GABAB-Receptor Agonist-Based Immunotherapy for Type 1 Diabetes in NOD Mice


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Mice
2.3. Lesogaberan Monotherapy
2.4. Combined Lesogaberan and Antigen-Specific Immunotherapy Treatment
2.5. Combined Lesogaberan and Low-Dose Anti-CD3 Treatment in Severely Diabetic NOD Mice
2.6. Intraperitoneal Glucose Tolerance (IPGT) Test
3. Results
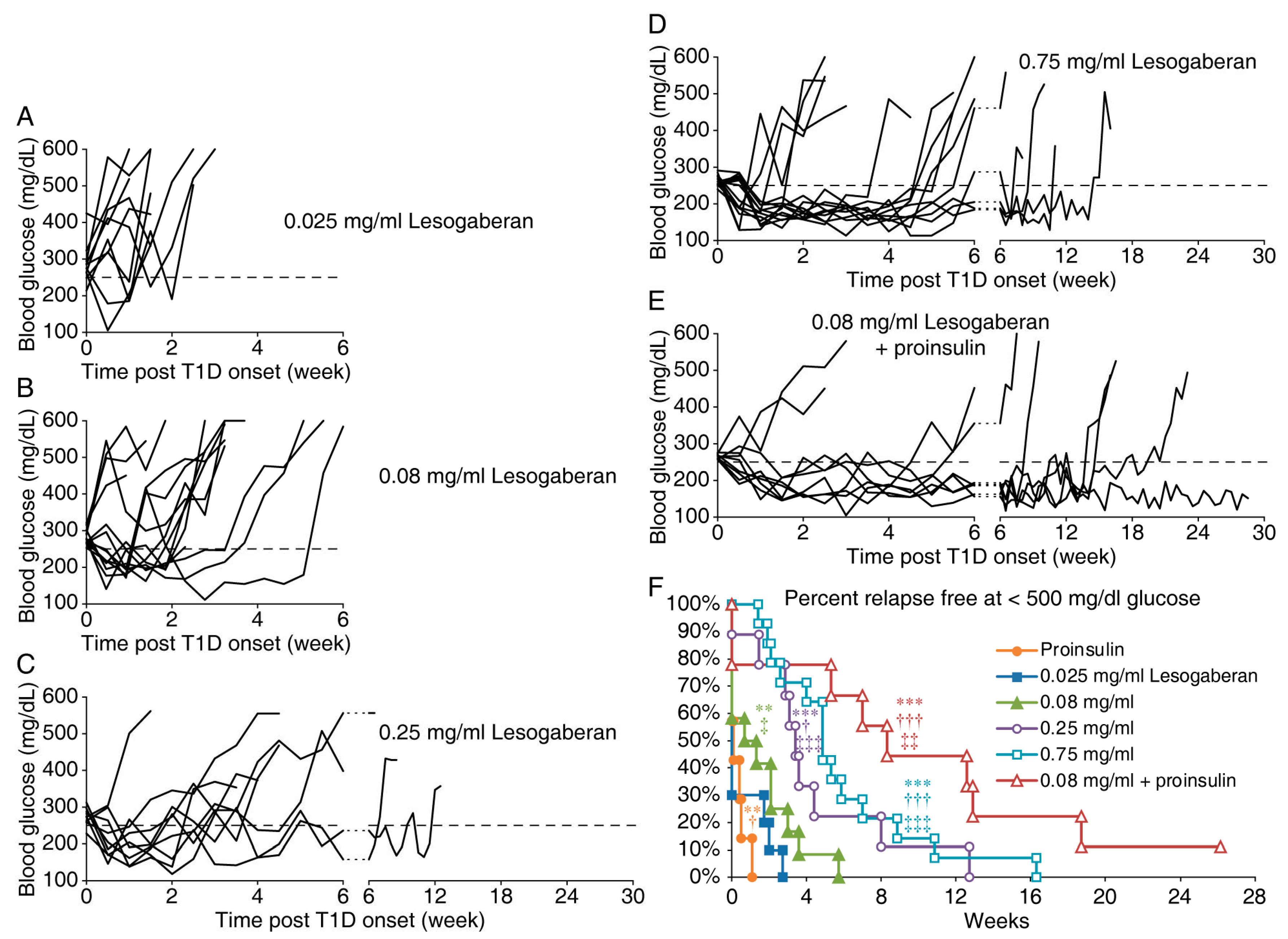
3.1. Lesogaberan Treatment Can Temporarily Correct Hyperglycemia in Newly Diabetic NOD Mice
3.2. The Combination of a Low Dose of Lesogaberan and Proinsulin/Alum Treatment Prolongs the Disease Remission Periods in Newly Diabetic NOD Mice
3.3. Combination of a Low Dose of Lesogaberan and Anti-CD3 Treatments Synergistically Increases the Frequency of Severely Diabetic NOD Mice with Diabetes Remission
3.4. Functional and Histological Assessments of Islets in Mice Given Low Doses of Anti-CD3 and Lesogaberan
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Olsen, R.W.; Sieghart, W. GABA A receptors: Subtypes provide diversity of function and pharmacology. Neuropharmacology 2009, 56, 141–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettler, B.; Kaupmann, K.; Mosbacher, J.; Gassmann, M. Molecular structure and physiological functions of GABA(B) receptors. Physiol. Rev. 2004, 84, 835–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.; Chau, C.; Hales, T.G.; Kaufman, D.L. GABA(A) receptors mediate inhibition of T cell responses. J. Neuroimmunol. 1999, 96, 21–28. [Google Scholar] [CrossRef]
- Mendu, S.K.; Bhandage, A.; Jin, Z.; Birnir, B. Different subtypes of GABA-A receptors are expressed in human, mouse and rat T lymphocytes. PLoS ONE 2012, 7, e42959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prud’homme, G.J.; Glinka, Y.; Hasilo, C.; Paraskevas, S.; Li, X.; Wang, Q. GABA protects human islet cells against the deleterious effects of immunosuppressive drugs and exerts immunoinhibitory effects alone. Transplantation 2013, 96, 616–623. [Google Scholar] [CrossRef]
- Alam, S.; Laughton, D.L.; Walding, A.; Wolstenholme, A.J. Human peripheral blood mononuclear cells express GABAA receptor subunits. Mol. Immunol. 2006, 43, 1432–1442. [Google Scholar] [CrossRef]
- Tian, J.; Lu, Y.; Zhang, H.; Chau, C.H.; Dang, H.N.; Kaufman, D.L. Gamma-aminobutyric acid inhibits T cell autoimmunity and the development of inflammatory responses in a mouse type 1 diabetes model. J. Immunol. 2004, 173, 5298–5304. [Google Scholar] [CrossRef]
- Bhandage, A.K.; Jin, Z.; Korol, S.V.; Shen, Q.; Pei, Y.; Deng, Q. GABA Regulates Release of Inflammatory Cytokines From Peripheral Blood Mononuclear Cells and CD4(+) T Cells and Is Immunosuppressive in Type 1 Diabetes. EBioMedicine 2018, 30, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Dionisio, L.; Jose De Rosa, M.; Bouzat, C.; Esandi Mdel, C. An intrinsic GABAergic system in human lymphocytes. Neuropharmacology 2011, 60, 513–519. [Google Scholar] [CrossRef]
- Wheeler, D.W.; Thompson, A.J.; Corletto, F.; Reckless, J.; Loke, J.C.; Lapaque, N. Anaesthetic impairment of immune function is mediated via GABA(A) receptors. PLoS ONE 2011, 6, e17152. [Google Scholar] [CrossRef] [Green Version]
