Mitochondrial Dysfunction and DNA Damage in the Context of Pathogenesis of Atherosclerosis

 , , , and
, , , and 
Abstract
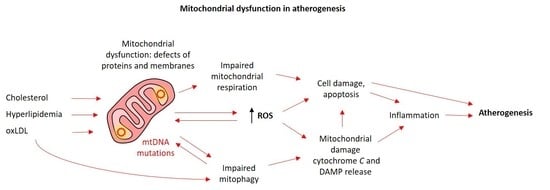
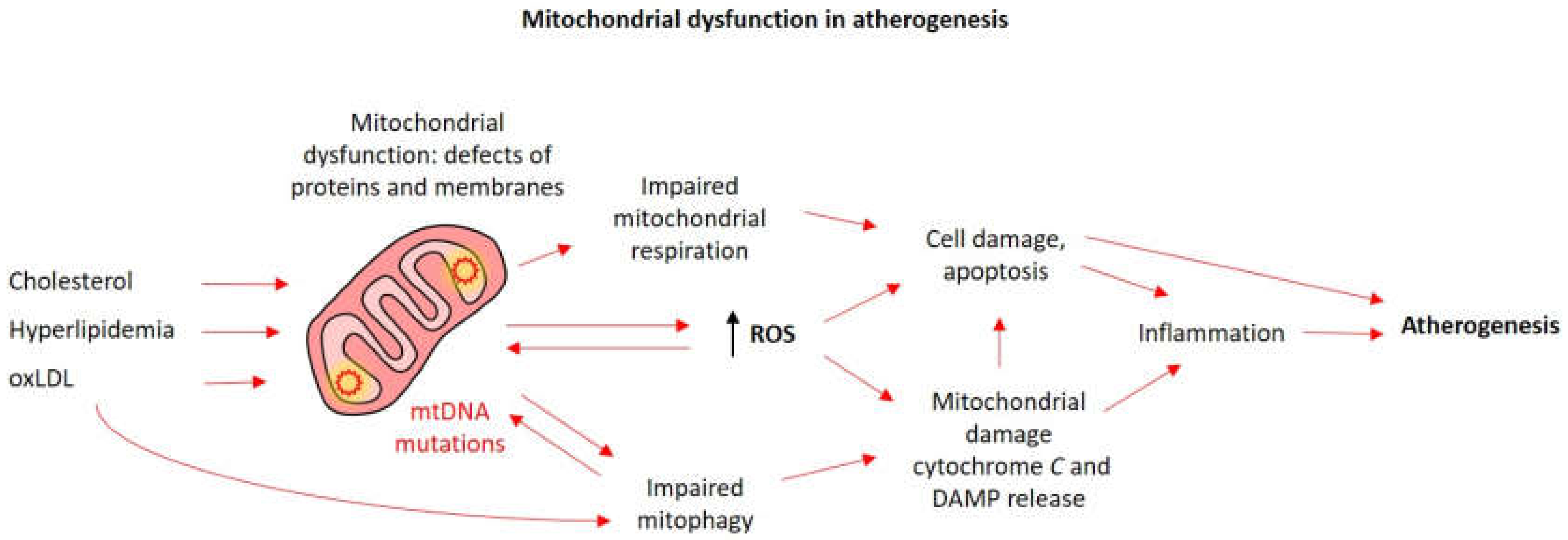
:
1. Introduction
2. Atherosclerosis as an Inflammatory Process
3. Oxidative Stress and Atherosclerosis
4. Mitochondrial Reactive Oxygen Species
5. Low Density Lipoproteins in the Pathogenesis of Atherosclerosis
6. Mitochondrial Dysfunction in Atherosclerosis
7. mtDNA Mutations
8. Endothelial Dysfunction in Atherosclerosis
9. Prospects for Development of Anti-Atherosclerosis Therapies
10. Conclusions
Funding
Conflicts of Interest
References
- WHO. Cardiovascular Diseases. Available online: https://www.who.int/health-topics/cardiovascular-diseases/#tab=tab_1 (accessed on 31 May 2020).
