1367. [Google Scholar] [CrossRef]
- Rothfuss, O.; Gasser, T.; Patenge, N. Analysis of differential DNA damage in the mitochondrial genome employing a semi-long run real-time PCR approach. Nucl. Acids Res. 2010, 38, e24. [Google Scholar] [CrossRef]
- Collins, A. Comparison of different methods of measuring 8- oxoguanine as a marker of oxidative DNA damage. Free Radic. Res. 2000, 32, 333–341. [Google Scholar] [CrossRef]
- Pilger, A.; Rüdiger, H.W. 8-Hydroxy-2′-deoxyguanosine as a marker of oxidative DNA damage related to occupational and environmental exposures. Int. Arch. Occup. Environ. Health 2006, 80, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mecocci, P.; Beal, M.F.; Cecchetti, R.; Polidori, M.C.; Cherubini, A.; Chionne, F.; Avellini, L.; Romano, G.; Senin, U. Mitochondrial membrane fluidity and oxidative damage to mitochondrial DNA in aged and AD human brain. Mol. Chem. Neuropathol. 1997, 31, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Barja, G.; Herrero, I. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J. 2000, 14, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Cao, G.; Hastings, T.; Feng, Y.; Pei, W.; O’Horo, C.; Chen, J. Age-dependent decline of DNA repair activity for oxidative lesions in rat brain mitochondria. J. Neurochem. 2002, 81, 1273–1284. [Google Scholar] [CrossRef]
- Imam, S.Z.; Karahalil, B.; Hogue, B.A.; Souza-Pinto, N.C.; Bohr, V.A. Mitochondrial and nuclear DNA-repair capacity of various brain regions in mouse is altered in an age-dependent manner. Neurobiol. Aging. 2006, 27, 1129–1136. [Google Scholar] [CrossRef]
- Gredilla, R.; Garm, C.; Holm, R.; Bohr, V.A.; Stevnsner, T. Differential age-related changes in mitochondrial DNA repair activities in mouse brain regions. Neurobiol. Aging 2010, 31, 993–1002. [Google Scholar] [CrossRef]
- Fukae, J.; Takanashi, M.; Kubo, S.-I.; Nishioka, K.-I.; Nakabeppu, Y.; Mori, H.; Mizuno, Y.; Hattori, N. Expression of 8-oxoguanine DNA glycosylase (OGG1) in Parkinson’s disease and related neurodegenerative disorders. Acta Neurophathol. 2005, 109, 256–262. [Google Scholar] [CrossRef]
- Sondheimer, N.; Glatz, C.E.; Tirone, J.E.; Deardorff, M.A.; Krieger, A.M.; Hakonarson, H. Neutral mitochondrial heteroplasmy and the influence of aging. Hum. Mol. Genet. 2011, 20, 1653–1659. [Google Scholar] [CrossRef]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef]
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006, 38, 518–520. [Google Scholar] [CrossRef]
- Dölle, C.; Flønes, I.; Nido, G.S.; Miletic, H.; Osuagwu, N.; Kristoffersen, S.; Lilleng, P.K.; Larsen, J.P.; Tysnes, O.B.; Haugarvoll, K.; et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat. Commun. 2016, 7, 13548. [Google Scholar] [CrossRef] [PubMed]
- Hebert, S.L.; Lanza, I.R.; Nair, K.S. Mitochondrial DNA alterations and reduced mitochondrial function in aging. Mech. Ageing Dev. 2010, 131, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Vives-Bauza, C.; Andreu, A.L.; Manfredi, G.; Beal, M.F.; Janetzky, B.; Gruenewald, T.H.; Lin, M.T. Sequence analysis of the entire mitochondrial genome in Parkinson’s disease. Biochem. Biophys. Res. Commun. 2002, 290, 1593–1601. [Google Scholar] [CrossRef] [PubMed]
- Gu, G.; Reyes, P.E.; Golden, G.T.; Woltjer, R.L.; Hulette, C.; Montine, T.J.; Zhang, J. Mitochondrial DNA deletions/rearrangements in parkinson disease and related neurodegenerative disorders. J. Neuropathol. Exp. Neurol. 2002, 61, 634–639. [Google Scholar] [CrossRef]
- Mayr-Wohlfart, U.; Rödel, G.; Henneberg, A. Mitochondrial tRNA(Gln) and tRNA(Thr) gene variants in Parkinson’s disease. Eur. J. Med. Res. 1997, 2, 111–113. [Google Scholar]
- Brown, M.D.; Shoffner, J.M.; Kim, Y.L.; Jun, A.S.; Graham, B.H.; Cabell, M.F.; Gurley, D.S.; Wallace, D.C. Mitochondrial DNA sequence analysis of four Alzheimer’s and Parkinson’s disease patients. Am. J. Med. Genet. 1996, 61, 283–289. [Google Scholar] [CrossRef]
