Clinical Aspects and Current Therapeutic Approaches for FOP


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Epidemiology
3. Pathophysiology
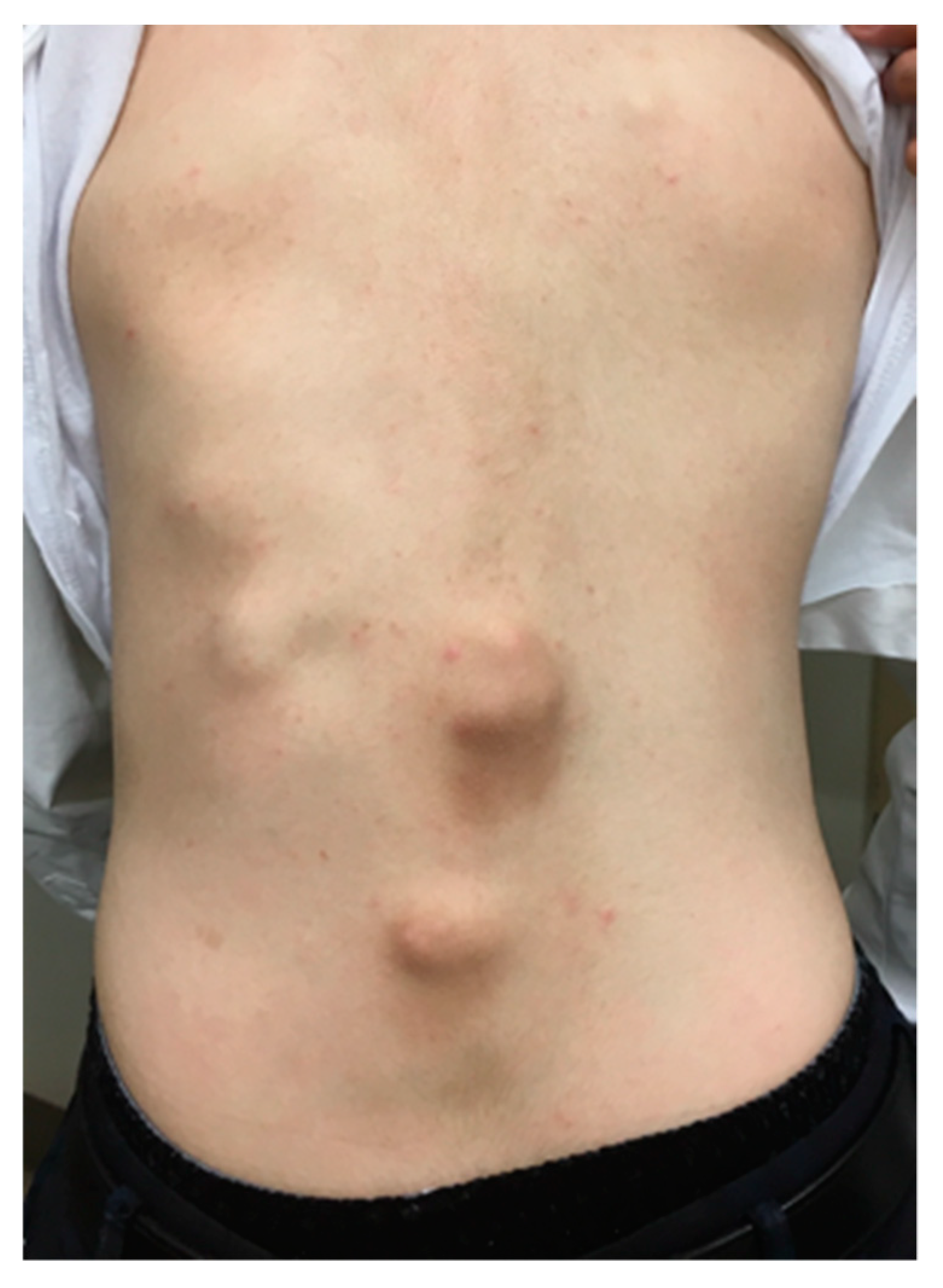
4. Natural Clinical Course
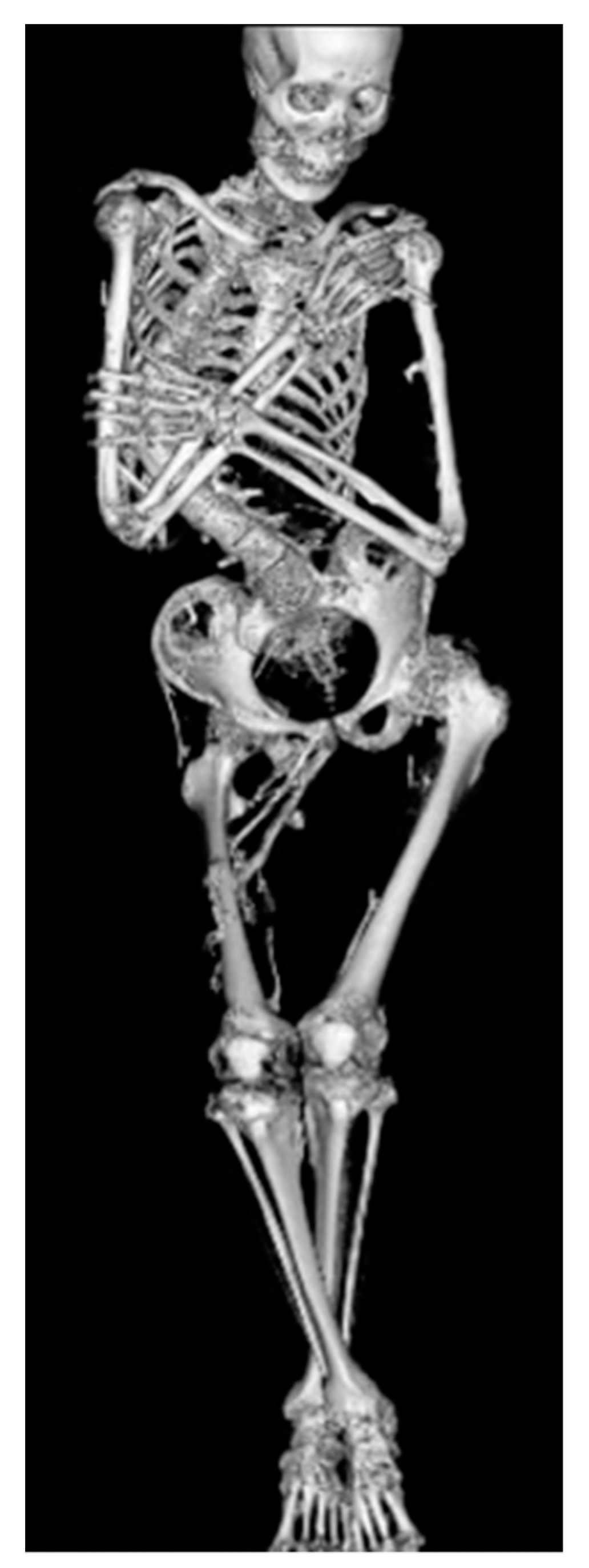
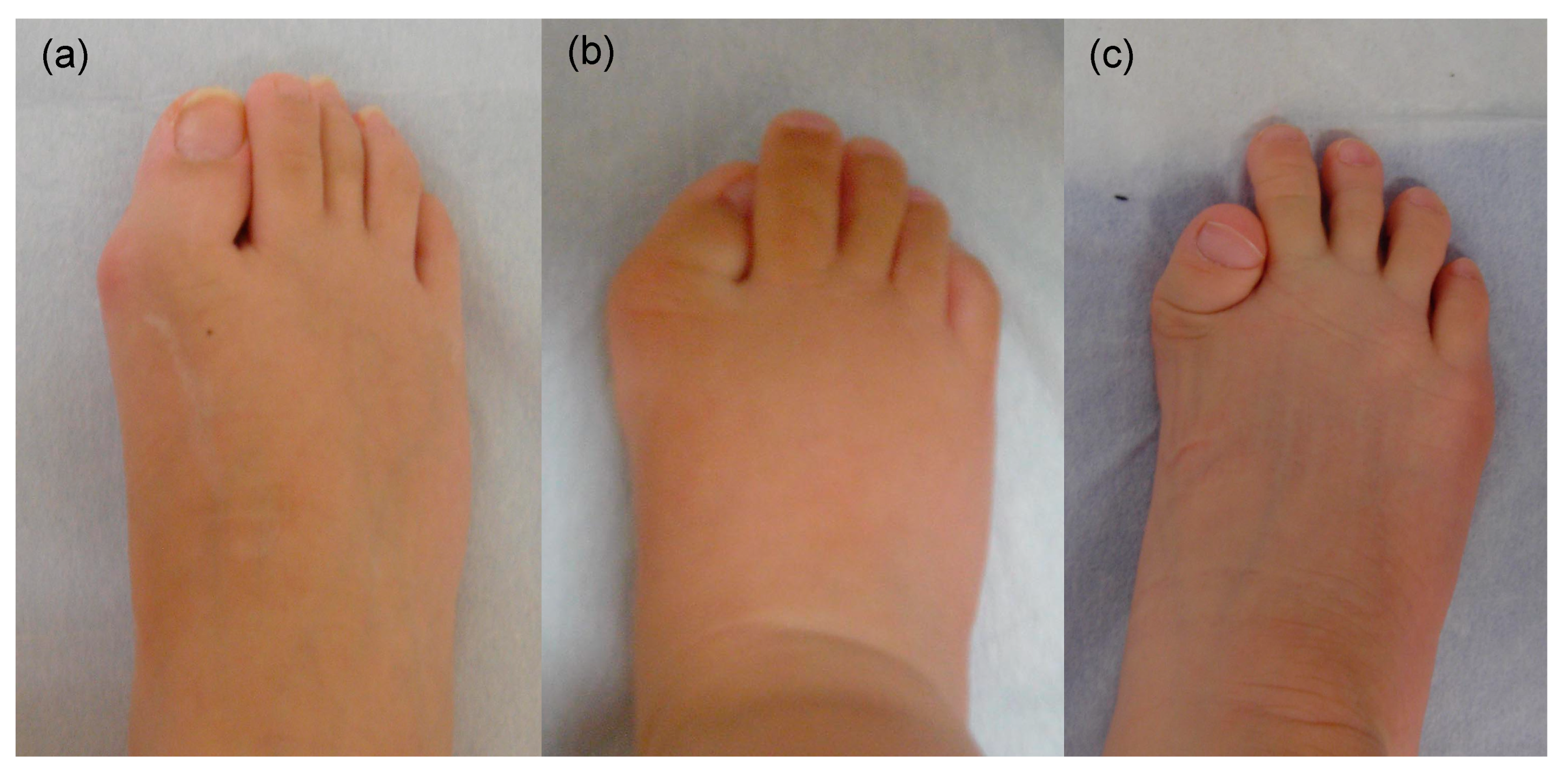
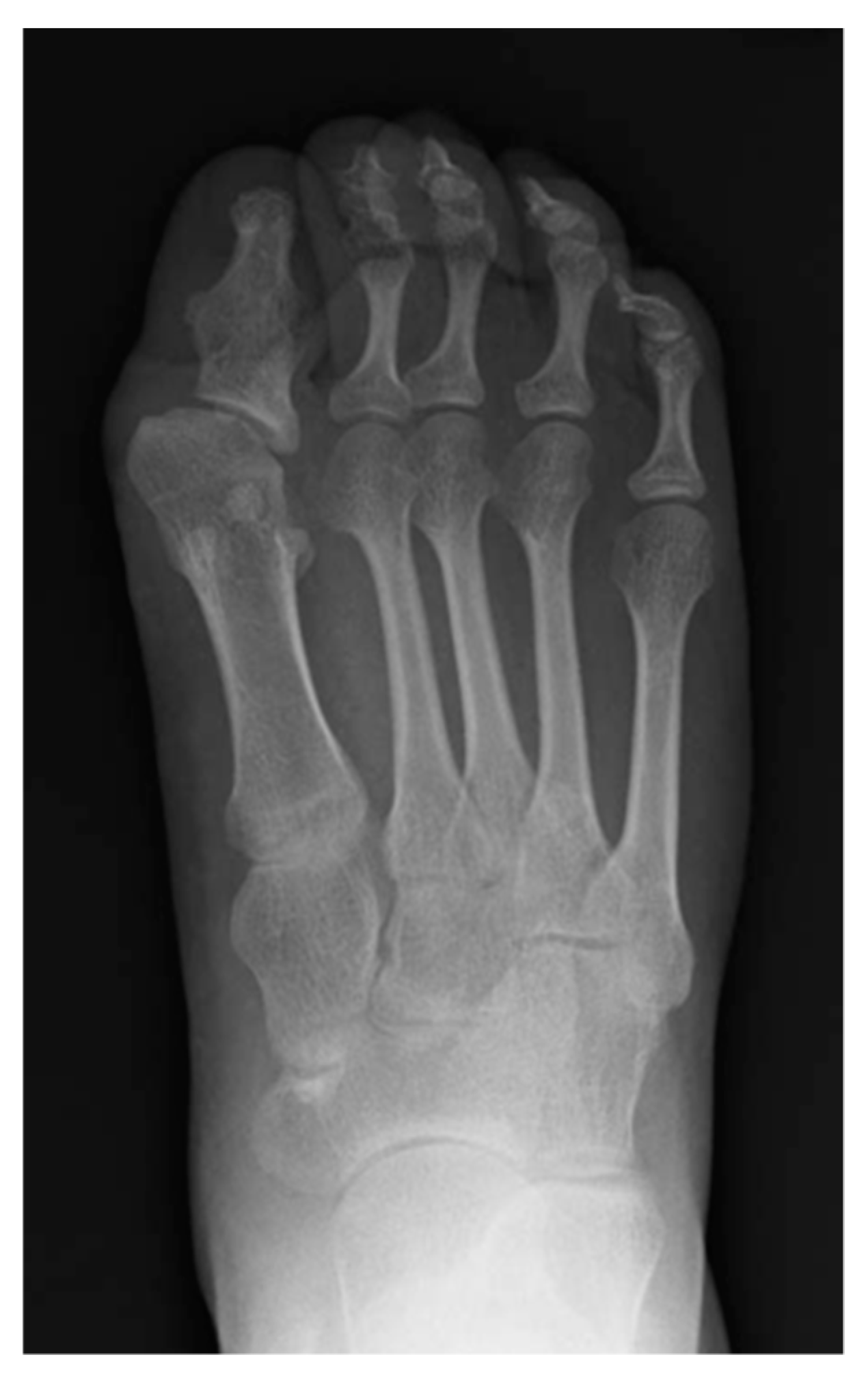
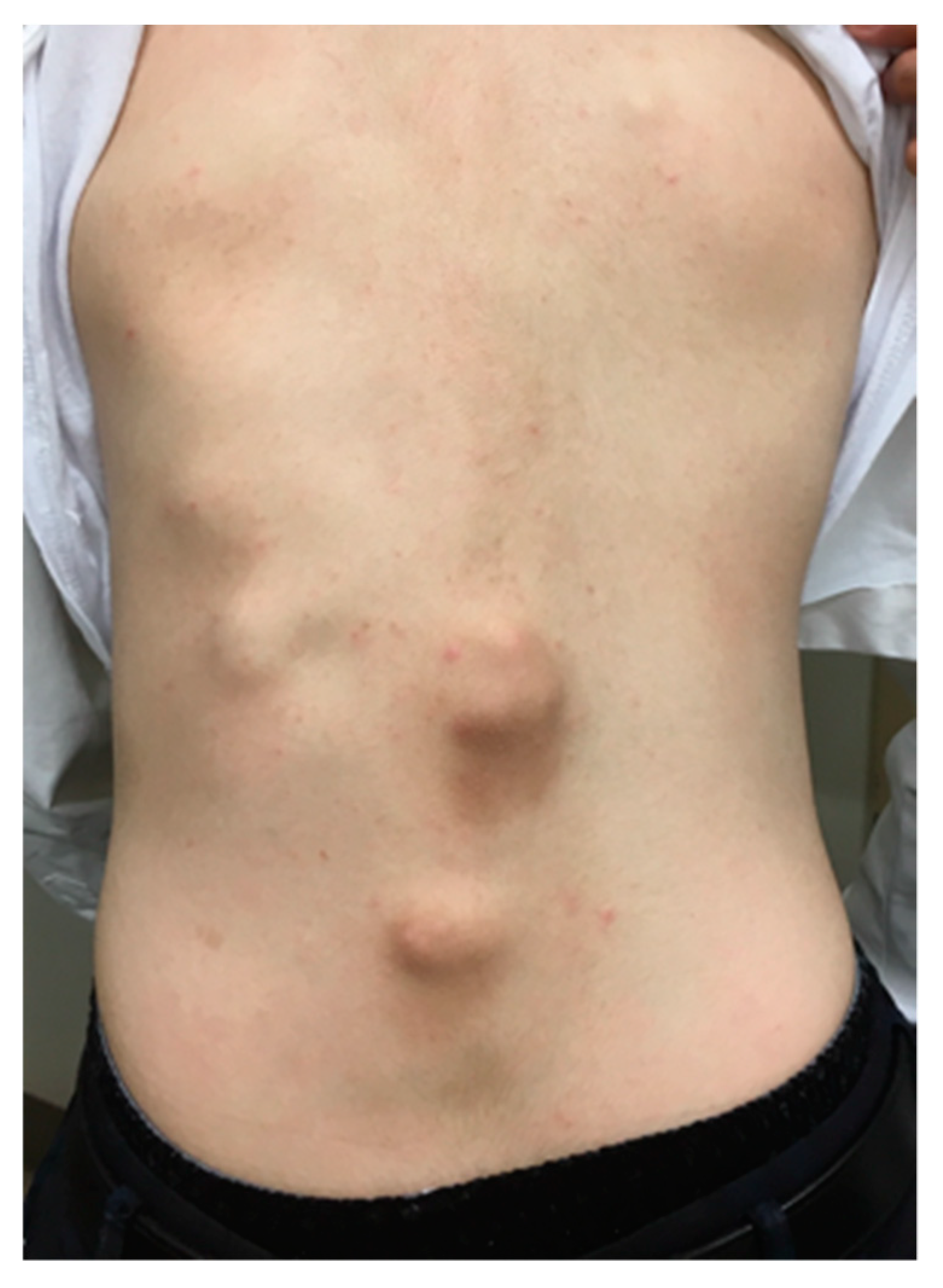
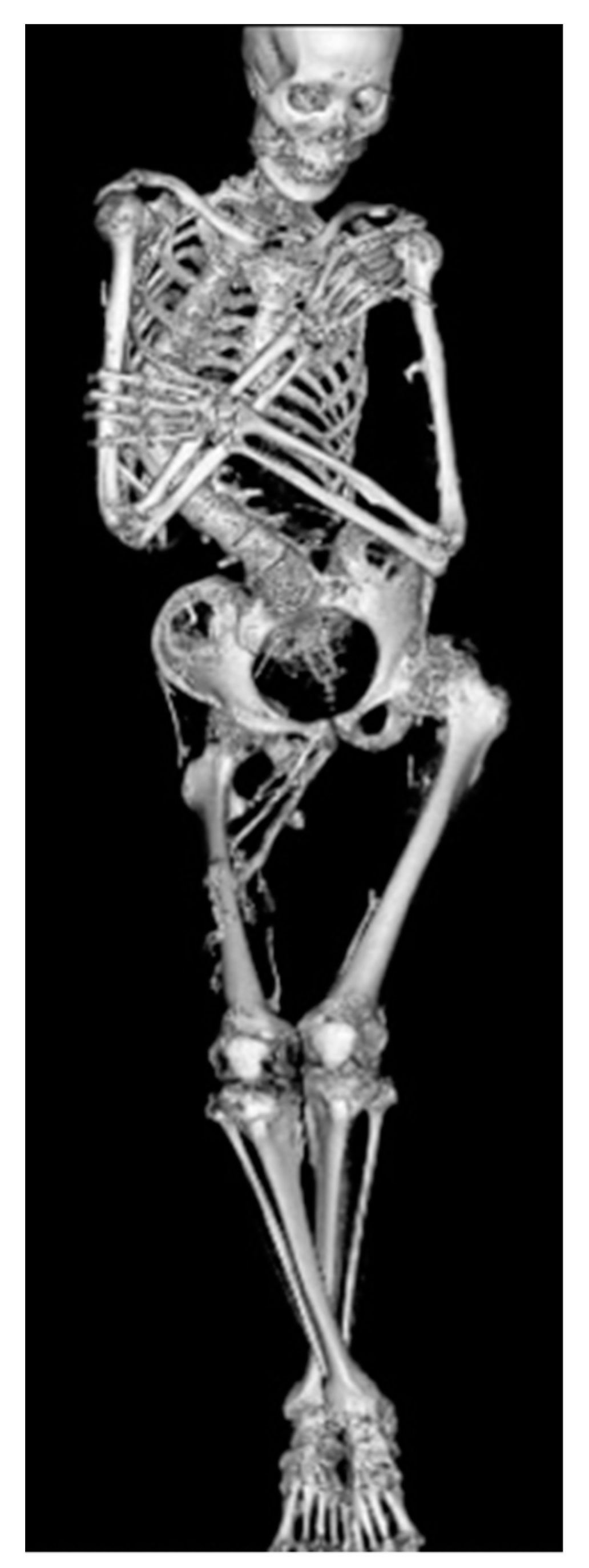
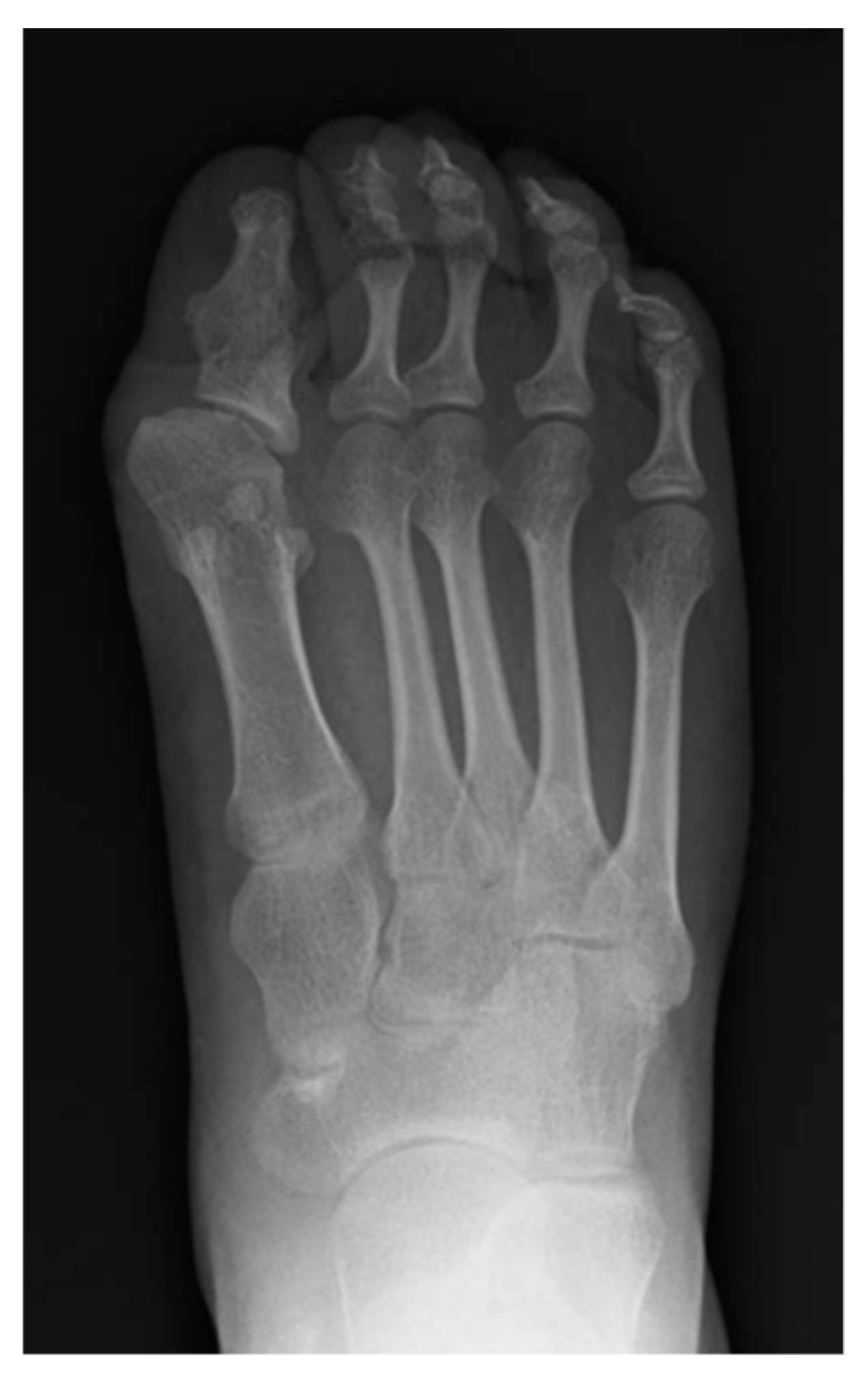
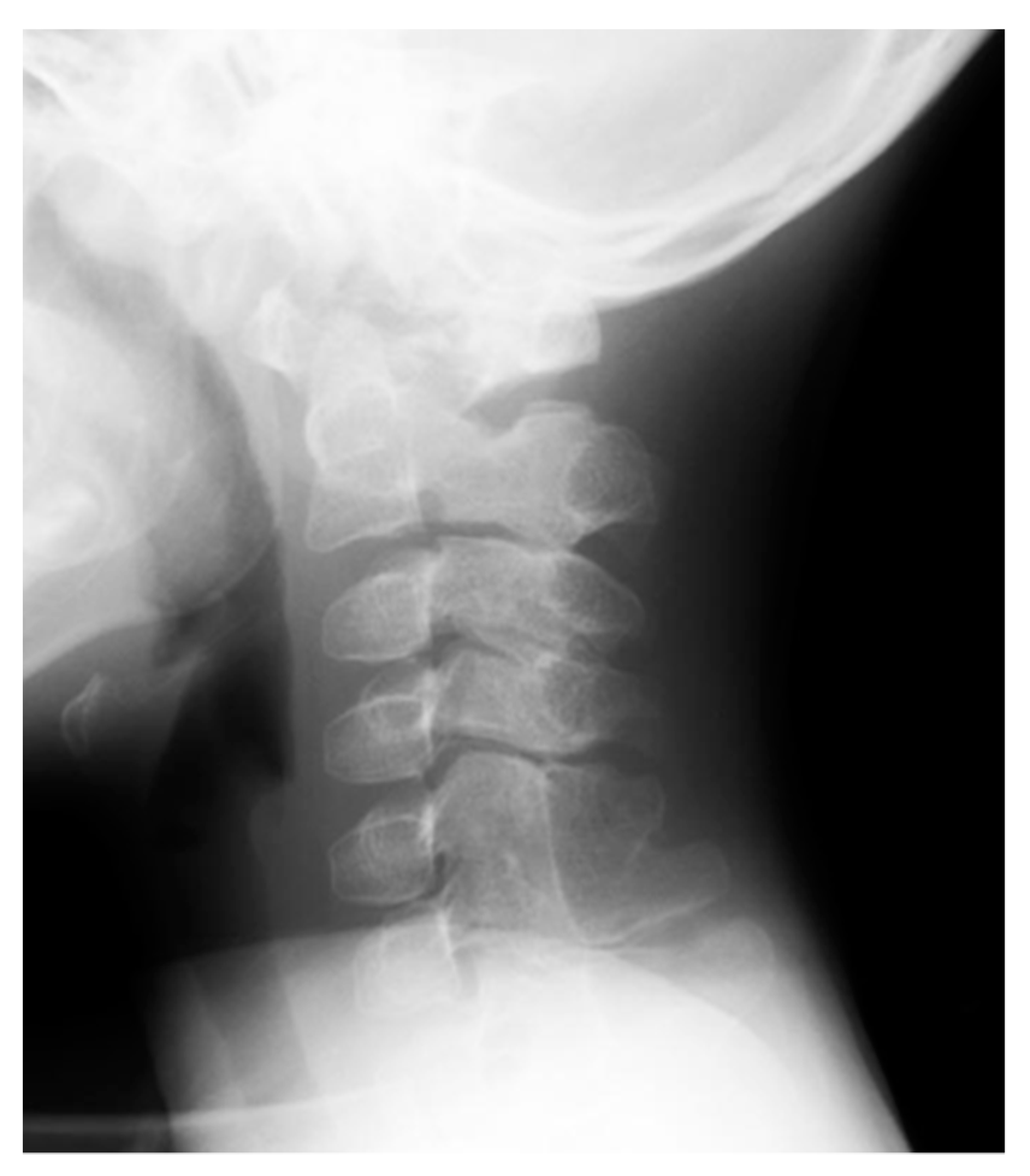
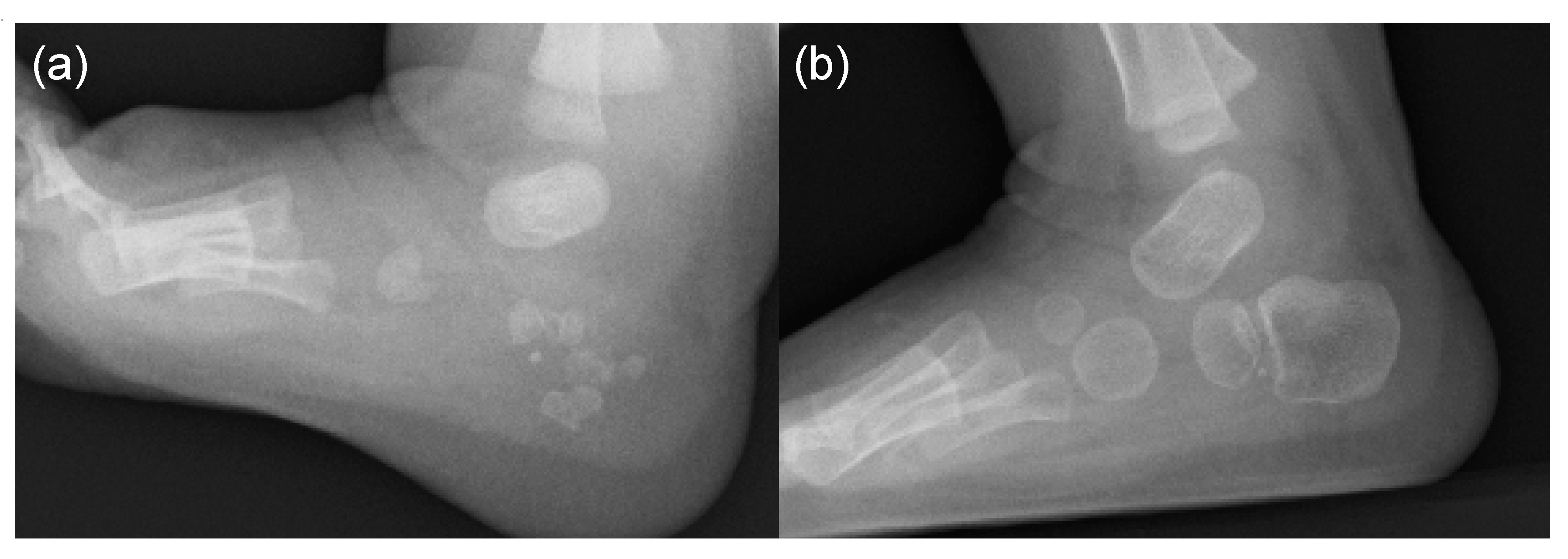
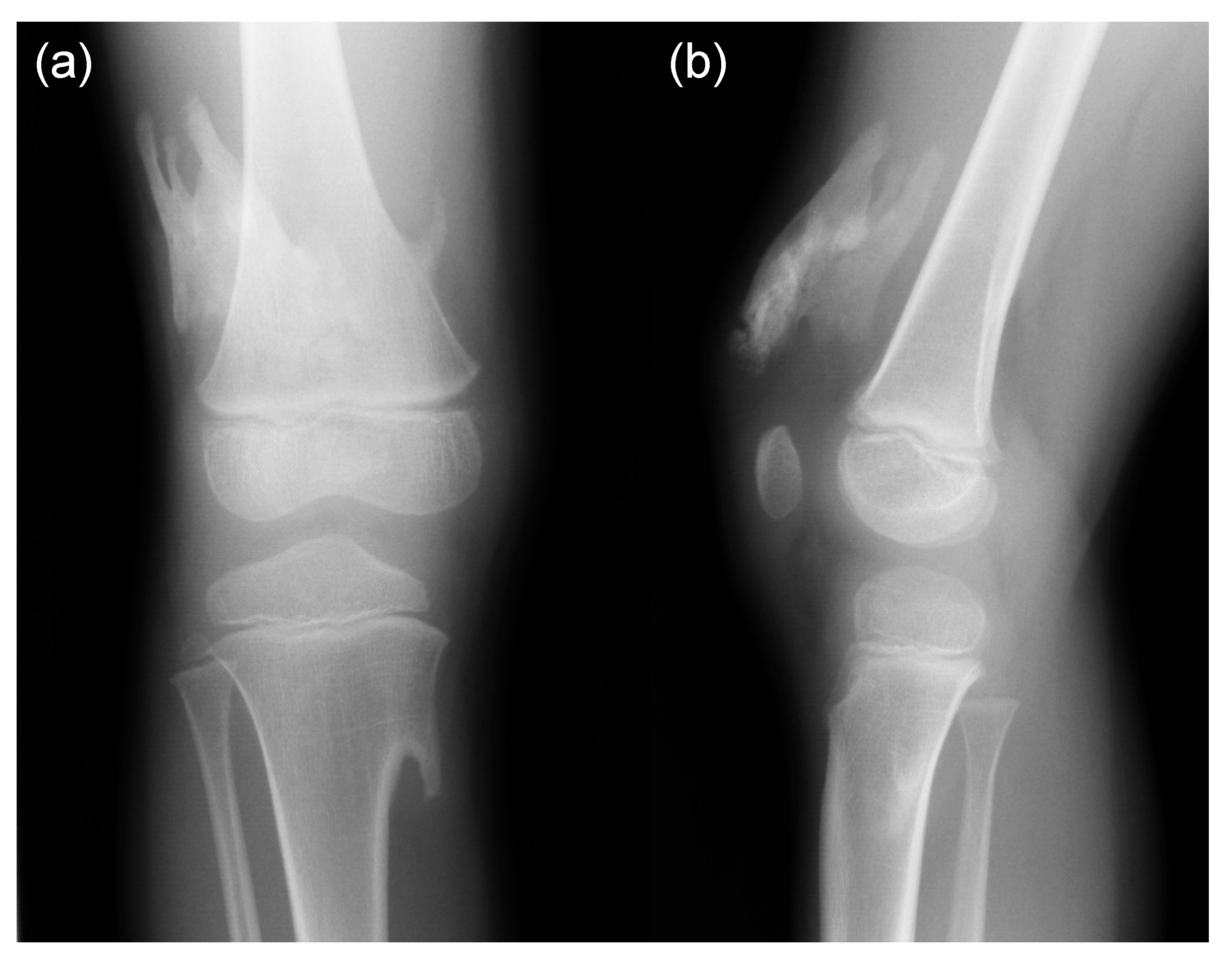
5. Skeletal Malformations
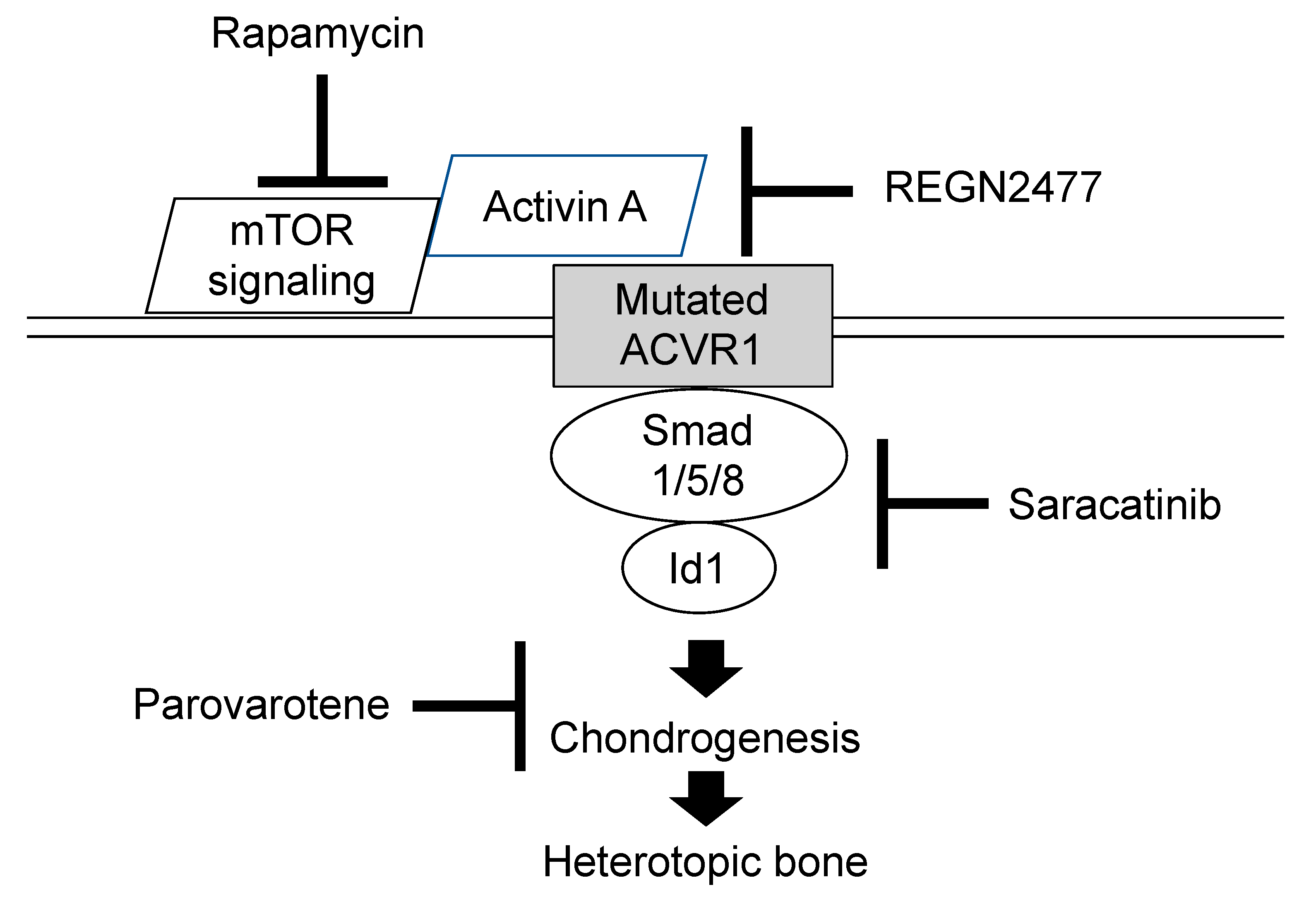
6. Managements and Treatments
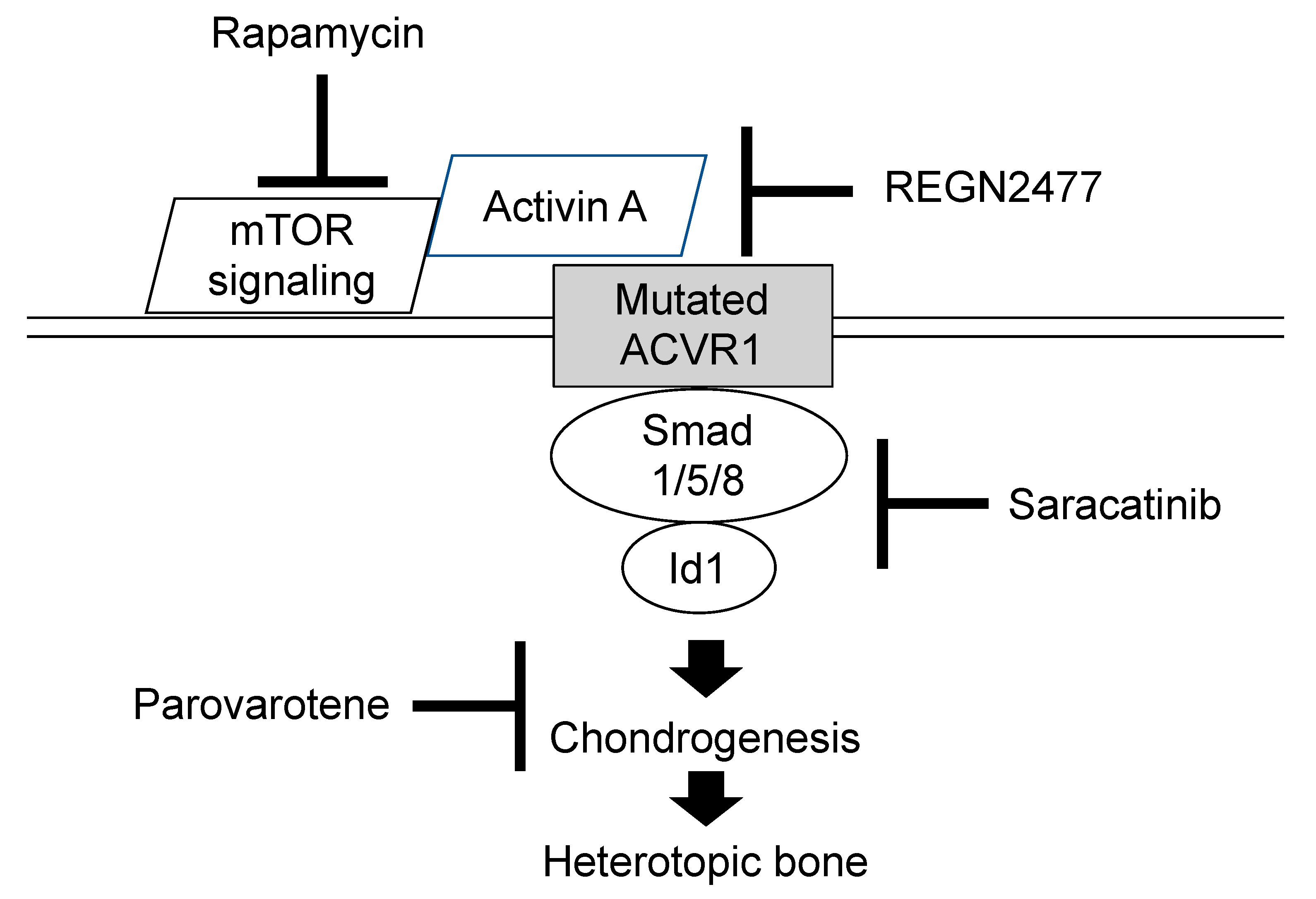
7. On-Going Clinical Trials for FOP (Phase 2 or Phase 3)
8. Conclusions
Funding
Conflicts of Interest
References
- Kitterman, J.A.; Kantanie, S.; Rocke, D.M.; Kaplan, F.S. Iatrogenic harm caused by diagnostic errors in fibrodysplasia ossificans progressiva. Pediatrics 2005, 116, e654–e661. [Google Scholar] [CrossRef] [Green Version]
- Zaghloul, K.A.; Heuer, G.G.; Guttenberg, M.D.; Shore, E.M.; Kaplan, F.S.; Storm, P.B. Lumbar puncture and surgical intervention in a child with undiagnosed fibrodysplasia ossificans progressiva. J. Neursurg. Pediatr. 2008, 1, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Xu, M.; Glaser, D.L.; Collins, F.; Connor, M.; Kitterman, J.; Sillence, D.; Zackai, E.; Ravitsky, V.; Zasloff, M.; et al. Early diagnosis of fibrodysplasia ossificans progressive. Pediatrics 2008, 151, e1295–e1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pignolo, R.J.; Shore, E.M.; Kaplan, F.S. Fibrodysplasia ossificans progressiva: Diagnosis, management, and therapeutic horizons. Pediatr. Endocrinol. Rev. 2013, 10, 437–448. [Google Scholar] [PubMed]
