
Tumor-Associated Mast Cells in Urothelial Bladder Cancer: Optimizing Immuno-Oncology


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. General Immunobiology of Mast Cells
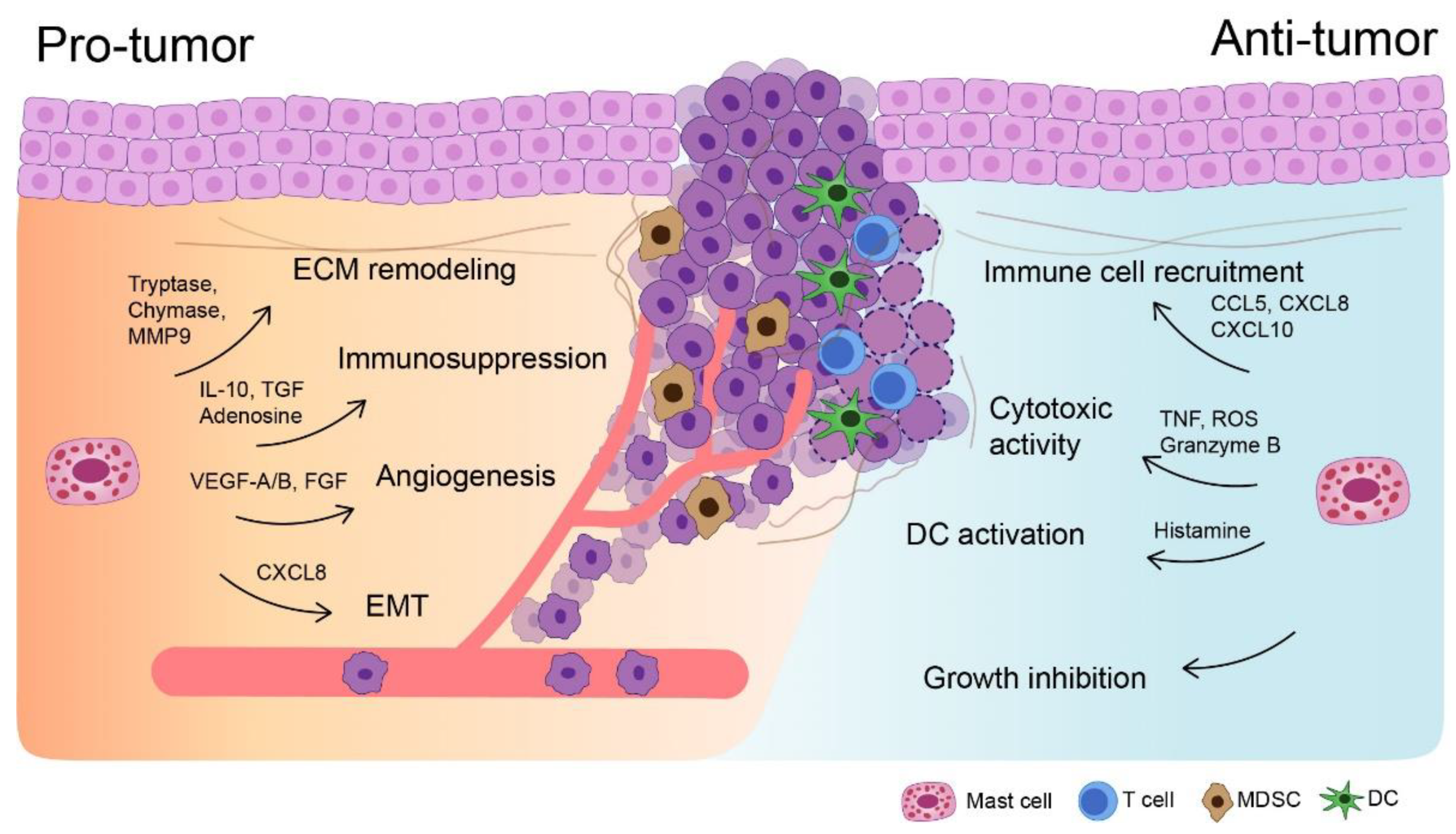
3. Mast Cells in UBC: A Double-Edged Sword
4. Evidence for Pro-Tumorigenic Roles of Mast Cells in Urothelial Bladder Cancer
5. Cytokine Milieu in the Tumor Microenvironment Generated by Mast Cells
6. Mast Cells Boost Therapeutic Efficacy against Non-Muscle Invasive Bladder Cancer
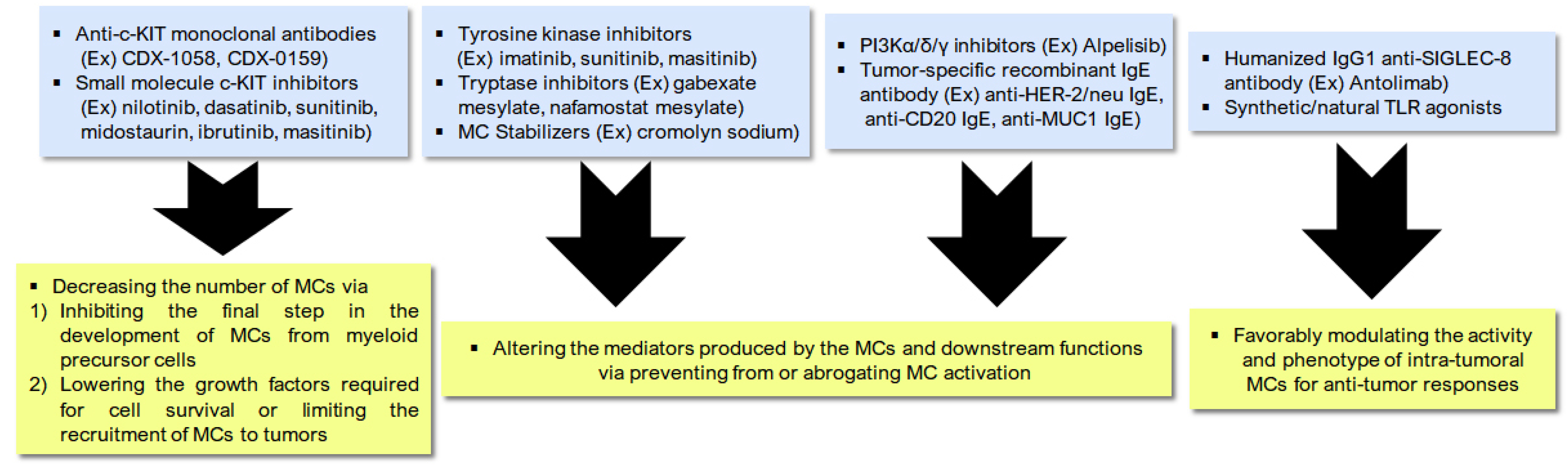
7. Manipulating Mast Cells to Boost Treatment-Induced Immunogenic Cell Death
8. Take-Home Messages and Challenges
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kaur, J.; Choi, W.; Geynisman, D.M.; Plimack, E.R.; Ghatalia, P. Role of immunotherapy in localized muscle invasive urothelial cancer. Ther. Adv. Med. Oncol. 2021, 13, 17588359211045858. [Google Scholar] [PubMed]
- Stecca, C.; Abdeljalil, O.; Sridhar, S.S. Metastatic Urothelial Cancer: A rapidly changing treatment landscape. Ther. Adv. Med. Oncol. 2021, 13. [Google Scholar] [CrossRef]
- Babjuk, M.; Burger, M.; Capoun, O.; Cohen, D.; Comperat, E.M.; Dominguez Escrig, J.L.; Gontero, P.; Liedberg, F.; Masson-Lecomte, A.; Mostafid, A.H.; et al. European Association of Urology Guidelines on Non-muscle-invasive Bladder Cancer (Ta, T1, and Carcinoma in Situ). Eur. Urol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Nelson, B.E.; Hong, A.; Jana, B. Elucidation of Novel Molecular Targets for Therapeutic Strategies in Urothelial Carcinoma: A Literature Review. Front. Oncol. 2021, 11, 705294. [Google Scholar] [CrossRef]
- Kim, I.H.; Lee, H.J. Perioperative Systemic Treatment for Muscle-Invasive Bladder Cancer: Current Evidence and Future Perspectives. Int. J. Mol. Sci. 2021, 22, 7201. [Google Scholar] [CrossRef]
- Renner, A.; Burotto, M.; Valdes, J.M.; Roman, J.C.; Walton-Diaz, A. Neoadjuvant immunotherapy for muscle invasive urothelial bladder carcinoma: Will it change current standards? Ther. Adv. Urol. 2021, 13. [Google Scholar] [CrossRef]
- Roviello, G.; Catalano, M.; Santi, R.; Palmieri, V.E.; Vannini, G.; Galli, I.C.; Buttitta, E.; Villari, D.; Rossi, V.; Nesi, G. Immune Checkpoint Inhibitors in Urothelial Bladder Cancer: State of the Art and Future Perspectives. Cancers 2021, 13, 4411. [Google Scholar] [CrossRef]