- Egensperger, R.; Kösel, S.; Schnopp, N.M.; Mehraein, P.; Graeber, M.B. Association of the mitochondrial tRNA(A4336G) mutation with Alzheimer’s and Parkinson’s diseases. Neuropathol. Appl. Neurobiol. 1997, 23, 315–321. [Google Scholar] [CrossRef]
- Richter, G.; Sonnenschein, A.; Grünewald, T.; Reichmann, H.; Janetzky, B. Novel mitochondrial DNA mutations in Parkinson’s disease. J. Neural Transm. 2002, 109, 721–729. [Google Scholar] [CrossRef]
- Huerta, C.; Castro, M.G.; Coto, E.; Blázquez, M.; Ribacoba, R.; Guisasola, L.M.; Salvador, C.; Martínez, C.; Lahoz, C.H.; Alvarez, V. Mitochondrial DNA polymorphisms and risk of Parkinson’s disease in Spanish population. J. Neurol. Sci. 2005, 236, 49–54. [Google Scholar] [CrossRef]
- García-Lozano, J.R.; Mir, P.; Alberca, R.; Aguilera, I.; Gil Néciga, E.; Fernández-López, O.; Cayuela, A.; Núñez-Roldan, A. Mitochondrial DNA A4336G mutation in Alzheimer’s and Parkinson’s diseases. Eur. Neurol. 2002, 48, 34–36. [Google Scholar] [CrossRef]
- Cavelier, L.; Erikson, I.; Tammi, M.; Jalonen, P.; Lindholm, E.; Jazin, E.; Smith, P.; Luthman, H.; Gyllensten, U. MtDNA mutations in maternally inherited diabetes: Presence of the 3397 ND1 mutation previously associated with Alzheimer’s and Parkinson’s disease. Hereditas 2001, 135, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Horvath, R.; Kley, R.A.; Lochmu¨ller, H.; Vorgerd, M. Parkinson syndrome, neuropathy, and myopathy caused by the mutation A8344G (MERRF) in tRNALys. Neurology 2007, 68, 56–58. [Google Scholar] [CrossRef] [PubMed]
- Shoffner, J.M.; Brown, M.D.; Torroni, A.; Lott, M.T.; Cabell, M.F.; Mirra, S.S.; Beal, M.F.; Yang, C.C.; Gearing, M.; Salvo, R.; et al. Mitochondrial DNA variants observed in Alzheimer disease and Parkinson disease patients. Genomics 1993, 17, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Thyagarajan, D.; Bressman, S.; Bruno, C.; Przedborski, S.; Shanske, S.; Lynch, T.; Fahn, S.; DiMauro, S. A novel mitochondrial 12SrRNA point mutation in parkinsonism, deafness, and neuropathy. Ann. Neurol. 2000, 48, 730–736. [Google Scholar] [CrossRef]
- Hudson, G.; Nalls, M.; Evans, J.R.; Breen, D.P.; Winder-Rhodes, S.; Morrison, K.E.; Morris, H.R.; Williams-Gray, C.H.; Barker, R.A.; Singleton, A.B.; et al. Two-stage association study and meta-analysis of mitochondrial DNA variants in Parkinson disease. Neurology 2013, 80, 2042–2048. [Google Scholar] [CrossRef] [PubMed]
- Ross, O.A.; McCormack, R.; Maxwell, L.D.; Duguid, R.A.; Quinn, D.J.; Barnett, Y.A.; Rea, I.M.; El-Agnaf, O.M.; Gibson, J.M.; Wallace, A.; et al. mt4216C variant in linkage with the mtDNA TJ cluster may confer a susceptibility to mitochondrial dysfunction resulting in an increased risk of Parkinson’s disease in the Irish. Exp. Gerontol. 2003, 38, 397–405. [Google Scholar] [CrossRef]
- Janetzky, B.; Schmid, C.; Bischof, F.; Frölich, L.; Gsell, W.; Kalaria, R.N.; Riederer, P.; Reichmann, H. Investigations on the point mutation at position 5460 of the mtDNA in different neurodegenerative and neuromuscular diseases. Eur. Neurol. 1996, 36, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.H.; Lin, R.; Wisniewski, H.M.; Hwang, Y.W.; Grundke Iqbal, I.; Healy Louie, G.; Iqbal, K. Detection of point mutations in codon 331 of mitochondrial NADH dehydrogenase subunit 2 in Alzheimer’s brains. Biochem. Biophys. Res. Commun. 1992, 182, 238–246. [Google Scholar] [CrossRef]
- Zhang, J.; Montine, T.J.; Smith, M.A.; Siedlak, S.L.; Gu, G.; Robertson, D.; Perry, G. The mitochondrial common deletion in Parkinson’s disease and related movement disorders. Parkinsonism Relat. Disord. 2002, 8, 165–170. [Google Scholar] [CrossRef]