- Pignolo, R.J.; Shore, E.M.; Kaplan, F.S. Fibrodysplasia ossificans progressiva: Clinical and genetic aspects. Orphanet J. Rare Dis. 2011, 6, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shore, E.M.; Feldman, G.J.; Xu, M.; Kaplan, F.S. The genetics of fibrodysplasia ossificans progressiva. Clin. Rev. Bone Miner. Metab. 2005, 3, 201–204. [Google Scholar] [CrossRef]
- Hebela, N.; Shore, E.M.; Kaplan, F.S. Three pairs of monozygotic twins with fibrodysplasia ossificans progressiva: The role of environment in the progression of heterotopic ossification. Clin. Rev. Bone Miner. Metab. 2005, 3, 205–208. [Google Scholar] [CrossRef]
- Shore, E.M.; Xu, M.; Feldman, G.J.; Fenstermacher, D.A.; Cho, T.-J.; Choi, I.H.; Connor, J.M.; Delai, P.; Glaser, D.L.; Le Merrer, M.; et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006, 38, 525–527. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Xu, M.; Seemann, P.; Connor, M.; Glaser, D.L.; Carroll, L.; Delai, P.; Fastnact-Urban, E.; Forman, S.J.; Gillessen-Kaesbach, G.; et al. Classical and atypical FOP phenotypes are caused by mutations in the BMP type I receptor ACVR1. Hum. Mutat. 2009, 30, 379–390. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, T.; Kanomata, K.; Nojima, J.; Kokabu, S.; Akita, M.; Ikebuchi, K.; Jimi, E.; Komori, T.; Maruki, Y.; Matsuoka, M.; et al. A unique mutation of ALK2, G356D, found in a patient with fibrodysplasia ossificans progressiva is a moderately activated BMP type I receptor. Biochem. Biophys. Res. Commum. 2008, 377, 905–909. [Google Scholar] [CrossRef]
- Katagiri, T.; Yamaguchi, A.; Komaki, M.; Abe, E.; Takahashi, N.; Ikeda, T.; Rosen, V.; Wozney, J.M.; Fujisawa-Sehara, A.; Suda, T. Bone morphogenetic protein-2 converts the differentiation pathway of C2C12 myoblasts into the osteoblast lineage. J. Cell Biol. 1994, 127, 1755–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatsell, S.J.; Idone, V.; Wolken, D.M.; Huang, L.; Kim, H.J.; Wang, L.; Wen, X.; Nannuru, K.C.; Jimenez, J.; Xie, L.; et al. Economides AN. ACVR1(R206H) receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci. Transl. Med. 2015, 7, 303ra137. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shore, E.M.; Pignolo, R.J.; Kaplan, F.S. Activin A amplifies dysregulated BMP signaling and induced chondro-osseous differentiation of primary connective tissue progenitor cells in patients with fibrodysplasia ossificans progressiva (FOP). Bone 2018, 109, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Billings, P.C.; Fiori, J.L.; Bentwood, J.L.; O’Connell, M.P.; Jiao, X.; Nussbaum, B.; Caron, R.J.; Shore, E.M.; Kaplan, F.S. Dysregulated BMP signaling and enhanced osteogenic differentiation of connective tissue progenitor cells from patients with fibrodysplasia ossificans progressiva (FOP). J. Bone Miner. Res. 2008, 23, 305–313. [Google Scholar] [CrossRef]
- Shen, Q.; Little, S.C.; Xu, M.; Haupt, J.; Ast, C.; Katagiri, T.; Mundlos, S.; Seemann, P.; Kaplan, F.S.; Mullins, M.C.; et al. The fibrodysplasia ossificans progressive R20H ACVR1 mutation activates BMP independent chondrogenesis and zebrafish embargo ventralization. J. Clin. Investig. 2009, 119, 3462–3472. [Google Scholar] [PubMed] [Green Version]
- Cohen, R.B.; Hahn, G.V.; Tabas, J.A.; Peeper, J.; Levitz, C.L.; Sando, A.; Sando, N.; Zasloff, M.; Kaplan, F.S. The natural history of heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. A study of forty-four patients. J. Bone Joint Surg. Am. 1993, 75, 215–219. [Google Scholar] [CrossRef]
- Nakahara, Y.; Kitoh, H.; Nakashima, Y.; Toguchida, J.; Haag, N. Longitudinal study of the activities of daily living and quality of life in Japanese patients with fibrodysplasia ossificans progressiva. Disabil. Rehabil. 2019, 41, 699–704. [Google Scholar] [CrossRef]
- Rocke, D.M.; Zasloff, M.; Peeper, J.; Cohen, R.B.; Kaplan, F.S. Age- and joint-specific risk of initial heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. Clin. Orthop. Relat. Res. 1994, 301, 243–248. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Al Mukaddam, M.; Pignolo, R.J. A cumulative analogue joint involvement scale for fibrodysplasia ossificans progressiva (FOP). Bone 2018, 101, 123–128. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Zasloff, M.A.; Kitterman, J.A.; Shore, E.M.; Hong, C.C.; Rocke, D.M. Early mortality and cardiorespiratory failure in patients with fibrodysplasia ossificans progressive. J. Bone Joint Surg. Am. 2010, 92, 686–691. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, F.S.; Glaser, D.L. Thoracic insufficiency syndrome in patients with fibrodysplasia ossificans progressiva. Clin. Rev. Bone Miner. Metab. 2005, 3, 213–216. [Google Scholar] [CrossRef]
- Nakashima, Y.; Haga, N.; Kitoh, H.; Kamizono, J.; Tozawa, K.; Katagiri, T.; Susami, T.; Fukushi, J.; Iwamoto, Y. Deformity of the great toe in fibrodysplasia ossificans progressive. J. Orthop. Sci. 2010, 15, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Kobori, J.A.; Orellana, C.; Calvo, I.; Rosello, M.; Martinez, F.; Lopez, B.; Xu, M.; Pignolo, R.J.; Shore, E.M.; et al. Multi-system involvement in a severe variant of fibrodysplasia ossificans progressiva (ACVR1c.772G>A.; R258G): A report of two patients. Am. J. Med. Genet. A 2015, 167, 2265–2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuya, H.; Ikezoe, K.; Wang, L.; Ohyagi, Y.; Motomura, K.; Fujii, N.; Kira, J.; Fukumaki, Y. A unique case of fibrodysplasia ossificans progressiva with an ACVR1mutation, G356D, other than the common mutation (R206H). Am. J. Med. Genet. 2008, 146, 459–463. [Google Scholar] [CrossRef]
- Karamboulas, K.; Dranse, H.J.; Underhill, T.M. Regulation of BMP-dependent chondrogenesis in early limb mesenchyme by TGFbeta signals. J. Cell Sci. 2010, 123, 2068–2076. [Google Scholar] [CrossRef] [Green Version]
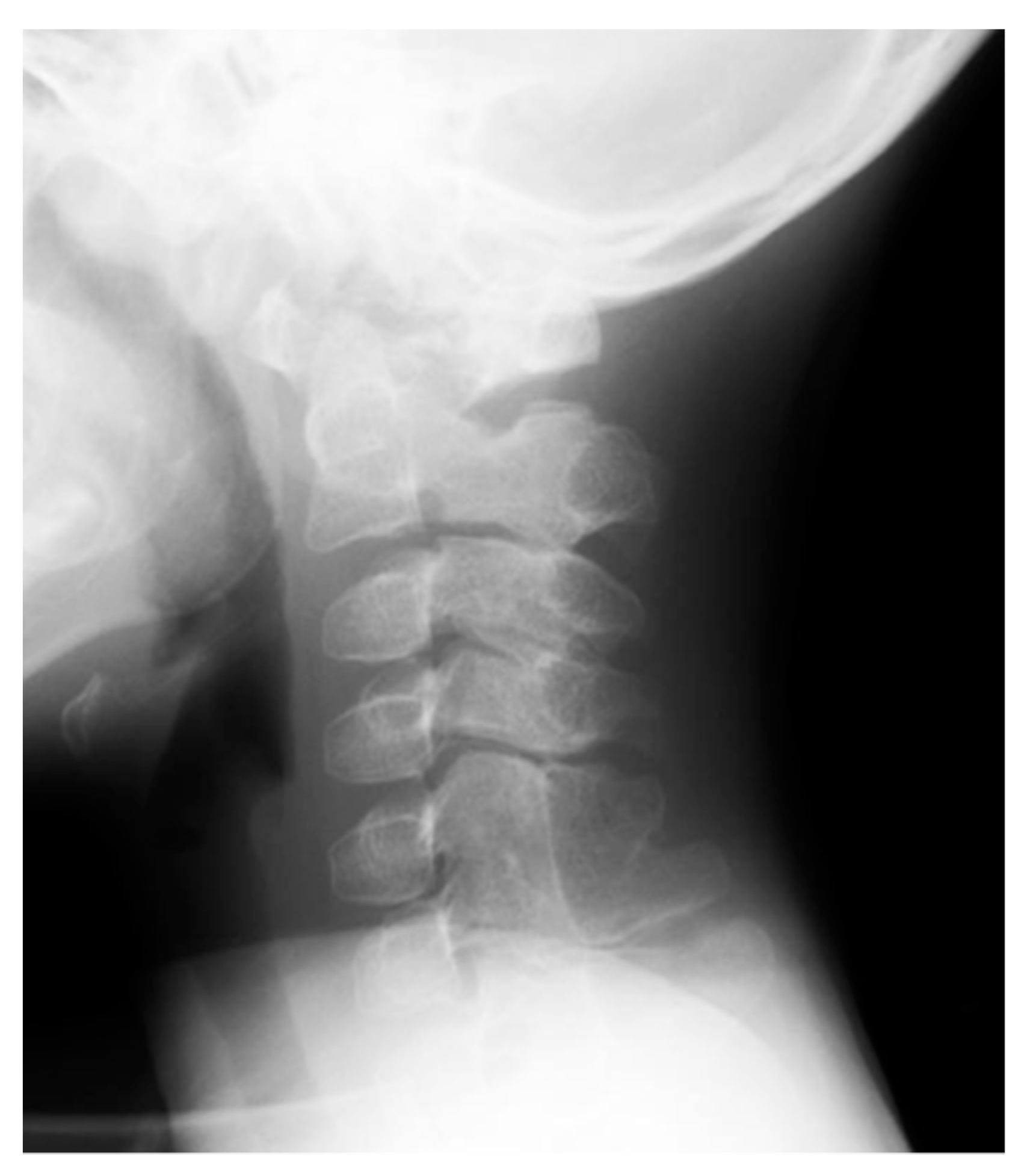
- Schaffer, A.A.; Kaplan, F.S.; Tracy, M.R.; O’Brien, M.L.; Dormans, J.P.; Shore, E.M.; Harland, R.M.; Kusumi, K. Developmental anomalies of the cervical spine in patients with fibrodysplasia ossificans progressiva are distinctly different from those in patients with Klippel-Feil syndrome. Spine 2005, 30, 1379–1385. [Google Scholar] [CrossRef]