- Yang, Z.; Xu, Y.; Bi, Y.; Zhang, N.; Wang, H.; Xing, T.; Bai, S.; Shen, Z.; Naz, F.; Zhang, Z.; et al. Immune escape mechanisms and immunotherapy of urothelial bladder cancer. J. Clin. Transl. Res. 2021, 7, 485–500. [Google Scholar]
- Bin Riaz, I.; Khan, A.M.; Catto, J.W.; Hussain, S.A. Bladder cancer: Shedding light on the most promising investigational drugs in clinical trials. Expert. Opin. Investig. Drugs 2021, 30, 837–855. [Google Scholar] [CrossRef]
- Chung, D.Y.; Kang, D.H.; Kim, J.W.; Ha, J.S.; Kim, D.K.; Cho, K.S. Comparison of Oncologic Outcomes of Dose-Dense Methotrexate, Vinblastine, Doxorubicin, and Cisplatin (ddMVAC) with Gemcitabine and Cisplatin (GC) as Neoadjuvant Chemotherapy for Muscle-Invasive Bladder Cancer: Systematic Review and Meta-Analysis. Cancers 2021, 13, 2770. [Google Scholar] [CrossRef]
- Suh, J.; Jung, J.H.; Kwak, C.; Kim, H.H.; Ku, J.H. Stratifying risk for multiple, recurrent, and large (>/=3 cm) Ta, G1/G2 tumors in non-muscle-invasive bladder cancer. Investig. Clin. Urol. 2021, 62, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Yassaie, O.; Chehroudi, C.; Black, P.C. Novel and emerging approaches in the management of non-muscle invasive urothelial carcinoma. Ther. Adv. Med. Oncol. 2021, 13. [Google Scholar] [CrossRef]
- Comperat, E.; Larre, S.; Roupret, M.; Neuzillet, Y.; Pignot, G.; Quintens, H.; Houede, N.; Roy, C.; Durand, X.; Varinot, J.; et al. Clinicopathological characteristics of urothelial bladder cancer in patients less than 40 years old. Virchows Arch. 2015, 466, 589–594. [Google Scholar] [CrossRef]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Derakhshani, A.; Vahidian, F.; Alihasanzadeh, M.; Mokhtarzadeh, A.; Lotfi Nezhad, P.; Baradaran, B. Mast cells: A double-edged sword in cancer. Immunol. Lett. 2019, 209, 28–35. [Google Scholar] [CrossRef]
- Domagala, M.; Laplagne, C.; Leveque, E.; Laurent, C.; Fournie, J.J.; Espinosa, E.; Poupot, M. Cancer Cells Resistance Shaping by Tumor Infiltrating Myeloid Cells. Cancers 2021, 13, 165. [Google Scholar] [CrossRef]
- Saxena, S.; Singh, A.; Singh, P. Tumor associated mast cells: Biological roles and therapeutic applications. Anat. Cell Biol. 2020, 53, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Gurish, M.F.; Boyce, J.A. Mast cell growth, differentiation, and death. Clin. Rev. Allergy Immunol. 2002, 22, 107–118. [Google Scholar] [CrossRef]
- Aller, M.A.; Arias, A.; Arias, J.I.; Arias, J. Carcinogenesis: The cancer cell-mast cell connection. Inflamm. Res. 2019, 68, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, S.C. Physiological and pathophysiological functions of intestinal mast cells. Semin. Immunopathol. 2009, 31, 185–205. [Google Scholar] [CrossRef]
- Christmas, T.J.; Rode, J. Characteristics of mast cells in normal bladder, bacterial cystitis and interstitial cystitis. Br. J. Urol. 1991, 68, 473–478. [Google Scholar] [CrossRef]
- Galli, S.J.; Tsai, M. Mast cells: Versatile regulators of inflammation, tissue remodeling, host defense and homeostasis. J. Dermatol. Sci. 2008, 49, 7–19. [Google Scholar] [CrossRef] [Green Version]
- Douaiher, J.; Succar, J.; Lancerotto, L.; Gurish, M.F.; Orgill, D.P.; Hamilton, M.J.; Krilis, S.A.; Stevens, R.L. Development of mast cells and importance of their tryptase and chymase serine proteases in inflammation and wound healing. Adv. Immunol. 2014, 122, 211–252. [Google Scholar] [PubMed] [Green Version]
- Moon, T.C.; St Laurent, C.D.; Morris, K.E.; Marcet, C.; Yoshimura, T.; Sekar, Y.; Befus, A.D. Advances in mast cell biology: New understanding of heterogeneity and function. Mucosal Immunol. 2010, 3, 111–128. [Google Scholar] [CrossRef] [Green Version]
- Komi, D.E.A.; Redegeld, F.A. Role of Mast Cells in Shaping the Tumor Microenvironment. Clin. Rev. Allergy Immunol. 2020, 58, 313–325. [Google Scholar] [CrossRef] [Green Version]
- Banstola, A.; Jeong, J.H.; Yook, S. Immunoadjuvants for cancer immunotherapy: A review of recent developments. Acta Biomater. 2020, 114, 16–30. [Google Scholar] [CrossRef]
- Azuma, I.; Seya, T. Development of immunoadjuvants for immunotherapy of cancer. Int. Immunopharmacol. 2001, 1, 1249–1259. [Google Scholar] [CrossRef]
- Gri, G.; Frossi, B.; D’Inca, F.; Danelli, L.; Betto, E.; Mion, F.; Sibilano, R.; Pucillo, C. Mast cell: An emerging partner in immune interaction. Front. Immunol. 2012, 3, 120. [Google Scholar] [CrossRef] [Green Version]
- Ribatti, D.; Tamma, R.; Crivellato, E. The dual role of mast cells in tumor fate. Cancer Lett. 2018, 433, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.J.; Nakae, S.; Tsai, M. Mast cells in the development of adaptive immune responses. Nat. Immunol. 2005, 6, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Rios, E.J.; Kalesnikoff, J. FcepsilonRI expression and dynamics on mast cells. Methods Mol. Biol. 2015, 1220, 239–255. [Google Scholar]
- Theoharides, T.C.; Alysandratos, K.D.; Angelidou, A.; Delivanis, D.A.; Sismanopoulos, N.; Zhang, B.; Asadi, S.; Vasiadi, M.; Weng, Z.; Miniati, A.; et al. Mast cells and inflammation. Biochim. Biophys. Acta 2012, 1822, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Ch’ng, S.; Wallis, R.A.; Yuan, L.; Davis, P.F.; Tan, S.T. Mast cells and cutaneous malignancies. Mod. Pathol. 2006, 19, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y. Heterogeneity of mast cells and phenotypic change between subpopulations. Annu. Rev. Immunol. 1989, 7, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, D.D.; Baram, D.; Mekori, Y.A. Mast cells. Physiol. Rev. 1997, 77, 1033–1079. [Google Scholar] [CrossRef]
- Williams, C.M.; Galli, S.J. The diverse potential effector and immunoregulatory roles of mast cells in allergic disease. J. Allergy Clin. Immunol. 2000, 105, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Oskeritzian, C.A. Mast cell plasticity and sphingosine-1-phosphate in immunity, inflammation and cancer. Mol. Immunol. 2015, 63, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.; Wang, S.; Cheng, P. Tumor-infiltrating tryptase(+) mast cells predict unfavorable clinical outcome in solid tumors. Int. J. Cancer 2018, 142, 813–821. [Google Scholar] [CrossRef] [Green Version]
- Cimpean, A.M.; Tamma, R.; Ruggieri, S.; Nico, B.; Toma, A.; Ribatti, D. Mast cells in breast cancer angiogenesis. Crit. Rev. Oncol. Hematol 2017, 115, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Guidolin, D.; Marzullo, A.; Nico, B.; Annese, T.; Benagiano, V.; Crivellato, E. Mast cells and angiogenesis in gastric carcinoma. Int. J. Exp. Pathol. 2010, 91, 350–356. [Google Scholar] [CrossRef]
- Zhao, Y.B.; Yang, S.H.; Shen, J.; Deng, K.; Li, Q.; Wang, Y.; Cui, W.; Ye, H. Interaction between regulatory T cells and mast cells via IL-9 and TGF-beta production. Oncol. Lett. 2020, 20, 360. [Google Scholar] [CrossRef]
- Aller, M.A.; Arias, J.I.; Arias, J. Pathological axes of wound repair: Gastrulation revisited. Theor. Biol. Med. Model. 2010, 7, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dazzi, F.; Krampera, M. Mesenchymal stem cells and autoimmune diseases. Best Pract. Res. Clin. Haematol. 2011, 24, 49–57. [Google Scholar] [CrossRef]
- Detoraki, A.; Granata, F.; Staibano, S.; Rossi, F.W.; Marone, G.; Genovese, A. Angiogenesis and lymphangiogenesis in bronchial asthma. Allergy 2010, 65, 946–958. [Google Scholar] [CrossRef]
- Diegelmann, R.F.; Evans, M.C. Wound healing: An overview of acute, fibrotic and delayed healing. Front. Biosci. 2004, 9, 283–289. [Google Scholar] [CrossRef]
- Lacerda Mariano, L.; Ingersoll, M.A. The immune response to infection in the bladder. Nat. Rev. Urol. 2020, 17, 439–458. [Google Scholar] [CrossRef]
- Singh, J.; Shah, R.; Singh, D. Targeting mast cells: Uncovering prolific therapeutic role in myriad diseases. Int. Immunopharmacol. 2016, 40, 362–384. [Google Scholar] [CrossRef] [PubMed]
- Melillo, R.M.; Guarino, V.; Avilla, E.; Galdiero, M.R.; Liotti, F.; Prevete, N.; Rossi, F.W.; Basolo, F.; Ugolini, C.; de Paulis, A.; et al. Mast cells have a protumorigenic role in human thyroid cancer. Oncogene 2010, 29, 6203–6215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, R.; Gu, S.; Zhang, Y.; Luo, A.; Jing, X.; Zhao, L.; Zhao, X.; Zhang, L. Estrogen promotes progression of hormone-dependent breast cancer through CCL2-CCR2 axis by upregulation of Twist via PI3K/AKT/NF-kappaB signaling. Sci. Rep. 2018, 8, 9575. [Google Scholar] [CrossRef] [Green Version]
- Sari, A.; Calli, A.; Cakalagaoglu, F.; Altinboga, A.A.; Bal, K. Association of mast cells with microvessel density in urothelial carcinomas of the urinary bladder. Ann. Diagn. Pathol. 2012, 16, 1–6. [Google Scholar] [CrossRef]
- Leal-Berumen, I.; O’Byrne, P.; Gupta, A.; Richards, C.D.; Marshall, J.S. Prostanoid enhancement of interleukin-6 production by rat peritoneal mast cells. J. Immunol. 1995, 154, 4759–4767. [Google Scholar]
- Nakayama, T.; Mutsuga, N.; Yao, L.; Tosato, G. Prostaglandin E2 promotes degranulation-independent release of MCP-1 from mast cells. J. Leukoc. Biol. 2006, 79, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varricchi, G.; Galdiero, M.R.; Loffredo, S.; Marone, G.; Iannone, R.; Marone, G.; Granata, F. Are Mast Cells MASTers in Cancer? Front. Immunol. 2017, 8, 424. [Google Scholar] [CrossRef] [Green Version]
- Elieh-Ali-Komi, D.; Cao, Y. Role of Mast Cells in the Pathogenesis of Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. Clin. Rev. Allergy Immunol. 2017, 52, 436–445. [Google Scholar] [CrossRef]
- Lichterman, J.N.; Reddy, S.M. Mast Cells: A New Frontier for Cancer Immunotherapy. Cells 2021, 10, 1270. [Google Scholar] [CrossRef]
- Wasiuk, A.; Dalton, D.K.; Schpero, W.L.; Stan, R.V.; Conejo-Garcia, J.R.; Noelle, R.J. Mast cells impair the development of protective anti-tumor immunity. Cancer Immunol. Immunother. 2012, 61, 2273–2282. [Google Scholar] [CrossRef] [Green Version]
- Gorzalczany, Y.; Sagi-Eisenberg, R. Role of Mast Cell-Derived Adenosine in Cancer. Int. J. Mol. Sci. 2019, 20, 2603. [Google Scholar] [CrossRef] [Green Version]
- