From Orai to E-Cadherin: Subversion of Calcium Trafficking in Cancer to Drive Proliferation, Anoikis-Resistance, and Metastasis



Abstract
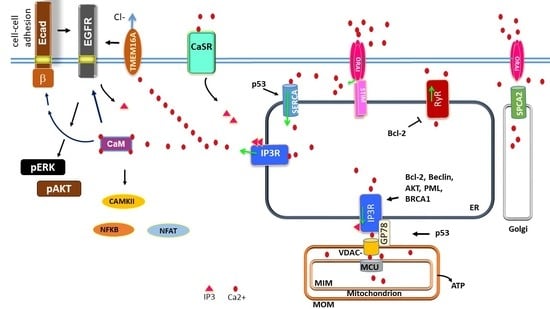
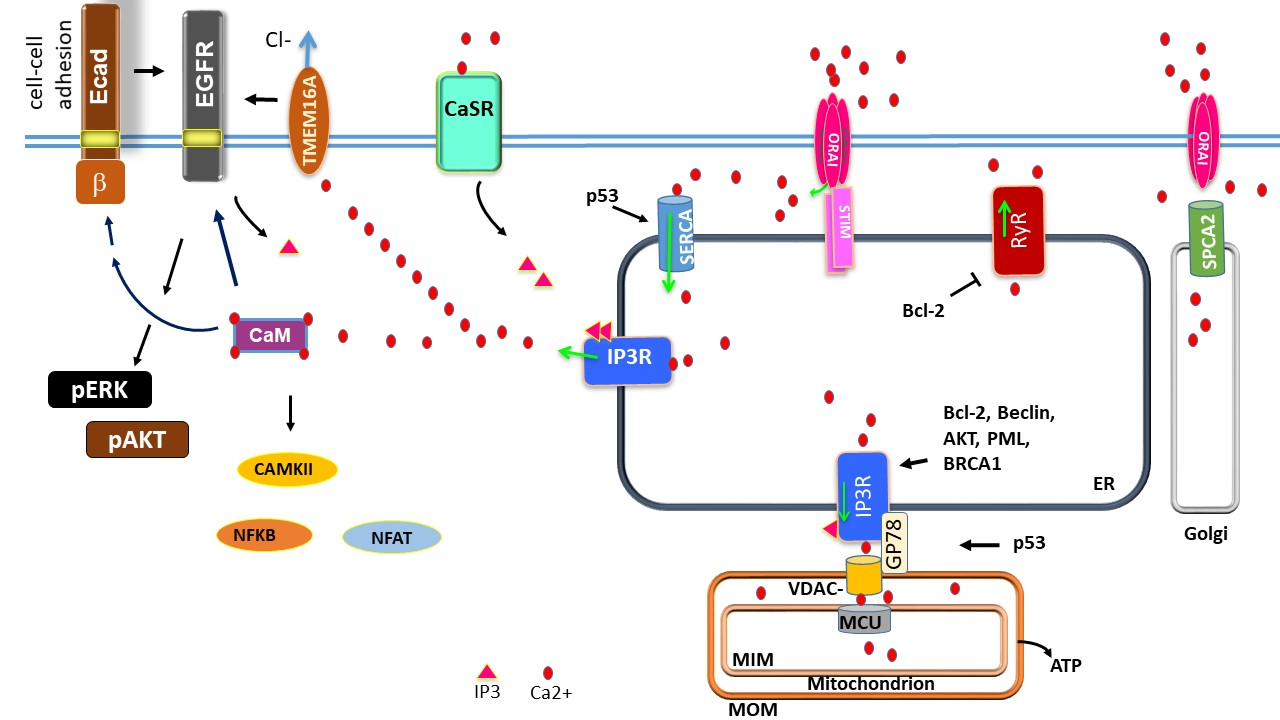{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Storage and Management of Intracellular Calcium in Normal and Cancer Cells
2.1. Intracellular Stores: IP3R, RyR, SERCA
2.2. Store-Operated Calcium Entry
2.3. Store-Independent Calcium Entry
3. Calcium Regulation of Epithelial Differentiation, E-Cadherin-Beta Catenin, and Cell–Cell Adhesion
4. Calcium Regulation of EGFR
4.1. Calcium Regulation of EGFR via Calmodulin
4.2. Calcium Regulation of EGFR via TMEM16A
5. Calcium, E-Cadherin, EGFR, and Routes to Anoikis-Resistance in Cancer
5.1. EMT vs. Retention of Epithelial Program in Circulating Tumor Cells
5.2. E-Cadherin Cooperativity with EGFR and Her2 in Anoikis-Resistance
5.3. Detachment, iCa2+, and ROS in Anoikis-Resistance
6. E-Cadherin, Invasion, and Metastasis
7. Conclusions
Funding
Conflicts of Interest
References
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signaling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.K. Intracellular Calcium. Wiley 2014, 1–38. [Google Scholar] [CrossRef]
- Carafoli, E.; Krebs, J. Why Calcium? How Calcium Became the Best Communicator. J. Biol. Chem. 2016, 40, 20849–20857. [Google Scholar] [CrossRef]
- Burk, S.E.; Lytton, J.; MacLennan, D.H.; Shull, G.E. cDNA Cloning, Functional Expression, and mRNA Tissue Distribution of a Third Organellar Ca2+ Pump. J. Biol. Chem. 1989, 264, 18561–18568. [Google Scholar]
- Foskett, J.K.; White, C.; Cheung, K.H.; Mak, D.O. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007, 87, 593–658. [Google Scholar] [CrossRef]
- Streb, H.; Irvine, R.F.; Berridge, M.J.; Schulz, I. Release of Ca2+from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-tris-phosphate. Nature 1983, 306, 67–69. [Google Scholar] [CrossRef]
- Lewis, R.S. Calcium signaling mechanisms in T lymphocytes. Ann. Rev. Immunol. 2001, 19, 497–521. [Google Scholar] [CrossRef]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef]
- Kaufman, R.J.; Malhotra, J.D. Calcium trafficking integrates endoplasmic reticulum function with mitochondrial bioenergetics. Biochim. Biophys. Acta 2014, 1843, 2233–2239. [Google Scholar] [CrossRef]
- Pinto, M.C.X.; Kihara, A.H.; Goulart, V.A.M.; Tonelli, F.M.P.; Gomes, K.N.; Ulrich, H.; Resende, R.R. Calcium signaling and cell proliferation. Cell Signal. 2015, 27, 2139–2149. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium signaling and cell proliferation. Bioessays 1995, 17, 491–500. [Google Scholar] [CrossRef]
- Szado, T.; Vanderheyden, V.; Parys, H.; De Smedt, J.B.; Rietdorf, K.; Kotelevets, L.; Chastre, E.; Khan, F.; Landegren, U.; Soderberg, O.; et al. Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 2427–2432. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, H.K.; Gerasimenko, J.V.; Thorne, C.; Ferdek, P.; Pozzan, T.; Tepikin, A.V.; Petersen, O.H.; Sutton, R.; Watson, A.J.; Gerasimenko, O.V. Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J. Biol. Chem. 2009, 284, 20796–20803. [Google Scholar] [CrossRef]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Eckenrode, E.F.; Yang, J.; Velmurugan, G.V.; Foskett, J.K.; White, C. Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1,4,5-trisphosphate receptor-dependent Ca2+ signaling. J. Biol Chem. 2010, 285, 13678–13684. [Google Scholar] [CrossRef]
- Monaco, G.; Beckers, M.; Ivanova, H.; Missiaen, L.; Parys, J.B.; De Smedt, H.; Bultynck, G. Profiling of the Bcl-2/Bcl-XL-binding sites on type 1 IP3 receptor. Biochem. Biophys. Res. Commun. 2012, 428, 31–35. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Li, C.; Yang, J.; Petrenko, N.B.; Madesh, M.; Thompson, C.B.; Foskett, J.K. The endoplasmic reticulum gateway to apoptosis by Bcl-XL modulation of the InsP3R. Nat. Cell Biol. 2005, 7, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Valencia, I.; Zhong, F.; McColl, K.S.; Roderick, H.L.; Bootman, M.D.; Berridge Michael, J.; Conway, J.S.; Holmes, A.B.; Mignery, G.A. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J. Cell Biol. 2004, 166, 193–203. [Google Scholar] [CrossRef]
- Yang, J.; Vais, H.; Gu, W.; Foskett, J.K. Biphasic regulation of InsP3 receptor gating by dual Ca2+ release channel BH3-like domains mediates Bcl-xL control of cell viability. Proc. Natl. Acad. Sci. USA 2016, 113, E1953–E1962. [Google Scholar] [CrossRef]
- Vervliet, T.; Clerix, E.; Seitaj, B.; Ivanova, H.; Monaco, G.; Bultynck, G. Modulation of Ca2+ Signaling by Anti-apoptotic B-Cell Lymphoma 2 Proteins at the Endoplasmic Reticulum–Mitochondrial Interface. Front. Oncol. 2017. [Google Scholar] [CrossRef]
- Akl, H.; Bultynck, G. Altered Ca(2+) signaling in cancer cells: Proto-oncogenes and tumor suppressors targeting IP3 receptors. Biochim. Biophys. Acta 2013, 1835, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Narita, M.; Tsujimoto, Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 1999, 399, 483–487. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Bononi, A.; Romagnoli, A.; Messina, A.; De Pinto, V.; Pinton, P.; Rizzuto, R. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ. 2012, 19, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.S.; Clements, A.; Williams, A.C.; Berger, C.N.; Frankel, G. Bax inhibitor 1 in apoptosis and disease. Oncogene 2011, 30, 2391–2400. [Google Scholar] [CrossRef]
- Li, C.; Wang, X.; Vais, H.; Thompson, C.B.; Foskett, J.K.; White, C. Apoptosis regulation by Bcl-x(L) modulation of mammalian inositol 1,4,5-trisphosphate receptor channel isoform gating. Proc. Natl. Acad. Sci. USA 2007, 104, 12565–12570. [Google Scholar] [CrossRef]
- Varadarajan, S.; Bampton, E.T.; Smalley, J.L.; Tanaka, K.; Caves, R.E.; Butterworth, M.; Wei, J.; Pellecchia, M.; Mitcheson, J.; Gant, T.W.; et al. A novel cellular stress response characterised by a rapid reorganisation of membranes of the endoplasmic reticulum. Cell Death Differ. 2012, 19, 1896–1907. [Google Scholar] [CrossRef]
- Wiestner, A. Ibrutinib and Venetoclax—Doubling Down on CLL. N. Engl. J. Med. 2019, 380, 2169–2171. [Google Scholar] [CrossRef]
- Giorgi, C.; Ito, K.; Lin, H.K.; Santangelo, C.; Wieckowski, M.R.; Lebiedzinska, M.; Bononi, A.; Bonora, M.; Duszynski, J.; Bernardi, R.; et al. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 2010, 330, 1247–1251. [Google Scholar] [CrossRef]
- Boehning, D.; Patterson, R.L.; Sedaghat, L.; Glebova, N.O.; Kurosaki, T.; Snyder, S.H. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium dependent apoptosis. Nat. Cell Biol. 2003, 5, 1051–1061. [Google Scholar] [CrossRef]
- Hedgepeth, S.C.; Garcia, M.I.; Wagner, L.E.; Rodriguez, A.M.; Chintapalli, S.V.; Snyder, R.R.; Hankins, G.D.; Henderson, B.R.; Brodie, K.M.; Yule, D.I.; et al. The BRCA1 tumor suppressor binds to inositol 1,4,5-trisphosphate receptors to stimulate apoptotic calcium release. J. Biol. Chem. 2015, 290, 7304–7313. [Google Scholar] [CrossRef]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, J.P.; Bultynck, G.; Parys, J.B. A dual role for Ca2+ in autophagy regulation. Cell Calcium 2011, 50, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, H.; Vervliet, T.; Missiaen, L.; Parys, J.B.; De Smedt, H.; Bultynck, G. Inositol 1,4,5-trisphosphate receptor-isoform diversity in cell death and survival? Biochim. Biophys. Acta 2014, 1843, 2164–2183. [Google Scholar] [CrossRef]
- Filadi, R.; Leal, N.S.; Schreiner, B.; Rossi, A.; Dentoni, G.; Pinho, C.M.; Wiehager, B.; Cieri, D.; Calì, T.; Pizzo, P.; et al. TOM70 Sustains Cell Bioenergetics by Promoting IP3R3-Mediated ER to Mitochondria Ca2+ Transfer. Curr Biol. 2018, 28, 369–382.e6. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R.J. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. Cell Biol. 2006, 2006 175, 901–911. [Google Scholar] [CrossRef]
- McCormack, J.G.; Denton, R.M. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem. J. 1979, 180, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.N.; Kelleher, J.K.; Fiskum, G. Submicromolar Ca2+ regulates phosphorylating respiration by normal rat liver and AS-30D hepatoma mitochondria by different mechanisms. J. Biol. Chem. 1990, 265, 10527–10534. [Google Scholar]
- Cárdenas, C.; Müller, M.; McNeal, A.; Lovy, A.; Jana, F.; Bustos, G.; Urra, F.; Smith, N.; Molgó, J.; Diehl, J.A.; et al. Selective Vulnerability of Cancer Cells by Inhibition of Ca(2+) Transfer from Endoplasmic Reticulum to Mitochondria. Cell Rep. 2016, 15, 219–220. [Google Scholar] [CrossRef]
- Van Petegem, F. Ryanodine receptors: Structure and function. J. Biol. Chem. 2012, 287, 31624–31632. [Google Scholar] [CrossRef]
- Denda, S.; Kumamoto, J.; Takei, K.; Tsutsumi, M.; Aoki, H.; Denda, M. Ryanodine receptors are expressed in epidermal keratinocytes and associated with keratinocyte differentiation and epidermal permeability barrier homeostasis. J. Invest. Dermatol. 2012, 132, 69–75. [Google Scholar] [CrossRef]