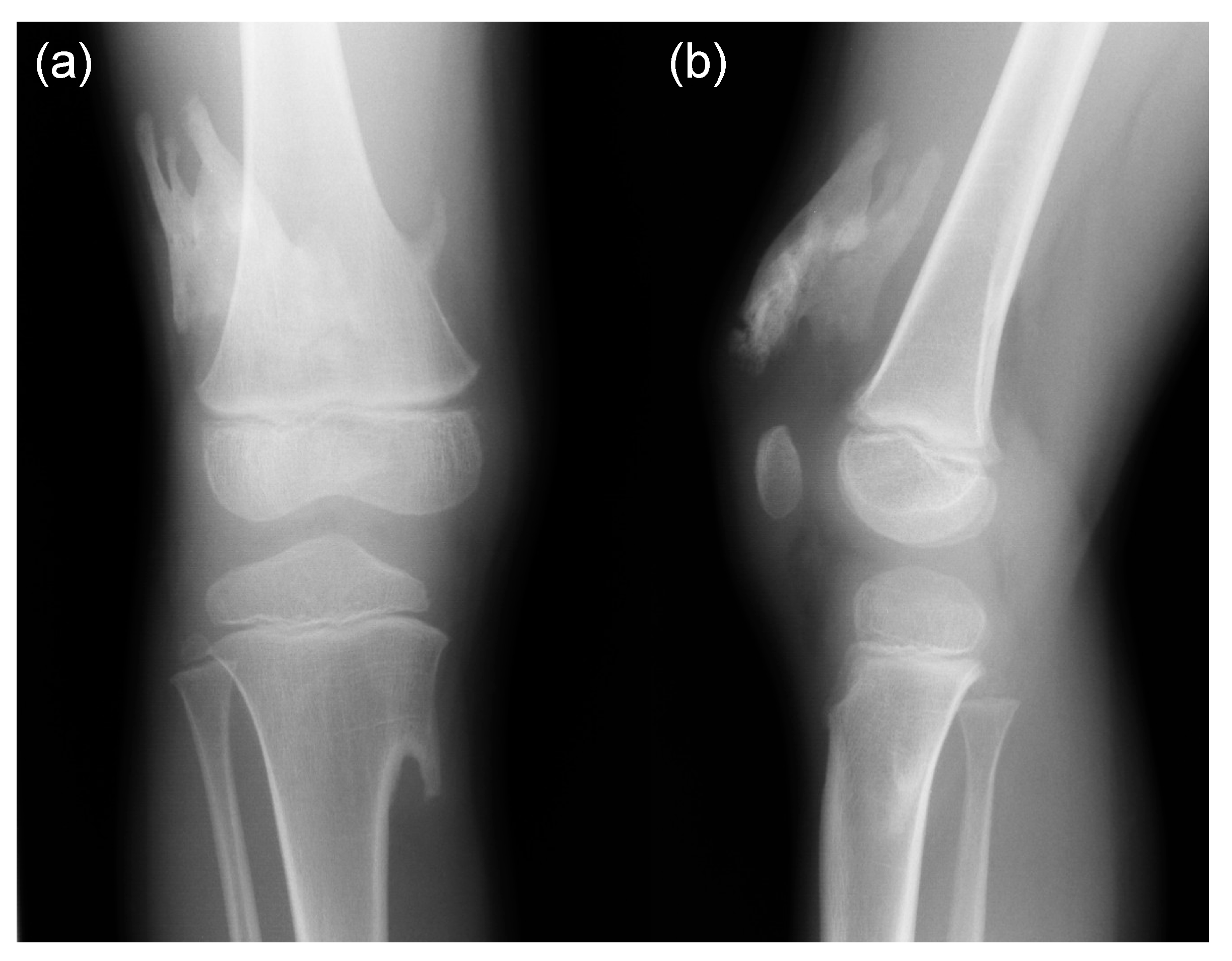
- Mishima, K.; Kitoh, H.; Katagiri, T.; Kaneko, H.; Ishiguro, N. Early clinical and radiographic characteristics in fibrodysplasia ossificans progressiva: A report of two cases. J. Bone Joint Surg. Am. 2011, 93, e52. [Google Scholar] [CrossRef]
- Monsoro-Burq, A.H.; Duprez, D.; Watanabe, Y.; Bontoux, M.; Vincent, C.; Brickell, P.; Le Douarin, N. The role of bone morphogenetic proteins in vertebral development. Development 1996, 122, 3607–3616. [Google Scholar]
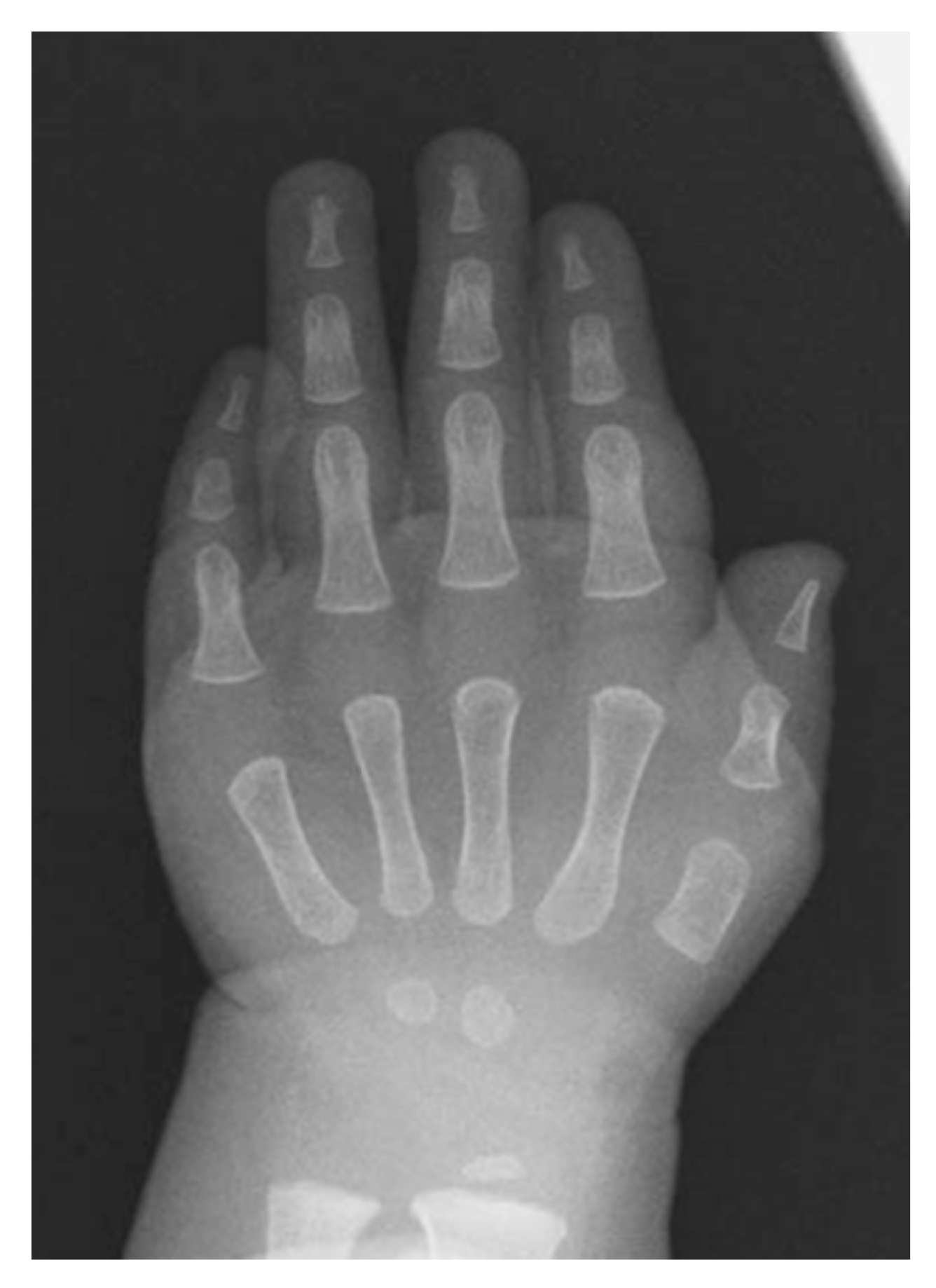
- Mishima, K.; Kitoh, H.; Haag, N.; Nakashima, Y.; Kamizono, J.; Katagiri, T.; Susami, T.; Matsushita, M.; Ishiguro, N. Radiographic characteristics of the hand and cervical spine in fibrodysplasia ossificans progressive. Intractable Rare Dis. Res. 2014, 3, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Oberg, K.C. Review of the molecular development of the thumb: Digit primera. Clin. Orthop. Relat. Res. 2014, 472, 1101–1105. [Google Scholar] [CrossRef] [Green Version]
- Deschamps, J. Tailored Hox gene transcription and the making of the thumb. Genes Dev. 2008, 22, 293–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Nie, S.; Chang, C.; Qiu, T.; Cao, X. Smads oppose Hox transcriptional activities. Exp. Cell Res. 2006, 312, 854–864. [Google Scholar] [CrossRef] [PubMed]
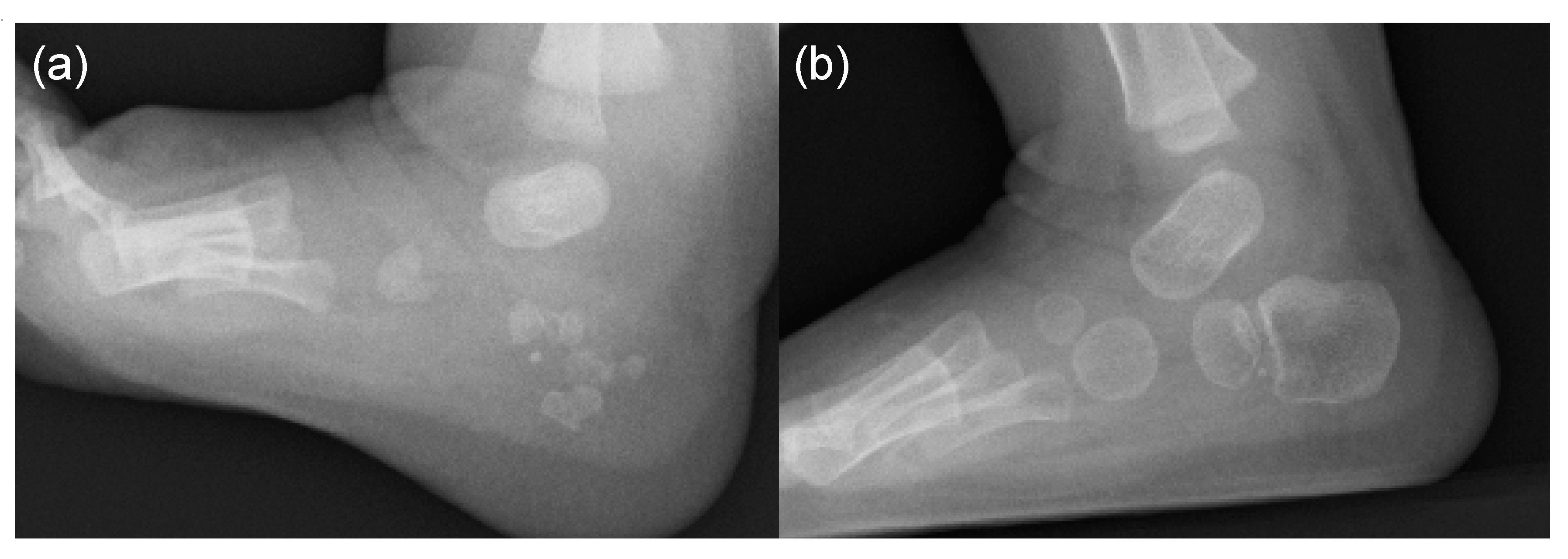
- Hasegawa, S.; Victoria, T.; Kayserili, H.; Zackai, E.; Nishimura, G.; Haga, N.; Nakashima, Y.; Miyazaki, O.; Kitoh, H. Characteristic calcaneal ossification: An additional early radiographic finding in infants with fibrodysplasia ossificans progressiva. Pediatr. Radiol. 2016, 46, 1568–1572. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Baek, H.J.; Karsenty, G.; Justice, M.J. Filamin B represses chondrocyte hypertrophy in a Runx2/Smad3-dependent manner. J. Cell Biol. 2007, 178, 121–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormick, C.; Leduc, Y.; Martindale, D.; Mattison, K.; Esford, L.E.; Dyer, A.P.; Tufaro, F. The putative tumour suppressor EXT1 alters the expression of cell-surface heparan sulfate. Nat. Genet. 1998, 19, 158–161. [Google Scholar] [CrossRef]