- Corina, D.-C. Atherosclerosis in the young adult: Fewer hypotheses, more facts. Med. Surg. J. 2016, 120, 768–776. [Google Scholar]
- McGill, H.C.; McMahan, A.; Zieske, A.W.; Sloop, G.D.; Walcott, J.F.; Troxclair, D.A.; Malcom, G.T.; Tracey, R.E.; Oalmann, M.C.; Strong, J.P. Pathobiological determinants of atherosclerosis in youth (PDAY) research group associations of coronary heart disease risk factors with the intermediate lesion of atherosclerosis in youth. Arterioscler. Throm. Basc. Biol. 2000, 20, 1998–2004. [Google Scholar]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting early atherosclerosis: a focus on oxidative stress and inflammation. Oxid. Med. Cell Longev. 2019, 2019, 8563845. [Google Scholar]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109, III-27–III-32. [Google Scholar]
- Wang, H.H.; Garruti, G.; Liu, M.; Portincasa, P.; Wang, D.Q. Cholesterol and lipoprotein metabolism and atherosclerosis: Recent advances in reverse cholesterol transport. Ann. Hepatol. 2017, 16, s27–s42. [Google Scholar]
- Capron, L.; Wyplosz, B. The infection theory in atherosclerosis. Arch. Maladies Coeur Vaiss. 1998, 91, 21–26. [Google Scholar]
- Mannarino, E.; Pirro, M. Endothelial injury and repair: A novel theory for atherosclerosis. Angiology 2008, 59, 69S–72S. [Google Scholar]
- Salvayre, R.; Negre-Salvayre, A.; Camaré, C. Oxidative theory of atherosclerosis and antioxidants. Biochimie 2016, 125, 281–296. [Google Scholar]
- Peterlin, A.; Petrovič, D.; Peterlin, B. Screening for rare genetic variants associated with atherosclerosis: Opportunity for personalized medicine. Curr. Vasc. Pharmacol. 2019, 17, 25–28. [Google Scholar]
- Freigang, S. The regulation of inflammation by oxidized phospholipids. Eur. J. Immunol. 2016, 46, 1818–1825. [Google Scholar]
- Farmer, J.A.; Torre-Amione, G. Atherosclerosis and inflammation. Curr. Atheroscl. Rep. 2002, 4, 92–98. [Google Scholar]
- Pelisek, J.; Wendorff, H.; Wendorff, C.; Kuehnl, A.; Eckstein, H. Age-associated changes in human carotid atherosclerotic plaques. Ann. Med. 2016, 48, 541–551. [Google Scholar]
- Gisterå, A.; Hansson, G.K. The immunology of atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368–380. [Google Scholar]
- Rosenfeld, M.E.; Ylä-Herttuala, S.; Lipton, B.A.; Ord, V.A.; Witztum, J.L.; Steinberg, D. Macrophage colony-stimulating factor mRNA and protein in atherosclerotic lesions of rabbits and humans. Am. J. Pathol. 1992, 140, 291. [Google Scholar]
- Tedgui, A.; Mallat, Z. Cytokines in atherosclerosis: Pathogenic and regulatory pathways. Physiol. Rev. 2006, 86, 515–581. [Google Scholar]
- Orekhov, A.N.; Sukhorukov, V.N.; Nikiforov, N.G.; Kubekina, M.V.; Sobenin, I.A.; Foxx, K.K.; Pintus, S.; Stegmaier, P.; Stelmashenko, D.; Kel, A. Signaling pathways potentially responsible for foam cell formation: cholesterol accumulation or inflammatory response—What is first? Int. J. Mol. Sci. 2020, 21, 2716. [Google Scholar]
- Hartman, J.; Frishman, W.H. Inflammation and atherosclerosis: A review of the role of interleukin-6 in the development of atherosclerosis and the potential for targeted drug therapy. Cardiol. Rev. 2014, 22, 147–151. [Google Scholar]
- Orekhov, A.N.; Poznyak, A.V.; Sobenin, I.A.; Nikifirov, N.N.; Ivanova, E.A. Mitochondrion as a selective target for treatment of atherosclerosis: Role of mitochondrial DNA mutations and defective mitophagy in the pathogenesis of atherosclerosis and chronic inflammation. Curr. Neuropharmacol. 2019. [Google Scholar] [CrossRef]
- Burtenshaw, D.; Kitching, M.; Redmond, E.M.; Megson, I.L.; Cahill, P.A. Reactive oxygen species (ROS), intimal thickening, and subclinical atherosclerotic disease. Front. Cardiovasc. Med. 2019, 6, 89. [Google Scholar]
- Steinhubl, S.R. Why have antioxidants failed in clinical trials? Am. J. Cardiol. 2008, 101, S14–S19. [Google Scholar]
- Vogiatzi, G.; Tousoulis, D.; Stefanadis, C. The role of oxidative stress in atherosclerosis. Hellenic J. Cardiol. 2009, 50, 402–409. [Google Scholar]
- Gimbrone, M.A. Jr.; García-Cardeña, G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar]
- Pescetelli, I.; Zimarino, M.; Ghirarduzzi, A.; De Caterina, R. Localizing factors in atherosclerosis. J. Cardiovasc. Med. (Hagerstown) 2015, 16, 824–830. [Google Scholar]
- Ragino, Y.I.; Polonskaya, Y.V.; Sadovski, E.V. Th-P15: 137 Parameters of oxidative stress and endothelial dysfunction in coronary atherosclerosis men. Atheroscler. Suppl. 2006, 7, 523. [Google Scholar]
- Sirisha, C.V.N.; Manohar, R.M. Study of antioxidant enzymes superoxide dismutase and glutathione peroxidase levels in tobacco chewers and smokers: A pilot study. J. Cancer Res. Ther. 2013, 9, 210. [Google Scholar]
- Kluge, M.A.; Fetterman, J.L.; Vita, J.A. Mitochondria and endothelial function. Circ. Res. 2013, 112, 1171–1188. [Google Scholar]
- Hulsmans, M.; Van Dooren, E.; Holvoet, P. Mitochondrial reactive oxygen species and risk of atherosclerosis. Curr. Atheroscler. Rep. 2012, 14, 264–276. [Google Scholar]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular mechanisms of angiotensin II–mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar]
- Peng, W.; Cai, G.; Xia, Y.; Chen, J.; Wu, P.; Wang, Z.; Li, G.; Wei, D. Mitochondrial dysfunction in atherosclerosis. DNA Cell Biol. 2019, 38, 597–606. [Google Scholar]
- Chistiakov, D.A.; Bobryshev, Y.V.; Orekhov, A.N. Macrophage-mediated cholesterol handling in atherosclerosis. J. Cell Mol. Med. 2016, 20, 17–28. [Google Scholar]
- Linton, M.F.; Yancey, P.G.; Davies, S.S.; Jerome, W.G.; Linton, E.F.; Song, W.L.; Doran, A.C.; Vickers, K.C. The role of lipids and lipoproteins in atherosclerosis. In Endotext [Internet]; MDText.com, Inc.: South Dartmouth, MA, USA, 2019. [Google Scholar]
- Ravnskov, U. High cholesterol may protect against infections and atherosclerosis. QJM 2003, 96, 927–934. [Google Scholar]
- Orekhov, A.N.; Tertov, V.V.; Mukhin, D.N.; Mikhailenko, I.A. Modification of low density lipoprotein by desialylation causes lipid accumulation in cultured cells: Discovery of desialylated lipoprotein with altered cellular metabolism in the blood of atherosclerotic patients. Biochem. Biophys. Res. Commun. 1989, 162, 206–211. [Google Scholar]
- Tatami, R.; Mabuchi, H.; Ueda, K.; Ueda, R.; Haba, T.; Kametani, T.; Ito, S.; Koizumi, J.; Ohta, M.; Miyamoto, S. Intermediate-density lipoprotein and cholesterol-rich very low density lipoprotein in angiographically determined coronary artery disease. Circulation 1981, 64, 1174–1184. [Google Scholar]
- Ivanova, E.A.; Bobryshev, Y.V.; Orekhov, A.N. LDL electronegativity index: A potential novel index for predicting cardiovascular disease. Vasc. Health Risk Manag. 2015, 11, 525. [Google Scholar]
- Tertov, V.V.; Bittolo-Bon, G.; Sobenin, I.A.; Cazzolato, G.; Orekhov, A.N.; Avogaro, P. Naturally occurring modified low density lipoproteins are similar if not identical: More electronegative and desialylated lipoprotein subfractions. Exp. Mol. Pathol. 1995, 62, 166–172. [Google Scholar]
- Hartley, A.; Haskard, D.; Khamis, R. Oxidized LDL and anti-oxidized LDL antibodies in atherosclerosis—Novel insights and future directions in diagnosis and therapy. Trends Cardiovasc. Med. 2019, 29, 22–26. [Google Scholar]
- Acton, S.L.; Scherer, P.E.; Lodish, H.F.; Krieger, M. Expression cloning of SR-BI, a CD36-related class B scavenger receptor. J. Biol. Chem. 1994, 269, 21003–21009. [Google Scholar]
- Silverstein, R.L.; Febbraio, M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal. 2009, 2, re3. [Google Scholar]
- Kosswig, N.; Rice, S.; Daugherty, A.; Post, S.R. Class A scavenger receptor-mediated adhesion and internalization require distinct cytoplasmic domains. J. Biol. Chem. 2003, 278, 34219–34225. [Google Scholar]
- Krieger, M. Scavenger receptor class B type I is a multiligand HDL receptor that influences diverse physiologic systems. J. Clin. Invest. 2001, 108, 793–797. [Google Scholar]
- Sobenin, I.A.; Salonen, J.T.; Zhelankin, A.V.; Melnichenko, A.A.; Kaikkonen, J.; Bobryshev, Y.V.; Orekhov, A.N. Low density lipoprotein-containing circulating immune complexes: Role in atherosclerosis and diagnostic value. BioMed Res. Int. 2014, 2014, 205697. [Google Scholar]
- Cimmino, G.; Cirillo, P.; Conte, S.; Pellegrino, G.; Barra, G.; Maresca, L.; Morello, A.; Calì, G.; Loffredo, F.; De Palma, R. Oxidized low-density lipoproteins induce tissue factor expression in T-lymphocytes via activation of lectin-like oxidized low-density lipoprotein receptor-1. Cardiovasc. Res. 2020, 116, 1125–1135. [Google Scholar]
- Zhong, S.; Li, L.; Shen, X.; Li, Q.; Xu, W.; Wang, X.; Tao, Y.; Yin, H. An update on lipid oxidation and inflammation in cardiovascular diseases. Free Radic. Biol. Med. 2019, 144, 266–278. [Google Scholar]
- Jin, H.; Ko, Y.S.; Park, S.W.; Kim, H.J. P2Y2R activation by ATP induces oxLDL-mediated inflammasome activation through modulation of mitochondrial damage in human endothelial cells. Free Radic. Biol. Med. 2019, 136, 109–117. [Google Scholar]
- Emma, P.K.; Bennett, M.R. The role of mitochondrial DNA damage in the development of atherosclerosis. Free Radic. Biol. Med. 2016, 100, 223–230. [Google Scholar]
- Yu, E.; Calvert, P.A.; Mercer, J.R.; Harrison, J.; Baker, L.; Figg, N.L.; Kumar, S.; Wang, J.C.; Hurst, L.A.; Obaid, D.R. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation 2013, 128, 702–712. [Google Scholar]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and mitophagy in cardiovascular disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar]
- Swaminathan, B.; Goikuria, H.; Vega, R.; Rodríguez-Antigüedad, A.; Medina, A.L.; del Mar Freijo, M.; Vandenbroeck, K.; Alloza, I. Autophagic marker MAP1LC3B expression levels are associated with carotid atherosclerosis symptomatology. PLoS ONE 2014, 9, e115176. [Google Scholar]
- Sergin, I.; Bhattacharya, S.; Emanuel, R.; Esen, E.; Stokes, C.J.; Evans, T.D.; Arif, B.; Curci, J.A.; Razani, B. Inclusion bodies enriched for p62 and polyubiquitinated proteins in macrophages protect against atherosclerosis. Sci. Signal. 2016, 9, ra2. [Google Scholar]
- Gottlieb, R.A.; Gustafsson, Å.B. Mitochondrial turnover in the heart. Biochim. Biophys. Acta. 2011, 1813, 1295–1301. [Google Scholar]
- Swiader, A.; Nahapetyan, H.; Faccini, J.; D’Angelo, R.; Mucher, E.; Elbaz, M.; Boya, P.; Vindis, C. Mitophagy acts as a safeguard mechanism against human vascular smooth muscle cell apoptosis induced by atherogenic lipids. Oncotarget 2016, 7, 28821. [Google Scholar]
- Grootaert, M.O.; Roth, L.; Schrijvers, D.M.; De Meyer, G.R.; Martinet, W. Defective autophagy in atherosclerosis: To die or to senesce? Oxid. Med. Cell Longev. 2018, 2018, 7687083. [Google Scholar]
- Morciano, G.; Patergnani, S.; Bonora, M.; Pedriali, G.; Tarocco, A.; Bouhamida, E.; Marchi, S.; Ancora, G.; Anania, G.; Wieckowski, M.R. Mitophagy in cardiovascular diseases. J. Clin. Med. 2020, 9, 892. [Google Scholar]
- Yu, S.; Zhang, L.; Liu, C.; Yang, J.; Zhang, J.; Huang, L. PACS2 is required for ox-LDL-induced endothelial cell apoptosis by regulating mitochondria-associated ER membrane formation and mitochondrial Ca2+ elevation. Exp. Cell Res. 2019, 379, 191–202. [Google Scholar]
- Golubenko, M.V.; Salakhov, R.R.; Makeeva, O.A.; Goncharova, I.A.; Kashtalap, V.V.; Barbarash, O.L.; Puzyrev, V.P. Association of mitochondrial DNA polymorphism with myocardial infarction and prognostic signs for atherosclerosis. Mol. Biol. 2015, 49, 867–874. [Google Scholar]
- Ballinger, S.W.; Patterson, C.; Knight-Lozano, C.A.; Burow, D.L.; Conklin, C.A.; Hu, Z.; Reuf, J.; Horaist, C.; Lebovitz, R.; Hunter, G.C. Mitochondrial integrity and function in atherogenesis. Circulation 2002, 106, 544–549. [Google Scholar]
- Suzuki, T.; Nagao, A.; Suzuki, T. Human mitochondrial tRNAs: Biogenesis, function, structural aspects, and diseases. Ann. Rev. Genet. 2011, 45, 299–329. [Google Scholar]
- Mohammed, S.A.; Ambrosini, S.; Lüscher, T.; Paneni, F.; Costantino, S. Epigenetic control of mitochondrial function in the vasculature. Front. Cardiovasc. Med. 2020, 7, 28. [Google Scholar]
- Sinyov, V.V.; Sazonova, M.A.; Ryzhkova, A.I.; Galitsyna, E.V.; Melnichenko, A.A.; Postnov, A.Y.; Orekhov, A.N.; Grechko, A.V.; Sobenin, I.A. Potential use of buccal epithelium for genetic diagnosis of atherosclerosis using mtDNA mutations. Vessel Plus 2017, 1, 145–150. [Google Scholar]
- Hu, H.; Lin, Y.; Xu, X.; Lin, S.; Chen, X.; Wang, S. The alterations of mitochondrial DNA in coronary heart disease. Exp. Mol. Pathol. 2020, 104412. [Google Scholar]
- Volobueva, A.; Grechko, A.; Yet, S.-F.; Sobenin, I.; Orekhov, A. Changes in mitochondrial genome associated with predisposition to atherosclerosis and related disease. Biomolecules 2019, 9, 377. [Google Scholar]
- Heidari, M.M.; Derakhshani, M.; Sedighi, F.; Foruzan-Nia, S.K. Mutation analysis of the mitochondrial tRNA genes in Iranian coronary atherosclerosis patients. Iran J. Public Health 2017, 46, 1379. [Google Scholar]
- Finsterer, J. Atherosclerosis can be mitochondrial: A review. Cureus 2020, 12, e6987. [Google Scholar]
- Mengel-From, J.; Thinggaard, M.; Dalgård, C.; Kyvik, K.O.; Christensen, K.; Christiansen, L. Mitochondrial DNA copy number in peripheral blood cells declines with age and is associated with general health among elderly. Human Genet. 2014, 133, 1149–1159. [Google Scholar]
- Baccarelli, A.A.; Byun, H.-M. Platelet mitochondrial DNA methylation: A potential new marker of cardiovascular disease. Clin. Epigenet. 2015, 7, 44. [Google Scholar]
- Frey, R.S.; Gao, X.; Javaid, K.; Siddiqui, S.S.; Rahman, A.; Malik, A.B. Phosphatidylinositol 3-kinase γ signaling through protein kinase Cζ induces NADPH oxidase-mediated oxidant generation and NF-κB activation in endothelial cells. J. Biol. Chem. 2006, 281, 16128–16138. [Google Scholar]
- Rush, J.W.; Denniss, S.G.; Graham, D.A. Vascular nitric oxide and oxidative stress: determinants of endothelial adaptations to cardiovascular disease and to physical activity. Can. J. Appl. Physiol. 2005, 30, 442–474. [Google Scholar]
- Paone, S.; Baxter, A.A.; Hulett, M.D.; Poon, I.K. Endothelial cell apoptosis and the role of endothelial cell-derived extracellular vesicles in the progression of atherosclerosis. Cell Mol. Life Sci. 2019, 76, 1093–1106. [Google Scholar]
- Sata, M.; Walsh, K. Oxidized LDL activates fas-mediated endothelial cell apoptosis. J. Clin. Invest. 1998, 102, 1682–1689. [Google Scholar]
- Sheu, M.L.; Ho, F.M.; Yang, R.S.; Chao, K.F.; Lin, W.W.; Lin-Shiau, S.Y.; Liu, S.-H. High glucose induces human endothelial cell apoptosis through a phosphoinositide 3-kinase–regulated cyclooxygenase-2 pathway. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 539–545. [Google Scholar]
- Gioscia-Ryan, R.A.; LaRocca, T.J.; Sindler, A.L.; Zigler, M.C.; Murphy, M.P.; Seals, D.R. Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. J. Physiol. 2014, 592, 2549–2561. [Google Scholar]
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; de la Mata, M.; Villanueva-Paz, M.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Suárez-Carrillo, A.; Talaverón-Rey, M.; Munuera, M. Atherosclerosis and coenzyme Q10. Int. J. Mol. Sci. 2019, 20, 5195. [Google Scholar]
- Poznyak, A.V.; Grechko, A.V.; Orekhova, V.A.; Chegodaev, Y.S.; Wu, W.-K.; Orekhov, A.N. Oxidative stress and antioxidants in atherosclerosis development and treatment. Biology 2020, 9, 60. [Google Scholar]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The essential medicinal chemistry of curcumin. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar]
- Chen, H.-I.; Hu, W.-S.; Hung, M.-Y.; Ou, H.-C.; Huang, S.-H.; Hsu, P.-T.; Day, C.-H.; Lin, K.-H.; Viswanadha, V.P.; Kuo, W.-W.; et al. Protective effects of luteolin against oxidative stress and mitochondrial dysfunction in endothelial cells. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 1032–1043. [Google Scholar]
- Sarparanta, J.; Garcia-Macia, M.; Singh, R. Autophagy and mitochondria in obesity and type 2 diabetes. Curr. Diabetes Rev. 2017, 13, 352–369. [Google Scholar]
- Bubb, K.J.; Drummond, G.R.; Figtree, G.A. New opportunities for targeting redox dysregulation in cardiovascular disease. Cardiovasc. Res. 2020, 116, 532–544. [Google Scholar]
- Jiang, L.; Wang, J.; Jiang, J.; Zhang, C.; Zhao, M.; Chen, Z.; Wang, N.; Hu, D.; Liu, X.; Peng, H.; et al. Sonodynamic therapy in atherosclerosis by curcumin nanosuspensions: Preparation design, efficacy evaluation, and mechanisms analysis. Eur. J. Pharm. Biopharm. 2020, 146, 101–110. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | mtDNA Variants | Reference(s) | ||
|---|---|---|---|---|
| Leukocytes and arterial wall tissue (post-mortem samples) from patients with atherosclerosis | m.204T>C | m.3243A>G | m.14459G>A | [63] |
| m.228G>A | m.12315G>A | m.8251G>A | ||
| m.16223C>T | m.3336T>C | m.9477G>A | ||
| m.1719G>A | m.5178C>A | m.ins8528A | ||
| m.3010G>A | m.12705C>T | m.14709G>A | ||
| m.3256C>T | m.13513G>A | m15059G>A | ||
| Peripheral blood cells from CAD patients | m.5725T>G | m.5568A>G | [64] | |
| m.12308A>G | m.5711A>G | |||
| Various sources | m.617G>A | m.8794T>C | m.3316G>A | [65] |
| m.3243A>G | m.8839G>C | |||
| Name | Description | Effect | Reference(s) |
|---|---|---|---|
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha - transcriptional coactivator | Favorable effect on the endothelial phenotype, stimulation of mitochondrial biogenesis | [27] |
| MitoQ | Modified mitochondria-targeted ubiquinone | Decreased vascular dysfunction, normalization of mitochondrial stress | [73,74] |
| Resveratrol | Antioxidant polyphenolic nature | Gene regulation, normalization of mitochondrial membrane potential, increased level of mitochondrial fusion proteins, inhibition of mitochondrial pharagmentation | [56,75] |
| Quercetin | Flavonoid antioxidant | Anti-inflammatory effect, effect on lipid metabolism, delayed development of atherosclerosis | [75] |
| Melatonin | Antioxidant | Disposal of ROS by activation of mitophagy in macrophages | [75] |
| Curcumin | Antioxidant polyphenolic nature | Decreased synthesis of proatherogenic cytokines, induction of anti-inflammatory polarization of macrophages | [75] |
| Luteolin | Flavonoid antioxidant | Protection against H2O2-induced oxidative stress, suppression of intracellular Ca2+ growth and cytochrome c release, normalization of mitochondrial membrane potential | [76] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shemiakova, T.; Ivanova, E.; Grechko, A.V.; Gerasimova, E.V.; Sobenin, I.A.; Orekhov, A.N. Mitochondrial Dysfunction and DNA Damage in the Context of Pathogenesis of Atherosclerosis. Biomedicines 2020, 8, 166. https://doi.org/10.3390/biomedicines8060166
Shemiakova T, Ivanova E, Grechko AV, Gerasimova EV, Sobenin IA, Orekhov AN. Mitochondrial Dysfunction and DNA Damage in the Context of Pathogenesis of Atherosclerosis. Biomedicines. 2020; 8(6):166. https://doi.org/10.3390/biomedicines8060166
Chicago/Turabian StyleShemiakova, Taisiia, Ekaterina Ivanova, Andrey V. Grechko, Elena V. Gerasimova, Igor A. Sobenin, and Alexander N. Orekhov. 2020. "Mitochondrial Dysfunction and DNA Damage in the Context of Pathogenesis of Atherosclerosis" Biomedicines 8, no. 6: 166. https://doi.org/10.3390/biomedicines8060166
APA StyleShemiakova, T., Ivanova, E., Grechko, A. V., Gerasimova, E. V., Sobenin, I. A., & Orekhov, A. N. (2020). Mitochondrial Dysfunction and DNA Damage in the Context of Pathogenesis of Atherosclerosis. Biomedicines, 8(6), 166. https://doi.org/10.3390/biomedicines8060166