- Vervliet, T.; Decrock, E.; Molgó, J.; Sorrentino, V.; Missiaen, L.; Leybaert, L.; De Smedt, H.; Kasri, N.N.; Parys, J.B.; Bultynck, G. Bcl-2 binds to and inhibits ryanodine receptors. J. Cell Sci. 2014, 127, 2782–2792. [Google Scholar] [CrossRef] [PubMed]
- Abdul, M.; Ramlal, S.; Hoosein, N. Ryanodine receptor expression correlates with tumor grade in breast cancer. Pathol. Oncol. Res. 2008, 14, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, Y.; Song, F.; Zheng, H.; Hu, L.; Lu, H.; Liu, P.; Hao, X.; Zhang, W.; Chen, K. Functional SNP in the microRNA-367 binding site in the 3′UTR of the calcium channel ryanodine receptor gene 3 (RYR3) affects breast cancer risk and calcification. Proc. Natl. Acad. Sci. USA 2011, 108, 13653–13658. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.M.; Parsonage, M.T.; Cabot, P.J.; Parat, M.O.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Assessment of gene expression of intracellular calcium channels, pumps and exchangers with epidermal growth factor-induced epithelial-mesenchymal transition in a breast cancer cell line. Cancer Cell Int. 2013, 13, 76. [Google Scholar] [CrossRef]
- Hamilton, S.; Terentyeva, R.; Kim, T.Y.; Bronk, P.; Clements, R.T.; O-Uchi, J.; Csordás, G.; Choi, B.R.; Terentyev, D. Pharmacological Modulation of Mitochondrial Ca(2+) Content Regulates Sarcoplasmic Reticulum Ca(2+) Release via Oxidation of the Ryanodine Receptor by Mitochondria-Derived Reactive Oxygen Species. Front. Physiol. 2018, 9, 1831. [Google Scholar] [CrossRef]
- Van Petegem, F. Ryanodine Receptors: Allosteric Ion Channel Giants. J. Mol. Biol. 2015, 427, 31–53. [Google Scholar] [CrossRef]
- Reddish, F.N.; Miller, C.L.; Gorkhali, R.; Yang, J.J. Calcium Dynamics Mediated by the Endoplasmic/Sarcoplasmic Reticulum and Related Diseases. Int. J. Mol. Sci. 2017, 18, 1024. [Google Scholar] [CrossRef]
- Taylor, C.W.; Tovey, S.C. IP(3) receptors: Toward understanding their activation. Cold Spring Harb. Persp. Biol. 2010, 2, a004010. [Google Scholar] [CrossRef]
- Kania, E.; Roest, G.; Vervliet, T.; Parys, J.B.; Bultynck, G. IP3 Receptor-Mediated Calcium Signaling and Its Role in Autophagy in Cancer. Front. Oncol. 2017, 7, 140. [Google Scholar] [CrossRef]
- Giorgi, C.; Bonora, M.; Sorrentino, G.; Missiroli, S.; Poletti, F.; Suski, J.M.; Galindo Ramirez, F.; Rizzuto, R.; Di Virgilio, F.; Zito, E.; et al. P53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. USA 2015, 112, 1779–1784. [Google Scholar] [CrossRef]
- Pierro, C.; Sneyers, F.; Bultynck, G.; Roderick, H.L. ER Ca(2+) release and store-operated Ca(2+) entry—Partners in crime or independent actors in oncogenic transformation? Cell Calcium 2019, 82, 102061. [Google Scholar] [CrossRef] [PubMed]
- Bergner, A.; Kellner, J.; Tufman, A.; Huber, R.M. Endoplasmic reticulum Ca2+-homeostasis is altered in Small and non-small Cell Lung Cancer cell lines. J. Exp. Clin. Cancer Res. 2009, 28, 25. [Google Scholar] [CrossRef] [PubMed]
- Pierro, C.; Cook, S.J.; Foets, T.C.; Bootman, M.D.; Roderick, H.L. Oncogenic K-Ras suppresses IP3-dependent Ca²+ release through remodelling of the isoform composition of IP3Rs and ER luminal Ca²? levels in colorectal cancer cell lines. J. Cell Sci. 2014, 127, 1607–1619. [Google Scholar] [CrossRef]
- Brouland, J.P.; Gélébart, P.; Kovàcs, T.; Enouf, J.; Grossmann, J.; Papp, B. The loss of sarco/endoplasmic reticulum calcium transport ATPase 3 expression is an early event during the multistep process of colon carcinogenesis. Am. J. Pathol. 2005, 167, 233–242. [Google Scholar] [CrossRef]
- Gélébart, P.; Kovács, T.; Brouland, J.P.; van Gorp, R.; Grossmann, J.; Rivard, N.; Panis, Y.; Martin, V.; Bredoux, R.; Enouf, J.; et al. Expression of endomembrane calcium pumps in colon and gastric cancer cells. Induction of SERCA3 expression during differentiation. J. Biol. Chem. 2002, 277, 26310–26320. [Google Scholar] [CrossRef] [PubMed]
- Dellis, O.; Arbabian, A.; Brouland, J.P.; Kovàcs, T.; Rowe, M.; Chomienne, C.; Joab, I.; Papp, B. Modulation of B-cell endoplasmic reticulum calcium homeostasis by Epstein-Barr virus latent membrane protein-1. Mol. Cancer 2009, 8, 59. [Google Scholar] [CrossRef]
- Roti, G.; Carlton, A.; Ross, K.N.; Markstein, M.; Pajcini, K.; Su, A.H.; Perrimon, N.; Pear, W.S.; Kung, A.L.; Blacklow, S.C.; et al. Complementary genomic screens identify SERCA as a therapeutic target in NOTCH1 mutated cancer. Cancer Cell 2013, 23, 390–405. [Google Scholar] [CrossRef]
- Chemaly, E.R.; Troncone, L.; Lebeche, D. SERCA control of cell death and survival. Cell Calcium 2018, 69, 46–61. [Google Scholar] [CrossRef]
- Denmeade, S.R.; Mhaka, A.M.; Rosen, D.M.; Brennen, W.N.; Dalrymple, S.; Dach, I.; Olesen, C.; Gurel, B.; DeMarzo, A.M.; Wilding, G.; et al. Engineering a prostate-specific membrane antigen-activated tumor endothelial cell prodrug for cancer therapy. Sci. Transl. Med. 2012, 4, 140ra86. [Google Scholar] [CrossRef]
- Van Coppenolle, F.; Vanden Abeele, F.; Slomianny, C.; Flourakis, M.; Hesketh, J.; Dewailly, E.; Prevarskaya, N. Ribosome-translocon complex mediates calcium leakage from endoplasmic reticulum stores. J. Cell Sci. 2004, 117, 4135–4142. [Google Scholar] [CrossRef]
- Hammadi, M.; Oulidi, A.; Gackière, F.; Katsogiannou, M.; Slomianny, C.; Roudbaraki, M.; Dewailly, E.; Delcourt, P.; Lepage, G.; Lotteau, S.; et al. Modulation of ER stress and apoptosis by endoplasmic reticulum calcium leak via translocon during unfolded protein response: Involvement of GRP78. FASEB J. 2013, 27, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Sterea, A.M.; Almasi, S.; El Hiani, Y. The hidden potential of lysosomal ion channels: A new era of oncogenes. Cell Calcium 2018, 72, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.E.; Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 2019, 21, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; de Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef]
- Parkash, J.; Asotra, K. Calcium wave signaling in cancer cells. Life Sci. 2010, 87, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, S.B.; Monteith, G.R. Orai channels and cancer. Cell Calcium 2018, 74, 160–167. [Google Scholar] [CrossRef]
- Tanwar, J.; Motiani, R.K. Role of SOCE architects STIM and Orai proteins in CellDeath. Cell Calcium 2018, 69, 19–27. [Google Scholar] [CrossRef]
- McAndrew, D.; Grice, D.M.; Peters, A.A.; Davis, F.M.; Stewart, T.; Rice, M.; Smart, C.E.; Brown, M.A.; Kenny, P.A.; Roberts-Thomson, S.J.; et al. Orai1-mediated calcium influx in lactation and in breast cancer. Mol. Cancer Ther. 2011, 10, 448–460. [Google Scholar] [CrossRef]
- Motiani, R.K.; Hyzinski-García, M.C.; Zhang, X.; Henkel, M.M.; Abdullaev, I.F.; Kuo, Y.H.; Matrougui, K.; Mongin, A.A.; Trebak, M. STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflugers Arch. Eur. J. Physiol. 2013, 465, 1249–1260. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef]
- Kim, J.H.; Lkhagvadorj, S.; Lee, M.R.; Hwang, K.H.; Chung, H.C.; Jung, J.H.; Cha, S.K.; Eom, M. Orai1 and STIM1 are critical for cell migration and proliferation of clear cell renal cell carcinoma. Biochem. Biophys. Res. Commun. 2014, 448, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhang, H.; Jin, F.; Fang, M.; Huang, M.; Yang, C.S.; Chen, T.; Fu, L.; Pan, Z. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget 2014, 5, 3455–3471. [Google Scholar] [CrossRef] [PubMed]
- Flourakis, M.; Lehenkyi, V.; Beck, B.; Raphael, M.; Vandenberghe, M.; Abeele, F.V.; Roudbaraki, M.; Lepage, G.; Mauroy, B.; Romanin, C.; et al. Orai1contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell Death Dis. 2010, 1, e75. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Cobos, J.C.; Zhang, X.; Zhang, W.; Ruhle, B.; Motiani, R.K.; Schindl, R.; Muik, M.; Spinelli, A.M.; Bisaillon, J.M.; Shinde, A.V.; et al. Store-Independent Orai1/3 channels activated by intracrine leukotriene C4: Role in neointimal hyperplasia. Circ. Res. 2013, 112, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.; Vanden Abeele, F.; Lehen’kyi, V.; Gkika, D.; Guarmit, B.; Lepage, G.; Slomianny, C.; Borowiec, A.S.; Bidaux, G.; Benahmed, M.; et al. Remodeling of channel-forming Orai proteins determines an oncogenic switch in prostate cancer. Cancer Cell. 2014, 26, 19–32. [Google Scholar] [CrossRef]
- Cui, C.; Chang, Y.; Zhang, X.; Choi, S.; Tran, H.; Penmetsa, K.V.; Viswanadha, S.; Fu, L.; Pan, Z. Targeting Orai1-mediated store-operated calcium entry by RP4010 for anti-tumor activity in esophagus squamous cell carcinoma. Cancer Lett. 2018, 432, 169–179. [Google Scholar] [CrossRef]
- Rahman, S.; Rahman, T. Unveiling some FDA-approved drugs as inhibitors of the store-operated Ca2+ entry pathway. Sci. Rep. 2017, 7, 12881. [Google Scholar] [CrossRef]
- Feng, M.Y.; Rao, R. New insights into store-independent Ca(2+) entry: Secretory pathway calcium ATPase 2 in normal physiology and cancer. Int. J. Oral Sci. 2013, 5, 71–74. [Google Scholar] [CrossRef]
- Dang, D.K.; Makena, M.R.; Llongueras, J.P.; Prasad, H.; Ko, M.; Bandral, M.; Rao, R. A Ca(2+)-ATPase Regulates E-cadherin Biogenesis and Epithelial-Mesenchymal Transition in Breast Cancer Cells. Mol. Cancer Res. 2019, 17, 1735–1747. [Google Scholar] [CrossRef]
- Makena, M.R.; Rao, R. Subtype specific targeting of calcium signaling in breast cancer. Cell Calcium 2020, 85, 102109. [Google Scholar] [CrossRef]
- Feng, M.; Grice, D.M.; Faddy, H.M.; Nguyen, N.; Leitch, S.; Wang, Y.; Muend, S.; Kenny, P.A.; Sukumar, S.; Roberts-Thomson, S.J.; et al. Store independent activation of Orai1 by SPCA2 in mammary tumors. Cell 2010, 143, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Smaardijk, S.; Chen, J.; Wuytack, F.; Vangheluwe, P. SPCA2 couples Ca(2+) influx via Orai1 to Ca(2þ) uptake into the Golgi/secretory pathway. Tissue Cell 2017, 49, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Hennings, H.; Michael, D.; Cheng, C.; Steinert, P.; Holbrook, K.; Yuspa, S.H. Calcium regulation of growth and differentiation of mouse epidermal cells in culture. Cell 1980, 19, 245–254. [Google Scholar] [CrossRef]
- Hyafil, F.; Babinet, C.; Jacob, F. Cell-cell interactions in early embryogenesis: A molecular approach to the role of calcium. Cell 1981, 26, 447–454. [Google Scholar] [CrossRef]
- Nagafuchi, A.; Shirayoshi, Y.; Okazaki, K.; Yasuda, K.; Takeichi, M. Transformation of cell adhesion properties by exogenously introduced E-cadherin cDNA. Nature 1987, 329, 341–343. [Google Scholar] [CrossRef]
- Li, L.; Tucker, R.W.; Hennings, H.; Yuspa, S.H. Chelation of intracellular Ca2+ inhibits murine keratinocyte differentiation in vitro. J. Cell Physiol. 1995, 163, 105–114. [Google Scholar] [CrossRef]
- Hennings, H.; Kruszewski, F.H.; Yuspa, S.H.; Tucker, R.W. Intracellular calcium alterations in response to increased external calcium in normal and neoplastic keratinocytes. Carcinogenesis 1989, 10, 777–780. [Google Scholar] [CrossRef]
- Tu, C.L.; Chang, W.; Bikle, D.D. The extracellular calcium-sensing receptor is required for Calcium-induced differentiation in human keratinocytes. J. Biol. Chem. 2001, 276, 41079–41085. [Google Scholar] [CrossRef]
- Tu, C.L.; Chang, W.; Xie, Z.; Bikle, D.D. Inactivation of the calcium sensing receptor inhibits E-cadherin-mediated cell-cell adhesion and calcium-induced differentiation in human epidermal keratinocytes. J. Biol. Chem. 2008, 283, 3519–3528. [Google Scholar] [CrossRef]
- Tu, C.L.; Chang, W.; Bikle, D.D. The role of the calcium sensing receptor in regulating intracellular calcium handling in human epidermal keratinocytes. J. Investig. Dermatol. 2007, 127, 1074–1083. [Google Scholar] [CrossRef]
- Li, L.; Tucker, R.W.; Hennings, H.; Yuspa, S.H. Inhibitors of the intracellular Ca(2+)-ATPase in cultured mouse keratinocytes reveal components of terminal differentiation that are regulated by distinct intracellular Ca2+ compartments. Cell Growth Differ. 1995, 6, 1171–1184. [Google Scholar] [PubMed]
- Li, Z.; Kim, S.H.; Higgins, J.M.; Brenner, M.B.; Sacks, D.B. IQGAP1 and calmodulin modulate E-cadherin function. J. Biol. Chem. 1999, 274, 37885–37892. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.M.; Hedman, A.C.; Sacks, D.B. IQGAPs choreograph cellular signaling from the membrane to the nucleus. Trends Cell Biol. 2015, 25, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Noritake, J.; Watanabe, T.; Sato, K.; Wang, S.; Kaibuchi, K. IQGAP1: A key regulator of adhesion and migration. J. Cell Sci. 2005, 118, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Noritake, J.; Fukata, M.; Sato, K.; Nakagawa, M.; Watanabe, T.; Izumi, N.; Wang, S.; Fukata, Y.; Kaibuchi, K. Positive role of IQGAP1, an effector of Rac1, in actin-meshwork formation at sites of cell-cell contact. Mol. Biol. Cell 2004, 15, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Suisse, A.; Treisman, J.E. Reduced SERCA Function Preferentially Affects Wnt Signaling by Retaining E-Cadherin in the Endoplasmic Reticulum. Cell Rep. 2019, 26, 322–329.e3. [Google Scholar] [CrossRef]
- Stuart, R.O.; Sun, A.; Bush, K.T.; Nigam, S.K. Dependence of epithelial intercellular junction biogenesis on thapsigargin-sensitive intracellular calcium stores. J. Biol. Chem. 1996, 271, 13636–13641. [Google Scholar] [CrossRef]
- Jouret, F.; Wu, J.; Hull, M.; Rajendran, V.; Mayr, B.; Schöfl, C.; Geibel, J.; Caplan, M.J. Activation of the Ca²+-sensing receptor induces deposition of tight junction components to the epithelial cell plasma membrane. J. Cell Sci. 2013, 126, 5132–5142. [Google Scholar] [CrossRef]
- Wakita, H.; Takigawa, M. Activation of epidermal growth factor receptor promotes late terminal differentiation of cell-matrix interaction-disrupted keratinocytes. J. Biol. Chem. 1999, 274, 37285–37291. [Google Scholar] [CrossRef]
- Sternlicht, M.D.; Sunnarborg, S.W. The ADAM17-amphiregulin-EGFR axis in mammary development and cancer. J. Mammary Gland Biol. Neoplasia 2008, 13, 181–194. [Google Scholar] [CrossRef]
- Schneider, M.R.; Wolf, E. The epidermal growth factor receptor ligands at a glance. J. Cell Physiol. 2009, 218, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.B.; Harris, R.C. Autocrine, paracrine and juxtacrine signaling by EGFR ligands. Cell Signal. 2005, 17, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.C.; Huang, D.Y.; Wu, N.L.; Kannagi, R.; Wang, L.F.; Lin, W.W. BLIMP1 transcriptionally induced by EGFR activation and post-translationally regulated by proteasome and lysosome is involved in keratinocyte differentiation, migration and inflammation. J. Dermatol. Sci. 2018, 92, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Casalini, P.; Iorio, M.V.; Galmozzi, E.; Ménard, S. Role of HER receptors family in development and differentiation. J. Cell Physiol. 2004, 200, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Ang, K.K.; Berkey, B.A.; Tu, X.; Zhang, H.Z.; Katz, R.; Hammond, E.H.; Fu, K.K.; Milas, L. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 2002, 62, 7350–7356. [Google Scholar] [CrossRef]
- Li, J.; Liang, R.; Song, C.; Xiang, Y.; Liu, Y. Prognostic significance of epidermal growth factor receptor expression in glioma patients. Onco Targets Ther. 2018, 11, 731–742. [Google Scholar] [CrossRef]
- Russo, A.; Franchina, T.; Ricciardi, G.R.R.; Smiroldo, V.; Picciotto, M.; Zanghì, M.; Rolfo, C.; Adamo, V. Third generation EGFR TKIs in EGFR-mutated NSCLC: Where are we now and where are we going. Crit. Rev. Oncol. Hematol. 2017, 117, 38–47. [Google Scholar] [CrossRef]
- Cassell, A.; Grandis, J.R. Investigational EGFR-targeted therapies in HNSCC. Exp. Opin. Invest. Drugs 2010, 19, 709–722. [Google Scholar] [CrossRef]
- Li, H.; Panina, S.; Kaur, A.; Ruano, M.J.; Sánchez-González, P.; la Cour, J.M.; Stephan, A.; Olesen, U.H.; Berchtold, M.W.; Villalobo, A. Regulation of the ligand-dependent activation of the epidermal growth factor receptor by calmodulin. J. Biol. Chem. 2012, 287, 3273–3281. [Google Scholar] [CrossRef]
- Stateva, S.R.; Salas, V.; Benguría, A.; Cossío, I.; Anguita, E.; Martín-Nieto, J.; Benaim, G.; Villalobo, A. The activating role of phospho-(Tyr)-calmodulin on the epidermal growth factor receptor. Biochem. J. 2015, 472, 195–204. [Google Scholar] [CrossRef]
- McLaughlin, S.; Smith, S.O.; Hayman, M.J.; Murray, D. An electrostatic engine model for autoinhibition and activation of the epidermal growth factor receptor (EGFR/ErbB) family. J. Gen. Physiol. 2005, 126, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sánchez-Torres, J.; Del Carpio, A.; Salas, V.; Villalobo, A. The ErbB2/Neu/HER2 receptor is a new calmodulin-binding protein. Biochem. J. 2004, 381, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, C.; Wasylyk, C.; Li, Y.; Thomas, E.; Marisa, L.; Robé, A.; Roux, M.; Abecassis, J.; de Reyniès, A.; Wasylyk, B. ANO1 amplification and expression in HNSCC with a high propensity for future distant metastasis and its functions in HNSCC cell lines. Br. J. Cancer 2010, 103, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Duvvuri, U.; Shiwarski, D.J.; Xiao, D.; Bertrand, C.; Huang, X.; Edinger, R.S.; Rock, J.R.; Harfe, B.D.; Henson, B.J.; Kunzelmann, K.; et al. TMEM16A induces MAPK and contributes directly to tumorigenesis and cancer progression. Cancer Res. 2012, 72, 3270–3281. [Google Scholar] [CrossRef]
- Ruiz, C.; Martins, J.R.; Rudin, F.; Schneider, S.; Dietsche, T.; Fischer, C.A.; Tornillo, L.; Terracciano, L.M.; Schreiber, R.; Bubendorf, L.; et al. Enhanced expression of ANO1 in head and neck squamous cell carcinoma causes cell migration and correlates with poor prognosis. PLoS ONE 2012, 7, e43265. [Google Scholar] [CrossRef]
- Britschgi, A.; Bill, A.; Brinkhaus, H.; Rothwell, C.; Clay, I.; Duss, S.; Rebhan, M.; Raman, P.; Guy, C.T.; Wetzel, K.; et al. Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc. Natl. Acad. Sci. USA 2013, 110, E1026–E1034. [Google Scholar] [CrossRef]
- Bill, A.; Gutierrez, A.; Kulkarni, S.; Kemp, C.; Bonenfant, D.; Voshol, H.; Duvvuri, U.; Gaither, L.A. ANO1/TMEM16A interacts with EGFR and correlates with sensitivity to EGFR-targeting therapy in head and neck cancer. Oncotarget 2015, 6, 9173–9188. [Google Scholar] [CrossRef]