- Stickens, D.; Brown, D.; Evans, G.A. EXT genes are differentially expressed in bone and cartilage during mouse embryogenesis. Dev. Dyn. 2000, 218, 452–464. [Google Scholar] [CrossRef]
- Zhang, D.; Schwarz, E.M.; Rosier, R.N.; Zuscik, M.J.; Puzas, J.E.; O’Keefe, R.J. ALK2 functions as a BMP type I receptor and induces Indian hedgehog in chondrocytes during skeletal development. J. Bone Miner. Res. 2003, 18, 1593–1604. [Google Scholar] [CrossRef] [Green Version]
- Shah, P.B.; Zasloff, M.A.; Drummond, D.; Kaplan, F.S. Spinal deformity in patients who have fibrodysplasia ossificans progressive. J. Bone Joint Surg. Am. 1994, 76, 705–712. [Google Scholar] [CrossRef]
- Haga, N.; Nakashima, Y.; Kitoh, H.; Kamizono, J.; Katagiri, T.; Saijo, H.; Tsukamoto, S.; Shinoda, Y.; Sawada, R.; Nakahara, Y. Fibrodysplasia ossificans progressive: Review and research activities in Japan. Pediatr. Int. 2020, 62, 3–13. [Google Scholar] [CrossRef]
- Rhen, T.; Cidlowski, J.A. Anti-inflammatory action of glucocorticoids—New mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [Green Version]
- Pignolo, R.J.; Bedford-Gay, C.; Liljesthrom, M.; Durbin-Johnson, B.P.; Shore, E.M.; Rocke, D.M.; Kaplan, F.S. The natural history of flare-ups in fibrodysplasia ossificans progressiva: A comprehensive global assessment. J. Bone Miner. Res. 2016, 31, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Brantus, J.F.; Meunier, P.J. Effects of intravenous etidronate and oral corticosteroids in fibrodysplasia ossificans progressiva. Clin. Orthop. 1998, 346, 117–120. [Google Scholar] [CrossRef]