- Bjurstom, H.; Wang, J.; Ericsson, I.; Bengtsson, M.; Liu, Y.; Kumar-Mendu, S. GABA, a natural immunomodulator of T lymphocytes. J. Neuroimmunol. 2008, 205, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Yong, J.; Dang, H.; Kaufman, D.L. Oral GABA treatment downregulates inflammatory responses in a mouse model of rheumatoid arthritis. Autoimmunity 2011, 44, 465–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, R.; Axtell, R.; Mitra, A.; Miranda, M.; Lock, C.; Tsien, R.W. Inhibitory role for GABA in autoimmune inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 2580–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soltani, N.; Qiu, H.; Aleksic, M.; Glinka, Y.; Zhao, F.; Liu, R. GABA exerts protective and regenerative effects on islet beta cells and reverses diabetes. Proc. Natl. Acad Sci. USA 2011, 108, 11692–11697. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Dang, H.; Wallner, M.; Olsen, R.; Kaufman, D.L. Homotaurine, a safe blood-brain barrier permeable GABAA-R-specific agonist, ameliorates disease in mouse models of multiple sclerosis. Sci. Rep. 2018, 8, 16555. [Google Scholar] [CrossRef]
- Tian, J.; Dang, H.; O’Laco, K.; Song, M.; Tiu, B.-C. Homotaurine treatment enhances CD4+ and CD8+ Treg responses and synergizes with low-dose anti-CD3 to enhance diabetes remission in type 1 diabetic mice. ImmuoHorizons 2019, 3, 498–510. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Milddleton, B.; Kaufman, D.L. GABA administration prevents severe illness and death following coronavirus infection in mice. bioRxiv 2020. [Google Scholar] [CrossRef]
- Huang, S.; Mao, J.; Wei, B.; Pei, G. The anti-spasticity drug baclofen alleviates collagen-induced arthritis and regulates dendritic cells. J. Cell Physiol. 2015, 230, 1438–1447. [Google Scholar] [CrossRef]
- Duthey, B.; Hubner, A.; Diehl, S.; Boehncke, S.; Pfeffer, J.; Boehncke, W.H. Anti-inflammatory effects of the GABA(B) receptor agonist baclofen in allergic contact dermatitis. Exp. Dermatol. 2010, 19, 661–666. [Google Scholar] [CrossRef]
- Beales, P.E.; Hawa, M.; Williams, A.J.; Albertini, M.C.; Giorgini, A.; Pozzilli, P. Baclofen, a gamma-aminobutyric acid-b receptor agonist, delays diabetes onset in the non-obese diabetic mouse. Acta Diabetol. 1995, 32, 53–56. [Google Scholar] [CrossRef]
- Crowley, T.; Fitzpatrick, J.M.; Kuijper, T.; Cryan, J.F.; O’Toole, O.; O’Leary, O.F. Modulation of TLR3/TLR4 inflammatory signaling by the GABAB receptor agonist baclofen in glia and immune cells: Relevance to therapeutic effects in multiple sclerosis. Front. Cell Neurosci. 2015, 9, 284. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.H.; Kurose, T.; Kato, S.; Masuda, K.; Tsuda, K.; Ishida, H. Suppressive effect of GABA on insulin secretion from the pancreatic beta-cells in the rat. Life Sci. 1993, 52, 687–694. [Google Scholar] [CrossRef]
- Rorsman, P.; Berggren, P.O.; Bokvist, K.; Ericson, H.; Mohler, H.; Ostenson, C.G. Glucose-inhibition of glucagon secretion involves activation of GABAA-receptor chloride channels. Nature 1989, 341, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Braun, M.; Ramracheya, R.; Bengtsson, M.; Clark, A.; Walker, J.N.; Johnson, P.R. Gamma-aminobutyric acid (GABA) is an autocrine excitatory transmitter in human pancreatic beta-cells. Diabetes 2010, 59, 1694–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, M.; Wendt, A.; Birnir, B.; Broman, J.; Eliasson, L.; Galvanovskis, J. Regulated exocytosis of GABA-containing synaptic-like microvesicles in pancreatic beta-cells. J. Gen. Physiol. 2004, 123, 191–204. [Google Scholar] [CrossRef] [Green Version]
- Taneera, J.; Jin, Z.; Jin, Y.; Muhammed, S.J.; Zhang, E.; Lang, S. Gamma-Aminobutyric acid (GABA) signalling in human pancreatic islets is altered in type 2 diabetes. Diabetologia 2012, 55, 1985–1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brice, N.L.; Varadi, A.; Ashcroft, S.J.; Molnar, E. Metabotropic glutamate and GABA(B) receptors contribute to the modulation of glucose-stimulated insulin secretion in pancreatic beta cells. Diabetologia 2002, 45, 242–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.; Dang, H.; Chen, Z.; Guan, A.; Jin, Y.; Atkinson, M.A. gamma-Aminobutyric acid regulates both the survival and replication of human beta-cells. Diabetes 2013, 62, 3760–3765. [Google Scholar] [CrossRef] [Green Version]
- Purwana, I.; Zheng, J.; Li, X.; Deurloo, M.; Son, D.O.; Zhang, Z. GABA promotes human beta-cell proliferation and modulates glucose homeostasis. Diabetes 2014, 63, 4197–4205. [Google Scholar] [CrossRef] [Green Version]
- Martin, P.; Massol, J.; Pichat, P.; Puech, A.J. Decreased central GABA B receptor binding sites in diabetic rats. Neuropsychobiology 1988, 19, 146–148. [Google Scholar] [CrossRef]
- Tian, J.; Dang, H. Combining Antigen-Based Therapy with GABA Treatment Synergistically Prolongs Survival of Transplanted ß-Cells in Diabetic NOD Mice. PLoS ONE 2011, 6, e25337. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, A.; Antonsson, M.; Holmberg, A.A.; Blackshaw, L.A.; Branden, L.; Brauner-Osborne, H. (R)-(3-amino-2-fluoropropyl) phosphinic acid (AZD3355), a novel GABAB receptor agonist, inhibits transient lower esophageal sphincter relaxation through a peripheral mode of action. J. Pharmacol. Exp. Ther. 2009, 331, 504–512. [Google Scholar] [CrossRef] [Green Version]
- Shaheen, N.J.; Denison, H.; Bjorck, K.; Karlsson, M.; Silberg, D.G. Efficacy and safety of lesogaberan in gastro-oesophageal reflux disease: A randomised controlled trial. Gut 2013, 62, 1248–1255. [Google Scholar] [CrossRef] [PubMed]
- Alstermark, C.; Amin, K.; Dinn, S.R.; Elebring, T.; Fjellstrom, O.; Fitzpatrick, K. Synthesis and pharmacological evaluation of novel gamma-aminobutyric acid type B (GABAB) receptor agonists as gastroesophageal reflux inhibitors. J. Med. Chem. 2008, 51, 4315–4320. [Google Scholar] [CrossRef] [PubMed]
- Niazi, M.; Skrtic, S.; Ruth, M.; Holmberg, A.A. Pharmacokinetic profile of lesogaberan (AZD3355) in healthy subjects: A novel GABA(B)-receptor agonist reflux inhibitor. Drugs R D 2011, 11, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Fransson, B.; Silberg, D.G.; Niazi, M.; Miller, F.; Ruth, M.; Holmberg, A.A. Effect of food on the bioavailability of lesogaberan given as an oral solution or as modified-release capsules in healthy male volunteers. Int. J. Clin. Pharmacol. Ther. 2012, 50, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Boeckxstaens, G.E.; Beaumont, H.; Mertens, V.; Denison, H.; Ruth, M.; Adler, J. Effects of lesogaberan on reflux and lower esophageal sphincter function in patients with gastroesophageal reflux disease. Gastroenterology 2010, 139, 409–417. [Google Scholar] [CrossRef]
- Tian, J.; Dang, H.; Hu, A.; Xu, W.; Kaufman, D.L. Repurposing Lesogaberan to Promote Human Islet Cell Survival and beta-Cell Replication. J. Diabetes Res. 2017, 2017, 6403539. [Google Scholar] [CrossRef] [Green Version]
- Shoda, L.K.; Young, D.L.; Ramanujan, S.; Whiting, C.C.; Atkinson, M.A.; Bluestone, J.A. A comprehensive review of interventions in the NOD mouse and implications for translation. Immunity 2005, 23, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Dang, H.; Nguyen, A.V.; Chen, Z.; Kaufman, D.L. Combined therapy with GABA and proinsulin/alum acts synergistically to restore long-term normoglycemia by modulating T-cell autoimmunity and promoting beta-cell replication in newly diabetic NOD mice. Diabetes 2014, 63, 3128–3134. [Google Scholar] [CrossRef] [Green Version]
- Herold, K.C.; Bundy, B.N.; Long, S.A.; Bluestone, J.A.; DiMeglio, L.A.; Dufort, M.J. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N. Engl. J. Med. 2019, 381, 603–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuks, J.M.; Arrighi, R.B.; Weidner, J.M.; Kumar Mendu, S.; Jin, Z.; Wallin, R.P. GABAergic signaling is linked to a hypermigratory phenotype in dendritic cells infected by Toxoplasma gondii. PLoS Pathog. 2012, 8, e1003051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatenoud, L.; Primo, J.; Bach, J.F. CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J. Immunol. 1997, 158, 2947–2954. [Google Scholar] [PubMed]
- Sherry, N.A.; Chen, W.; Kushner, J.A.; Glandt, M.; Tang, Q.; Tsai, S. Exendin-4 improves reversal of diabetes in NOD mice treated with anti-CD3 monoclonal antibody by enhancing recovery of beta-cells. Endocrinology 2007, 148, 5136–5144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bresson, D.; Togher, L.; Rodrigo, E.; Chen, Y.; Bluestone, J.A.; Herold, K.C. Anti-CD3 and nasal proinsulin combination therapy enhances remission from recent-onset autoimmune diabetes by inducing Tregs. J. Clin. Investig. 2006, 116, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Chatenoud, L.; Thervet, E.; Primo, J.; Bach, J.F. Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc. Natl. Acad. Sci. USA 1994, 91, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Mottram, P.L.; Murray-Segal, L.J.; Han, W.; Maguire, J.; Stein-Oakley, A.N. Remission and pancreas isograft survival in recent onset diabetic NOD mice after treatment with low-dose anti-CD3 monoclonal antibodies. Transpl. Immunol. 2002, 10, 63–72. [Google Scholar] [CrossRef]
- Bonaventura, M.M.; Crivello, M.; Ferreira, M.L.; Repetto, M.; Cymeryng, C.; Libertun, C. Effects of GABAB receptor agonists and antagonists on glycemia regulation in mice. Eur. J. Pharmacol. 2012, 677, 188–196. [Google Scholar] [CrossRef]
- Tian, J.; Dang, H.N.; Yong, J.; Chui, W.S.; Dizon, M.P.; Yaw, C.K. Oral treatment with gamma-aminobutyric acid improves glucose tolerance and insulin sensitivity by inhibiting inflammation in high fat diet-fed mice. PLoS ONE 2011, 6, e25338. [Google Scholar] [CrossRef] [Green Version]
- Boeckxstaens, G.E.; Beaumont, H.; Hatlebakk, J.G.; Silberg, D.G.; Bjorck, K.; Karlsson, M. A novel reflux inhibitor lesogaberan (AZD3355) as add-on treatment in patients with GORD with persistent reflux symptoms despite proton pump inhibitor therapy: A randomised placebo-controlled trial. Gut 2011, 60, 1182–1188. [Google Scholar] [CrossRef]
- Niazi, M.; Silberg, D.G.; Miller, F.; Ruth, M.; Holmberg, A.A. Evaluation of the pharmacokinetic interaction between lesogaberan (AZD3355) and esomeprazole in healthy subjects. Drugs R D 2010, 10, 243–251. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rane, M.J.; Gozal, D.; Butt, W.; Gozal, E.; Pierce, W.M., Jr.; Guo, S.Z. Gamma-amino butyric acid type B receptors stimulate neutrophil chemotaxis during ischemia-reperfusion. J. Immunol. 2005, 174, 7242–7249. [Google Scholar] [CrossRef] [PubMed]
- Kaupmann, K.; Huggel, K.; Heid, J.; Flor, P.J.; Bischoff, S.; Mickel, S.J. Expression cloning of GABA(B) receptors uncovers similarity to metabotropic glutamate receptors. Nature 1997, 386, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Prosser, H.M.; Gill, C.H.; Hirst, W.D.; Grau, E.; Robbins, M.; Calver, A. Epileptogenesis and enhanced prepulse inhibition in GABA(B1)-deficient mice. Mol. Cell Neurosci. 2001, 17, 1059–1070. [Google Scholar] [CrossRef]
- Schuler, V.; Luscher, C.; Blanchet, C.; Klix, N.; Sansig, G.; Klebs, K. Epilepsy, hyperalgesia, impaired memory, and loss of pre- and postsynaptic GABA(B) responses in mice lacking GABA(B(1)). Neuron 2001, 31, 47–58. [Google Scholar] [CrossRef]
- Robbins, M.J.; Calver, A.R.; Filippov, A.K.; Hirst, W.D.; Russell, R.B.; Wood, M.D. GABA(B2) is essential for g-protein coupling of the GABA(B) receptor heterodimer. J. Neurosci. 2001, 21, 8043–8052. [Google Scholar] [CrossRef] [Green Version]
- Monaco, G.; Lee, B.; Xu, W.; Mustafah, S.; Hwang, Y.Y.; Carre, C. RNA-Seq Signatures Normalized by mRNA Abundance Allow Absolute Deconvolution of Human Immune Cell Types. Cell Rep. 2019, 26, 1627–1640. [Google Scholar] [CrossRef] [Green Version]
- Soghomonian, J.J.; Martin, D.L. Two isoforms of glutamate decarboxylase: Why? Trends Pharmacol Sci. 1998, 19, 500–505. [Google Scholar]
- Seiler, N. On the role of GABA in vertebrate polyamine metabolism. Physiol. Chem. Phys. 1980, 12, 411–429. [Google Scholar]
- Caron, P.C.; Kremzner, L.T.; Cote, L.J. GABA and its relationship to putrescine metabolism in the rat brain and pancreas. Neurochem. Int. 1987, 10, 219–229. [Google Scholar] [CrossRef]
- Lee, S.; Yoon, B.E.; Berglund, K.; Oh, S.J.; Park, H.; Shin, H.S. Channel-mediated tonic GABA release from glia. Science 2010, 330, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.J.; Lee, C.J. Distribution and Function of the Bestrophin-1 (Best1) Channel in the Brain. Exp. Neurobiol. 2017, 26, 113–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhandage, A.K.; Cunningham, J.L.; Jin, Z.; Shen, Q.; Bongiovanni, S.; Korol, S.V. Depression, GABA, and Age Correlate with Plasma Levels of Inflammatory Markers. Int. J. Mol. Sci. 2019, 20, 6172. [Google Scholar] [CrossRef] [Green Version]
- David, M.; Richer, M.; Mamarbachi, A.M.; Villeneuve, L.R.; Dupre, D.J.; Hebert, T.E. Interactions between GABA-B1 receptors and Kir 3 inwardly rectifying potassium channels. Cell Signal. 2006, 18, 2172–2181. [Google Scholar] [CrossRef] [PubMed]
- Richer, M.; David, M.; Villeneuve, L.R.; Trieu, P.; Ethier, N.; Petrin, D. GABA-B(1) receptors are coupled to the ERK1/2 MAP kinase pathway in the absence of GABA-B(2) subunits. J. Mol. Neurosci. 2009, 38, 67–79. [Google Scholar] [CrossRef]
- Baloucoune, G.A.; Chun, L.; Zhang, W.; Xu, C.; Huang, S.; Sun, Q. GABAB receptor subunit GB1 at the cell surface independently activates ERK1/2 through IGF-1R transactivation. PLoS ONE 2012, 7, e39698. [Google Scholar] [CrossRef]
- Olsen, R.W. GABAA receptor: Positive and negative allosteric modulators. Neuropharmacology 2018, 136, 10–22. [Google Scholar] [CrossRef]
- Shimada, S.; Cutting, G.; Uhl, G.R. Gamma-Aminobutyric acid A or C receptor? gamma-Aminobutyric acid rho 1 receptor RNA induces bicuculline-, barbiturate-, and benzodiazepine-insensitive gamma-aminobutyric acid responses in Xenopus oocytes. Mol. Pharmacol. 1992, 41, 683–687. [Google Scholar]
- Enz, R.; Cutting, G.R. Molecular composition of GABAC receptors. Vis. Res. 1998, 38, 1431–1441. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Dang, H.; Karashchuk, N.; Xu, I.; Kaufman, D.L. A Clinically Applicable Positive Allosteric Modulator of GABA Receptors Promotes Human beta-Cell Replication and Survival as well as GABA’s Ability to Inhibit Inflammatory T Cells. J. Diabetes Res. 2019, 2019, 5783545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhandage, A.K.; Olivera, G.C.; Kanatani, S.; Thompson, E.; Lore, K.; Varas-Godoy, M. A motogenic GABAergic system of mononuclear phagocytes facilitates dissemination of coccidian parasites. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Hales, T.G.; Tyndale, R.F. Few cell lines with GABAA mRNAs have functional receptors. J. Neurosci. 1994, 14, 5429–5436. [Google Scholar] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, J.; Middleton, B.; Lee, V.S.; Park, H.W.; Zhang, Z.; Kim, B.; Lowe, C.; Nguyen, N.; Liu, H.; Beyer, R.S.; et al. GABAB-Receptor Agonist-Based Immunotherapy for Type 1 Diabetes in NOD Mice. Biomedicines 2021, 9, 43. https://doi.org/10.3390/biomedicines9010043
Tian J, Middleton B, Lee VS, Park HW, Zhang Z, Kim B, Lowe C, Nguyen N, Liu H, Beyer RS, et al. GABAB-Receptor Agonist-Based Immunotherapy for Type 1 Diabetes in NOD Mice. Biomedicines. 2021; 9(1):43. https://doi.org/10.3390/biomedicines9010043
Chicago/Turabian StyleTian, Jide, Blake Middleton, Victoria Seunghee Lee, Hye Won Park, Zhixuan Zhang, Bokyoung Kim, Catherine Lowe, Nancy Nguyen, Haoyuan Liu, Ryan S. Beyer, and et al. 2021. "GABAB-Receptor Agonist-Based Immunotherapy for Type 1 Diabetes in NOD Mice" Biomedicines 9, no. 1: 43. https://doi.org/10.3390/biomedicines9010043