- Wei, Y.H. Mitochondrial DNA alterations as ageing-associated molecular events. Mutat. Res. 1992, 275, 145–155. [Google Scholar] [CrossRef]
- Smigrodzki, R.; Parks, J.; Parker, W.D. High frequency of mitochondrial complex I mutations in Parkinson’s disease and aging. Neurobiol. Aging 2004, 25, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Taravari, A.; Panov, S.; Petrov, I.; Petrova, V.; Medziti, F.; Haliti, G. Delta deletion 4977 in mitochondrial DNA in patients with idiopathic Parkinson’s disease. Bratisl. Lek. Listy. 2014, 115, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Gusdon, A.M.; Fang, F.; Chen, J.; Mathews, C.E.; Li, W.; Chu, C.T.; Ding, J.Q.; Chen, S.D. Association of the mt-ND2 5178A/C polymorphism with Parkinson’s disease. Neurosci. Lett. 2015, 587, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Vives-Bauza, C.; Gonzalo, R.; Manfredi, G.; Garcia-Arumi, E.; Andreu, A.L. Enhanced ROS production and antioxidant defenses in cybrids harbouring mutations in mtDNA. Neurosci. Lett. 2006, 391, 136–141. [Google Scholar] [CrossRef]
- Bruno, C.; Martinuzzi, A.; Tang, Y.; Andreu, A.L.; Pallotti, F.; Bonilla, E.; Shanske, S.; Fu, J.; Angelini, C.; DiMauro, S.; et al. A stop-codon mutation in the human mtDNA cytochrome c oxidase I gene disrupts the functional structure of complex IV. Am. J. Hum. Genet. 1999, 65, 611–620. [Google Scholar] [CrossRef][Green Version]
- D’Aurelio, M.; Palloti, F.; Barrientos, A.; Gajewski, C.D.; Kwong, J.Q.; Bruno, C.; Beal, M.F.; Manfredi, G. In vivo regulation of oxidative phosphorylation in cells harboring a stop-codon mutation in mitochondrial DNA-encoded cytochrome c oxidase subunit I. J. Biol. Chem. 2001, 276, 46925–46932. [Google Scholar] [CrossRef]
- Van der Walt, J.M.; Nicodemus, K.K.; Martin, E.R.; Scott, W.K.; Nance, M.A.; Watts, R.L.; Hubble, J.P.; Haines, J.L.; Koller, W.C.; Lyons, K.; et al. Mitochondrial polymorphisms significantly reduce the risk of Parkinson disease. Am. J. Hum. Genet. 2003, 72, 804–811. [Google Scholar] [CrossRef]
- Autere, J.; Moilanen, J.S.; Finnilä, S.; Soininen, H.; Mannermaa, A.; Hartikainen, P.; Hallikainen, M.; Majamaa, K. Mitochondrial DNA polymorphisms as risk factors for Parkinson’s disease and Parkinson’s disease dementia. Hum. Genet. 2004, 115, 29–35. [Google Scholar]
- Parker, W.D.; Parks, J.K. Mitochondrial ND5 mutations in idiopathic Parkinson’s disease. Biochem. Biophys. Res. Commun. 2005, 326, 667–669. [Google Scholar] [CrossRef]
- Gonzalo, R.; Garcia-Arumi, E.; Llige, D.; Martia, R.; Solano, A.; Montoya, J.; Arenasd, J.; Andreua, A.L. Free radicals-mediated damage in transmitochondrial cells harboring the T14487C mutation in the ND6 gene of mtDNA. FEBS Lett. 2005, 579, 6909–6913. [Google Scholar] [CrossRef]
- Solano, A.; Roig, M.; Vives-Bauza, C.; Hernandez-Pena, J.; Garcia-Arumi, E.; Playan, A.; Lopez-Perez, M.J.; Andreu, A.L.; Montoya, J. Bilateral Striatal Necrosis Associated with a Novel Mutation in the Mitochondrial ND6 Gene. Ann. Neurol. 2003, 54, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Keogh, M.J.; Wilson, I.; Coxhead, J.; Ryan, S.; Rollinson, S.; Griffin, H.; Kurzawa-Akanbi, M.; Santibanez-Koref, M.; Talbot, K.; et al. Mitochondrial DNA point mutations and relative copy number in 1363 disease and control human brains. Acta Neuropathol. Commun. 2017, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.K.; Lin, M.T.; Zheng, L.; Liu, G.J.; Ahn, C.H.; Kim, L.M.; Mauck, W.M.; Twu, F.; Beal, M.F.; Johns, D.R. Somatic mitochondrial DNA mutations in cortex and substantia nigra in aging and Parkinson’s disease. Neurobiol. Aging. 2004, 25, 71–81. [Google Scholar] [CrossRef]
- Alam, Z.I.; Jenner, A.; Daniel, S.E.; Lees, A.J.; Cairns, N.; Marsden, D.; Jenner, P.; Halliwell, B.J. Oxidative DNA damage in the parkinsonian brain: An apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J. Neurochem. 1997, 69, 1196–1203. [Google Scholar] [CrossRef] [PubMed]
- Nido, G.S.; Dölle, C.; Flønes, I.; Tuppen, H.A.; Alves, G.; Tysnes, O.B.; Haugarvoll, K.; Tzoulis, C. Ultradeep mapping of neuronal mitochondrial deletions in Parkinson’s disease. Neurobiol. Aging 2018, 63, 120–127. [Google Scholar] [CrossRef]
- Podlesniy, P.; Puigròs, M.; Serra, N.; Fernández-Santiago, R.; Ezquerra, M.; Tolosa, E.; Trullas, R. Accumulation of mitochondrial 7S DNA in idiopathic and LRRK2 associated Parkinson’s disease. EBioMedicine 2019, 48, 554–567. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria in cybrids containing mtDNA from persons with mitochondriopathies. J. Neurosci. Res. 2007, 85, 3416–3428. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Carl, S.M.; Swerdlow, R.H. Cytoplasmic hybrid (cybrid) cell lines as a practical model for mitochondriopathies. Redox Biol. 2014, 2, 619–631. [Google Scholar] [CrossRef]
- Arduíno, D.M.; Esteves, A.R.; Swerdlow, R.H.; Cardoso, S.M. A cybrid cell model for the assessment of the link between mitochondrial deficits and sporadic Parkinson’s disease. Methods Mol. Biol. 2015, 1265, 415–424. [Google Scholar]
- Swerdlow, R.H.; Parks, J.K.; Miller, S.W.; Tuttle, J.B.; Trimmer, P.A.; Sheehan, J.P.; Bennett, J.P., Jr.; Davis, R.E.; Parker, W.D., Jr. Origin and functional consequences of the complex I defect in Parkinson’s disease. Ann. Neurol. 1996, 40, 663–671. [Google Scholar] [CrossRef]
- Gu, M.; Cooper, J.M.; Taanman, J.W.; Schapira, A.H. Mitochondrial DNA transmission of the mitochondrial defect in Parkinson’s disease. Ann. Neurol. 1998, 44, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Domingues, A.F.; Ferreira, I.L.; Januario, C.; Swerdlow, R.H.; Oliveira, C.R.; Cardoso, S.M. Mitochondrial function in Parkinson’s disease cybrids containing an nt2 neuron-like nuclear background. Mitochondrion 2008, 8, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Arduino, D.M.; Swerdlow, R.H.; Oliveira, C.R.; Cardoso, S.M. Oxidative stress involvement in alpha-synuclein oligomerization in Parkinsons disease cybrids. Antioxid. Redox Signal. 2009, 11, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Arduíno, D.M.; Swerdlow, R.H.; Oliveira, C.R.; Cardoso, S.M. Microtubule depolymerization potentiates alpha-synuclein oligomerization. Front. Aging Neurosci. 2010, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Arduino, D.M.; Swerdlow, R.H.; Oliveira, C.R.; Cardoso, S.M. Dysfunctional mitochondria uphold calpain activation: Contribution to Parkinson’s disease pathology. Neurobiol. Dis. 2010, 37, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Lu, J.; Rodova, M.; Onyango, I.; Lezi, E.; Dubinsky, R.; Lyons, K.E.; Pahwa, R.; Burns, J.M.; Cardoso, S.M.; et al. Mitochondrial respiration and respiration-associated proteins in cell lines created through Parkinson’s subject mitochondrial transfer. J. Neurochem. 2010, 113, 674–682. [Google Scholar] [CrossRef]
- Keeney, P.M.; Dunham, L.D.; Quigley, C.K.; Morton, S.L.; Bergquist, K.; Bennett, J.P., Jr. Cybrid models of Parkinson’s disease show variable mitochondrial biogenesis and genotype respiration relationships. Exp. Neurol. 2009, 220, 374–382. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Parks, J.K.; Cassarino, D.S.; Binder, D.R.; Bennett, J.P., Jr.; Di Iorio, G.; Golbe, L.I.; Parker, W.D., Jr. Biochemical analysis of cybrids expressing mitochondrial DNA from Contursi kindred Parkinson’s subjects. Exp. Neurol. 2001, 169, 479–485. [Google Scholar] [CrossRef]
- Aomi, Y.; Chen, C.S.; Nakada, K.; Ito, S.; Isobe, K.; Murakami, H.; Kuno, S.Y.; Tawata, M.; Matsuoka, R.; Mizusawa, H.; et al. Cytoplasmic transfer of platelet mtDNA from elderly patients with Parkinson’s disease to mtDNA-less HeLa cells restores complete mitochondrial respiratory function. Biochem. Biophys. Res. Commun. 2001, 280, 265–273. [Google Scholar] [CrossRef]
- Spinazzola, A.; Zeviani, M. Mitochondrial diseases: A cross-talk between mitochondrial and nuclear genomes. Adv. Exp. Med. Biol. 2009, 652, 69–84. [Google Scholar]
- Maret, G.; Testa, B.; Jenner, P.; el Tayar, N.; Carrupt, P.A. The MPTP story: MAO activates tetrahydropyridine derivatives to toxins causing parkinsonism. Drug Metab. Rev. 1990, 22, 291–332. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M.R. The biochemistry of Parkinson’s disease. Annu. Rev. Biochem. 2005, 74, 9–52. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, A.E.; Buneeva, O.A.; Kopylov, A.T.; Tikhonova, O.V.; Medvedeva, M.V.; Nerobkova, L.N.; Kapitsa, I.G.; Zgoda, V.G. The brain mitochondrial subproteome of Rpn10-binding proteins and its changes induced by the neurotoxin MPTP and the neuroprotector isatin. Biochemistry (Moscow) 2017, 82, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Buneeva, O.; Kopylov, A.; Kapitsa, I.; Ivanova, E.; Zgoda, V.; Medvedev, A. The Effect of Neurotoxin MPTP and Neuroprotector Isatin on the Profile of Ubiquitinated Brain Mitochondrial Proteins. Cells 2018, 7, 91. [Google Scholar] [CrossRef]
- Mandavilli, B.S.; Ali, S.F.; Van Houten, B. DNA damage in brain mitochondria caused by aging and MPTP treatment. Brain Res. 2000, 885, 45–52. [Google Scholar] [CrossRef]
- Chen, L.J.; Gao, Y.Q.; Li, X.J.; Shen, D.H.; Sun, F.Y. Melatonin protects against MPTP/MPP+ -induced mitochondrial DNA oxidative damage in vivo and in vitro. J. Pineal. Res. 2005, 39, 34–42. [Google Scholar] [CrossRef]
- Chin, M.H.; Qian, W.J.; Wang, H.; Petyuk, V.A.; Bloom, J.S.; Sforza, D.M.; Laćan, G.; Liu, D.; Khan, A.H.; Cantor, R.M.; et al. Mitochondrial dysfunction, oxidative stress, and apoptosis revealed by proteomic and transcriptomic analyses of the striata in two mouse models of Parkinson’s disease. J. Proteome Res. 2008, 7, 666–677. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, J.Y.; Chin, M.H.; Schepmoes, A.A.; Petyuk, V.A.; Weitz, K.K.; Petritis, B.O.; Monroe, M.E.; Camp, D.G.; Wood, S.A.; et al. Region-specific protein abundance changes in the brain of MPTP-induced Parkinson’s disease mouse model. J. Proteome Res. 2010, 9, 1496–1509. [Google Scholar] [CrossRef][Green Version]
- Cannon, J.R.; Tapias, V.; Na, H.M.; Honick, A.S.; Drolet, R.E.; Greenamyre, J.T. A highly reproducible rotenone model of Parkinson’s disease. Neurobiol. Dis. 2009, 34, 279–290. [Google Scholar] [CrossRef]
- Sanders, L.H.; McCoy, J.; Hu, X.; Mastroberardino, P.G.; Dickinson, B.C.; Chang, C.J.; Chu, C.T.; Van Houten, B.; Greenamyre, J.T. Mitochondrial DNA damage: Molecular marker of vulnerable nigral neurons in Parkinson’s disease. Neurobiol. Dis. 2014, 70, 214–223. [Google Scholar] [CrossRef]
- Heinz, S.; Freyberger, A.; Lawrenz, B.; Schladt, L.; Schmuck, G.; Ellinger-Ziegelbauer, H. Mechanistic Investigations of the Mitochondrial Complex I Inhibitor Rotenone in the Context of Pharmacological and Safety Evaluation. Sci. Rep. 2017, 7, 45465. [Google Scholar] [CrossRef] [PubMed]
- Karthikkeyan, G.; Najar, M.A.; Pervaje, R.; Pervaje, S.K.; Modi, P.K.; Prasad, T.S.K. Identification of Molecular Network Associated with Neuroprotective Effects of Yashtimadhu (Glycyrrhiza glabra L.) by Quantitative Proteomics of Rotenone-Induced Parkinson’s Disease Model. ACS Omega 2020, 5, 26611–26625. [Google Scholar] [CrossRef] [PubMed]
- Hance, N.; Ekstrand, M.I.; Trifunovic, A. Mitochondrial DNA polymerase gamma is essential for mammalian embryogenesis. Hum. Mol. Genet. 2005, 14, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly-Y, M.; Gidlöf, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Vermulst, M.; Wanagat, J.; Kujoth, G.C.; Bielas, J.H.; Rabinovitch, P.S.; Prolla, T.A.; Loeb, L.A. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat. Genet. 2008, 40, 392–394. [Google Scholar] [CrossRef] [PubMed]
- Hiona, A.; Sanz, A.; Kujoth, G.C.; Pamplona, R.; Seo, A.Y.; Hofer, T.; Someya, S.; Miyakawa, T.; Nakayama, C.; Samhan-Arias, A.K.; et al. Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS ONE 2010, 5, e11468. [Google Scholar] [CrossRef]
- Edgar, D.; Shabalina, I.; Camara, Y.; Wredenberg, A.; Calvaruso, M.A.; Nijtmans, L.; Nedergaard, J.; Cannon, B.; Larsson, N.G.; Trifunovic, A. Random point mutations with major effects on protein-coding genes are the driving force behind premature aging in mtDNA mutator mice. Cell Metab. 2009, 10, 131–138. [Google Scholar] [CrossRef]
- Dai, Y.; Clark, J.; Zheng, K.; Kujoth, G.C.; Prolla, T.A.; Simon, D.K. Somatic mitochondrial DNA mutations do not increase neuronal vulnerability to MPTP in young POLG mutator mice. Neurotoxicol. Teratol. 2014, 46, 62–67. [Google Scholar] [CrossRef]
- Hauser, D.N.; Primiani, C.T.; Langston, R.G.; Kumaran, R.; Cookson, M.R. The Polg Mutator Phenotype Does Not Cause Dopaminergic Neurodegeneration in DJ-1-Deficient Mice. eNeuro 2015, 2. [Google Scholar] [CrossRef]
- Ahlqvist, K.J.; Hämäläinen, R.H.; Yatsuga, S.; Uutela, M.; Terzioglu, M.; Götz, A.; Forsström, S.; Salven, P.; Angers-Loustau, A.; Kopra, O.H.; et al. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell Metab. 2012, 15, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Logan, A.; Shabalina, I.G.; Prime, T.A.; Rogatti, S.; Kalinovich, A.V.; Hartley, R.C.; Budd, R.C.; Cannon, B.; Murphy, M.P. In vivo levels of mitochondrial hydrogen peroxide increase with age in mtDNA mutator mice. Aging Cell 2014, 13, 765–768. [Google Scholar] [CrossRef] [PubMed]
- Hauser, D.N.; Dillman, A.A.; Ding, J.; Li, Y.; Cookson, M.R. Post-translational decrease in respiratory chain proteins in the Polg mutator mouse brain. PLoS ONE 2014, 9, e94646. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Turnbull, D.M.; Reeve, A.K. Mitochondrial Dysfunction in Parkinson’s Disease-Cause or Consequence? Biology 2019, 8, 38. [Google Scholar] [CrossRef]
- Müller-Nedebock, A.C.; Brennan, R.R.; Venter, M.; Pienaar, I.S.; van der Westhuizen, F.H.; Elson, J.L.; Ross, O.A.; Bardien, S. The unresolved role of mitochondrial DNA in Parkinson’s disease: An overview of published studies, their limitations, and future prospects. Neurochem. Int. 2019, 129, 104495. [Google Scholar] [CrossRef]
- Lin, X.; Shi, M.; Masilamoni, J.G.; Dator, R.; Movius, J.; Aro, P.; Smith, Y.; Zhang, J. Proteomic profiling in MPTP monkey model for early Parkinson disease biomarker discovery. Biochim. Biophys Acta 2015, 1854, 779–787. [Google Scholar] [CrossRef]
- Sharma, L.K.; Lu, J.; Bai, Y. Mitochondrial respiratory complex I: Structure, function and implication in human diseases. Curr Med. Chem. 2009, 16, 1266–1277. [Google Scholar] [CrossRef]
- Kirby, D.M.; McFarland, R.; Ohtake, A.; Dunning, C.; Ryan, M.T.; Wilson, C.; Ketteridge, D.; Turnbull, D.M.; Thorburn, D.R.; Taylor, R.W. Mutations of the mitochondrial ND1 gene as a cause of MELAS. J. Med. Genet. 2004, 41, 784–789. [Google Scholar] [CrossRef]
- McFarland, R.; Kirby, D.M.; Fowler, K.J.; Ohtake, A.; Ryan, M.T.; Amor, D.J.; Fletcher, J.M.; Dixon, J.W.; Collins, F.A.; Turnbull, D.M.; et al. De novo mutations in the mitochondrial ND3 gene as a cause of infantile mitochondrial encephalopathy and complex I deficiency. Ann. Neurol. 2004, 55, 58–64. [Google Scholar] [CrossRef]
- Deng, J.H.; Li, Y.; Park, J.S.; Wu, J.; Hu, P.; Lechleiter, J.; Bai, Y. Nuclear suppression of mitochondrial defects in cells without the ND6 subunit. Mol. Cell. Biol. 2006, 26, 1077–1086. [Google Scholar] [CrossRef][Green Version]
- Das, S.; Ferlito, M.; Kent, O.A.; Fox-Talbot, K.; Wang, R.; Liu, D.; Raghavachari, N.; Yang, Y.; Wheelan, S.J.; Murphy, E.; et al. Nuclear miRNA Regulates the Mitochondrial Genome in the Heart. Circ. Res. 2012, 110, 1596–1603. [Google Scholar] [CrossRef] [PubMed]
- Passamonti, M.; Calderone, M.; Delpero, M.; Plazzi, F. Clues of in vivo nuclear gene regulation by mitochondrial short non-coding RNAs. Sci. Rep. 2020, 10, 8219. [Google Scholar] [CrossRef] [PubMed]
- Castegna, A.; Iacobazzi, V.; Infantino, V. The mitochondrial side of epigenetics. Physiol. Genom. 2015, 47, 299–307. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Complexes [21,22,23,24,25] | Catalytic Activity (EC) | Total Number of Subunits | Subunits Encoded by the Mitochondrial Genome |
|---|---|---|---|
| Complex I | NADH:ubiquinone reductase (EC 7.1.1.2) | 44 | ND1, ND2, ND3, ND4, ND4L, ND5, ND6 |
| Complex II | Succinate dehydrogenase (EC 1.3.5.1) | 4 | 0 |
| Complex III | Ubiquinol—cytochrome-c reductase (EC 7.1.1.8) | 11 | 1 (CYB) |
| Complex IV | Cytochrome c oxidase (EC 7.1.1.9) | 13 | COXI, COXII, COXIII |
| Complex V | ATP synthase (H+-transporting two-sector ATPase; EC 7.1.2.2) | 14 | ATP6, ATP8 |
| Gene Encoding | Nucleotide Position in mtDNA Genome | Mutation Location | Detected Effect | References |
|---|---|---|---|---|
| tRNA threonine tRNA (Thr) | 15888..15953 | nt.15927 and nt.15928 | Frequent point mutations | [95] |
| tRNA glutamine tRNA (Gln) | 4329..4400 | nt.A4336G | Loss of the HpaII site, increased frequency in PD-women The activity of complex I may be decreased | [95,96,97,98,99,100] |
| tRNA leucine 1 (UUA/G) tRNA (Leu) | 3230..3304 | nt.G3243A | point mutation, heteroplasmic state | [101] |
| tRNA lysine tRNA (Lys) | 8295..8364 | nt.A8344G | point mutation | [102] |
| 12S ribosomal RNA (RNR1) | 648..1601 |
nt. 956-965, nt. T1095C |
5-nucleotide insertion, point mutation | [103,104] |
| 16S RNA (RNR2) | 1671..3229 |
nt.T2158C nt.3196 |
associated with reduced risk of PD heteroplasmic 16S rRNA variant | [103,105] |
| NADH dehydrogenase, subunit 1 (ND1) | 3307..4262 |
nt.A3397G nt.T4216C | polymorphism | [101,106] |
| NADH dehydrogenase, subunit 2 (ND2) | 4470..5511 | nt.G5460A nt.C5178A nt.4977 p.A5T, p.A5V, p.M187T, p.M187I, p.I239M, p.I239H | point mutation point mutation common deletion amino acid substitutions | [98] [107,108] [109,110] [111,112,113] |
| Cytochrome c oxidase subunit I | 5904..7445 | nt.G6930A | Point mutation, causing enhanced ROS production a | [114,115,116] |
| NADH dehydrogenase, subunit 3 (ND3) | 10059..10404 | nt.A10398G | point mutation Haplogroup I, J, or K had a slightly decreased risk of PD but an increased risk of PDD Protective effect for women | [99,117,118] |
| NADH dehydrogenase, subunit 4L (ND4L) | 10470..10766 | p.L77F | amino acid substitution | [111] |
| NADH dehydrogenase, subunit 4 (ND4) | 10760..12137 | nt.A11251G | point mutation associated with reduced risk of PD | [105] |
| NADH dehydrogenase, subunit 5 (ND5) | 12337..14148 | p.E145G, pE145V, p.E145D p.124-145 |
amino acid substitution, deletion of 30 nts | [111] [119] |
| 12S ribosomal RNA (RNR1) | 648..1601 |
nt. 956-965, nt. T1095C |
5-nucleotide insertion point mutation | [103,104] |
| 16S ribosomal RNA (RNR2) | 1671..3229 |
nt.T2158C nt.3196 |
associated with reduced risk of PD heteroplasmic 16S rRNA variant | [105] [103] |
| NADH dehydrogenase, subunit 6 (ND6) | 14149..14673 | nt.T14487C | Point mutation causing free radical damage of cells b | [120,121] |
| mtDNA | complete genome 16569 | heteroplasmy | [122] | |
| mtDNA | complete genome 16569 bp | Transversions G: C → T: A and T: A → G: C in point mutations | all point mutations increase with age in the frontal cortex (FCtx) | [123] |
| Source of PD mtDNA | Cell Line Used to Generate Cybrids | Mitochondrial Changes | Extramitochondrial Changes | Reference |
|---|---|---|---|---|
| Platelets from sporadic PD patients | SHSY5Y neuroblastoma | Decreased complex I activity and increased ROS production | Increased susceptibility MPP-induced programmed cell death | [130] |
| PD patients with low platelet complex I activity | A549 lung adenocarcinoma | combined complex I and IV deficiencies | [131] | |
| PD patients with reduced platelet complex I activity | NT2 teratocarcinoma cells | Decreased Complex I-IV activities | Increased LDH release, increased caspase-3 activity, increased MPP+-induced activation of caspase-9 and caspase-3 | [132] |
| Platelets from sporadic PD patients | NT2 teratocarcinoma cells | Decreased Complex I activity and ATP level | Higher ROS production, Increased number of protein carbonyl groups, microtubule alteration, α-synuclein oligomerization | [133] |
| Platelets from PD patients without any nuclear DNA mutation | NT2 teratocarcinoma cells | Increased protein ubiquitination, microtubule depolymerization, and α-synuclein oligomerization | [134] | |
| Platelets from PD patients | NT2 teratocarcinoma cells | Decreased mitochondrial calcium | Increased cytosolic calcium, increased calpain expression, and activation | [135] |
| PD patients with reduced platelet complex I activity | NT2 teratocarcinoma cells | Decreased Complex I activity, lower ATP, depolarized mitochondria, slightly increased ATP-independent proton leak | Decreased levels of PGC1α Decreased levels of SIRT1 phosphorylation Higher transcriptional activity of NF-κB | [136] |
| Platelets from individuals with idiopathic (sporadic) Parkinson’s disease (sPD) | SH-SY5Y Neuroblastoma cells | Insignificant trend for reduction of Complex I respiration, unaltered level of ETC subunit proteins mtDNA levels varied | mtDNA levels varied and correlated with expression of PGC-1α | [137] |
| Platelets from Contursi kindred PD subjects | SH-SY5Y Neuroblastoma cells | Lack of significant changes in Complex I and IV activities | Increased glutathione peroxidase | [138] |
| Platelets from elderly PD patients | HeLa cells | mtDNA transfer restored mitochondrial respiration of HeLa cells No significant changes were found between control and PD cybrids | [139] | |
| Platelets from a patient with mtDNA mutation T14487C mutant | human osteosarcoma 143B cells | Overproduction of ROS causing increased oxidation of lipids and mtDNA | Increased lipid oxidation Insignificant changes in catalase and SOD | [121] [120] |
| Enucleated cells from patients with mtDNA mutationsA3243G in tRNALeuUUR and A8344G in tRNALys | human osteosarcoma cells (A3243G in tRNALeuUUR and A8344G in tRNALys) | both mutations showed severe deficits of complexes I, III, and IV | Increased ROS production with a parallel increase in the antioxidant enzyme activities (SOD, catalase, glutathione peroxidase) | [114] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buneeva, O.; Fedchenko, V.; Kopylov, A.; Medvedev, A. Mitochondrial Dysfunction in Parkinson’s Disease: Focus on Mitochondrial DNA. Biomedicines 2020, 8, 591. https://doi.org/10.3390/biomedicines8120591
Buneeva O, Fedchenko V, Kopylov A, Medvedev A. Mitochondrial Dysfunction in Parkinson’s Disease: Focus on Mitochondrial DNA. Biomedicines. 2020; 8(12):591. https://doi.org/10.3390/biomedicines8120591
Chicago/Turabian StyleBuneeva, Olga, Valerii Fedchenko, Arthur Kopylov, and Alexei Medvedev. 2020. "Mitochondrial Dysfunction in Parkinson’s Disease: Focus on Mitochondrial DNA" Biomedicines 8, no. 12: 591. https://doi.org/10.3390/biomedicines8120591
APA StyleBuneeva, O., Fedchenko, V., Kopylov, A., & Medvedev, A. (2020). Mitochondrial Dysfunction in Parkinson’s Disease: Focus on Mitochondrial DNA. Biomedicines, 8(12), 591. https://doi.org/10.3390/biomedicines8120591