- Gannon, F.H.; Glaser, D.; Caron, R.; Thompson, L.D.R.; Shore, E.M.; Kaplan, F.S. Mast cell involvement in fibrodysplasia ossificans progressiva. Hum. Pathol. 2001, 32, 842–848. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, F.S.; Andolina, J.R.; Adamson, P.C.; Teachey, D.T.; Finklestein, J.Z.; Ebb, D.H.; Whitehead, B.; Jacobs, B.; Siegel, D.M.; Keen, R.; et al. Early clinical observations on the use of imatinib mesylate in FOP: A report of seven cases. Bone 2018, 109, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Cappato, S.; Giacopelli, F.; Ravazzolo, R.; Bocciardi, R. The horizon of a therapy for rare genetic diseases: A “druggable” future for fibrodysplasia ossificans progressiva. Int. J. Mol. Sci. 2018, 19, 989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katagiri, T.; Tsukamoto, S.; Nakachi, Y.; Kuratani, M. Recent topics in fibrodysplasia ossificans progressiva. Endocrinol. Metab. 2018, 33, 331–338. [Google Scholar] [CrossRef]
- Wentworth, K.L.; Masharani, U.; Hsiao, E.C. Therapeutic advanced for blocking heterotopic ossification in fibrodysplasia ossificans progressiva. Br. J. Clin. Pharmacol. 2019, 85, 1180–1187. [Google Scholar] [CrossRef]
- Pacifici, M.; Cossu, G.; Molinaro, M.; Tato, F. Vitamin A inhibits chondrogenesis but not myogenesis. Exp. Cell Res. 1980, 129, 469–474. [Google Scholar] [CrossRef]
- Shimono, K.; Tung, W.-E.; Macolino, C.; Chi, A.H.T.; Didizian, J.H.; Mundy, C.; Chandraratna, R.A.; Mishina, Y.; Enomoto-Iwamoto, M.; Pacifici, M.; et al. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-g agonists. Nat. Med. 2011, 17, 454–460. [Google Scholar] [CrossRef] [Green Version]
- Chakkalakal, S.A.; Uchibe, K.; Convente, M.R.; Zhang, D.; Economides, A.N.; Kaplan, F.S.; Pacifici, M.; Iwamoto, M.; Shore, E.M. Palovarotene inhibits heterotopic ossification and maintains limb mobility and growth in mice with the human ACVR1R206H fibrodysplasia ossificans progressiva (FOP) Mutation. J. Bone Miner. Res. 2016, 31, 1666–1675. [Google Scholar] [CrossRef] [Green Version]
- Lee-Shepard, J.B.; Nicholas, S.A.; Stoessel, S.J.; Devarakonda, P.M.; Schneider, M.J.; Yamamoto, M.; Goldhamer, D.J. Palovarotene reduced heterotopic ossification in juvenile FOP mice but exhibits pronounced skeletal toxicity. eLife 2018, 7, e40814. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Ikeya, M.; Horigome, K.; Matsumoto, Y.; Ebise, H.; Nishio, M.; Sekiguchi, K.; Shibata, M.; Nagata, S.; Matsuda, S.; et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc. Natl. Acad. Sci. USA 2015, 112, 15438–15443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hino, K.; Zhao, C.; Horigome, K.; Nishio, M.; Okanishi, Y.; Nagata, S.; Komura, S.; Yamada, Y.; Toguchida, J.; Ohta, A.; et al. An mTOR signaling modulator suppressed heterotopic ossification of fibrodysplasia ossificans progressiva. Stem Cell Rep. 2018, 11, 1106–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hino, K.; Horigome, K.; Nishio, M.; Komura, S.; Nagata, S.; Zhao, C.; Jin, Y.; Kawakami, K.; Yamada, Y.; Ohta, A.; et al. Activin-A enhances mTOR signaling to promote aberrant chondrogenesis in fibrodysplasia ossificans progressive. J. Clin. Investig. 2017, 127, 3339–3352. [Google Scholar] [CrossRef] [Green Version]
- Lewis, T.C.; Prywes, R. Serum regulation of Id1 expression by a BMP pathway and BMP responsive element. Biochim. Biophys. Acta 2013, 1829, 1147–1159. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitoh, H. Clinical Aspects and Current Therapeutic Approaches for FOP. Biomedicines 2020, 8, 325. https://doi.org/10.3390/biomedicines8090325
Kitoh H. Clinical Aspects and Current Therapeutic Approaches for FOP. Biomedicines. 2020; 8(9):325. https://doi.org/10.3390/biomedicines8090325
Chicago/Turabian StyleKitoh, Hiroshi. 2020. "Clinical Aspects and Current Therapeutic Approaches for FOP" Biomedicines 8, no. 9: 325. https://doi.org/10.3390/biomedicines8090325
APA StyleKitoh, H. (2020). Clinical Aspects and Current Therapeutic Approaches for FOP. Biomedicines, 8(9), 325. https://doi.org/10.3390/biomedicines8090325