- Bill, A.; Hall, M.L.; Borawski, J.; Hodgson, C.; Jenkins, J.; Piechon, P.; Popa, O.; Rothwell, C.; Tranter, P.; Tria, S.; et al. Small Molecule-Facilitated Degradation of ANO1 Protein: A New Targeting Approach for Anticancer Therapeutics. J. Biol. Chem. 2014, 289, 11029–11041. [Google Scholar] [CrossRef]
- Jin, X.; Shah, S.; Liu, Y.; Zhang, H.; Lees, M.; Fu, Z.; Lippiat, J.D.; Beech, D.J.; Sivaprasadarao, A.; Baldwin, S.A.; et al. Activation of the Cl- channel ANO1 by localized calcium signals in nociceptive sensory neurons requires coupling with the IP3 receptor. Sci. Signal. 2013, 6, ra73. [Google Scholar] [CrossRef]
- Concepcion, A.R.; Vaeth, M.; Wagner, L.E.; Eckstein, M.; Hecht, L.; Yang, J.; Crottes, D.; Seidl, M.; Shin, H.P.; Weidinger, C.; et al. Store-operated Ca 2+ entry regulates Ca 2+-activated chloride channels and eccrine sweat gland function. J. Clin. Investig. 2016, 126, 4303–4318. [Google Scholar] [CrossRef]
- Sharma, A.; Ramena, G.; Yin, Y.; Premkumar, L.; Elble, R.C. CLCA2 is a positive regulator of store-operated calcium entry and TMEM16A. PLoS ONE 2011, 13, e0196512. [Google Scholar] [CrossRef] [PubMed]
- Crottès, D.; Lin, Y.T.; Peters, C.J.; Gilchrist, J.M.; Wiita, A.P.; Jan, Y.N.; Jan, L.Y. TMEM16A controls EGF-induced calcium signaling implicated in pancreatic cancer prognosis. Proc. Natl. Acad. Sci. USA 2019, 116, 13026–13035. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Duan, L.; He, Q.; Zhang, Z.; Zhou, Y.; Wu, D.; Pan, J.; Pei, D.; Ding, K. Identification of Niclosamide as a New Small-Molecule Inhibitor of the STAT3 Signaling Pathway. ACS Med. Chem. Lett. 2010, 1, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Weinberg, R.A. Epithelial-to-mesenchymal transition in cancer:complexity and opportunities. Front. Med. 2018, 12, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Ocana, O.H.; Corcoles, R.; Fabra, A.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef]
- Cheung, K.J.; Ewald, A.J. A collective route to metastasis: Seeding by tumor cell clusters. Science 2016, 352, 167–169. [Google Scholar] [CrossRef]
- Cheung, K.J.; Padmanaban, V.; Silvestri, V.; Schipper, K.; Cohen, J.D.; Fairchild, A.N.; Gorin, M.A.; Verdone, J.E.; Pienta, K.J.; Bader, J.S.; et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA 2016, 113, E854–E863. [Google Scholar] [CrossRef]
- Kulasinghe, A.; Schmidt, H.; Perry, C.; Whitfield, B.; Kenny, L.; Nelson, C.; Warkiani, M.E.; Punyadeera, C. A Collective Route to Head and Neck Cancer Metastasis. Sci. Rep. 2018, 8, 746. [Google Scholar] [CrossRef] [PubMed]
- Pece, S.; Gutkind, J.S. Signaling from E-cadherins to the MAPK pathway by the recruitment and activation of epidermal growth factor receptors upon cell-cell contact formation. J. Biol. Chem. 2000, 275, 41227–41233. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Kramer, R.H. Adhesion-mediated squamous cell carcinoma survival through ligand-independent activation of epidermal growth factor receptor. Am. J. Pathol. 2004, 165, 1315–1329. [Google Scholar] [CrossRef]
- Rayavarapu, R.R.; Heiden, B.; Pagani, N.; Shaw, M.M.; Shuff, S.; Zhang, S.; Schafer, Z.T. The role of multicellular aggregation in the survival of ErbB2-positive breast cancer cells during extracellular matrix detachment. J. Biol. Chem. 2015, 290, 8722–8733. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Tennenbaum, T.; Yuspa, S.H. Suspension-induced murine keratinocyte differentiation is mediated by calcium. J. Investig. Dermatol. 1996, 106, 254–260. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef]
- Hawk, M.A.; Schafer, Z.T. Mechanisms of redox metabolism and cancer cell survival during extracellular matrix detachment. J. Biol. Chem. 2018, 293, 7531–7537. [Google Scholar] [CrossRef]
- Hempel, N.; Trebak, M. Crosstalk between Calcium and Reactive Oxygen Species Signaling in Cancer. Cell Calcium 2017, 63, 70–96. [Google Scholar] [CrossRef]
- Takahashi, N.; Chen, H.Y.; Harris, I.S.; Stover, D.G.; Selfors, L.M.; Bronson, R.T.; Deraedt, T.; Cichowski, K.; Welm, A.L.; Mori, Y.; et al. Cancer Cells Co-opt the Neuronal Redox-Sensing Channel TRPA1 to Promote Oxidative-Stress Tolerance. Cancer Cell 2018, 33, 985–1003.e7. [Google Scholar] [CrossRef]
- Lehen’kyi, V.; Flourakis, M.; Skryma, R.; Prevarskaya, N. TRPV6 channel controls prostate cancer cell proliferation via Ca(2+)/NFAT-dependent pathways. Oncogene 2007, 26, 7380–7385. [Google Scholar] [CrossRef]
- Fu, S.; Hirte, H.; Welch, S.; Ilenchuk, T.T.; Lutes, T.; Rice, C.; Fields, N.; Nemet, A.; Dugourd, D.; Piha-Paul, S.; et al. First-in-human phase I study of SOR-C13, a TRPV6 calcium channel inhibitor, in patients with advanced solid tumors. Investig. New Drug 2017, 35, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for K-Ras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed]
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyürek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308re8. [Google Scholar] [CrossRef] [PubMed]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 221ra15. [Google Scholar] [CrossRef] [PubMed]
- Breau, M.; Houssaini, A.; Lipskaia, L.; Abid, S.; Born, E.; Marcos, E.; Czibik, G.; Attwe, A.; Beaulieu, D.; Palazzo, A.; et al. The antioxidant N-acetylcysteine protects from lung emphysema but induces lung adenocarcinoma in mice. JCI Insight 2019, 4, 127647. [Google Scholar] [CrossRef]
- Kamarajugadda, S.; Cai, Q.; Chen, H.; Nayak, S.; Zhu, J.; He, M.; Jin, Y.; Zhang, Y.; Ai, L.; Martin, S.S.; et al. Manganese superoxide dismutase promotes anoikis resistance and tumor metastasis. Cell Death Dis. 2013, e504. [Google Scholar] [CrossRef]
- Klein, E.A.; Thompson, I.M.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the risk of prostate cancer: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef]
- Lappe, J.; Watson, P.; Travers-Gustafson, D.; Recker, R.; Garland, C.; Gorham, E.; Baggerly, K.; McDonnell, S.L. Effect of Vitamin D and Calcium Supplementation on Cancer Incidence in Older Women: A Randomized Clinical Trial. JAMA 2017, 137, 1234–1243. [Google Scholar] [CrossRef]
- Barry, E.L.; Peacock, J.L.; Rees, J.R.; Bostick, R.M.; Robertson, D.J.; Bresalier, R.S.; Baron, J.A. Vitamin D Receptor Genotype, Vitamin D3 Supplementation, and Risk of Colorectal Adenomas: A Randomized Clinical Trial. JAMA Oncol. 2017, 3, 628–635. [Google Scholar] [CrossRef]
- Padmanaban, V.; Krol, I.; Suhail, Y.; Szczerba, B.M.; Aceto, N.; Bader, J.S.; Ewald, A.J. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 2019, 573, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Watson, J. Oxidants, antioxidants and the current incurability of metastatic cancers. Open Biol. 2013, 3, 120144. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R. Cancer: Context Is Key for E-cadherin in Invasion and Metastasis. Curr. Biol. 2019, 29, R1140–R1142. [Google Scholar] [CrossRef] [PubMed]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.J.; Ewald, A.J. Invasive leader cells: Metastatic oncotarget. Oncotarget 2014, 5, 1390–1391. [Google Scholar] [CrossRef] [PubMed]
- Shamir, E.R.; Pappalardo, E.; Jorgens, D.M.; Coutinho, K.; Tsai, W.T.; Aziz, K.; Auer, M.; Tran, P.T.; Bader, J.S.; Ewald, A.J. Twist1-induced dissemination preserves epithelial identity and requires E-cadherin. J. Cell Biol. 2014, 204, 839–856. [Google Scholar] [CrossRef]
- Yu, Y.; Elble, R.C. Homeostatic Signaling by Cell-Cell Junctions and Its Dysregulation during Cancer Progression. J. Clin. Med. 2016, 5, 26. [Google Scholar] [CrossRef]
- Kourtidis, A.; Ngok, S.P.; Pulimeno, P.; Feathers, R.W.; Carpio, L.R.; Baker, T.R.; Carr, J.M.; Yan, I.K.; Borges, S.; Perez, E.A.; et al. Distinct E-cadherin-based complexes regulate cell behaviour through miRNA processing or Src and p120 catenin activity. Nat. Cell Biol. 2015, 17, 1145–1157. [Google Scholar] [CrossRef]
- Cui, C.; Merritt, R.; Fu, L.; Pan, Z. Targeting calcium signaling in cancer therapy. Acta Pharm. Sin. B 2017, 7, 3–17. [Google Scholar] [CrossRef]
- Noyer, L.; Lemonnier, L.; Mariot, P.; Gkika, D. Partners in Crime: Towards New Ways of Targeting Calcium Channels. Int. J. Mol. Sci. 2019, 20, 6344. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, A.; Elble, R.C. From Orai to E-Cadherin: Subversion of Calcium Trafficking in Cancer to Drive Proliferation, Anoikis-Resistance, and Metastasis. Biomedicines 2020, 8, 169. https://doi.org/10.3390/biomedicines8060169
Sharma A, Elble RC. From Orai to E-Cadherin: Subversion of Calcium Trafficking in Cancer to Drive Proliferation, Anoikis-Resistance, and Metastasis. Biomedicines. 2020; 8(6):169. https://doi.org/10.3390/biomedicines8060169
Chicago/Turabian StyleSharma, Aarushi, and Randolph C. Elble. 2020. "From Orai to E-Cadherin: Subversion of Calcium Trafficking in Cancer to Drive Proliferation, Anoikis-Resistance, and Metastasis" Biomedicines 8, no. 6: 169. https://doi.org/10.3390/biomedicines8060169
APA StyleSharma, A., & Elble, R. C. (2020). From Orai to E-Cadherin: Subversion of Calcium Trafficking in Cancer to Drive Proliferation, Anoikis-Resistance, and Metastasis. Biomedicines, 8(6), 169. https://doi.org/10.3390/biomedicines8060169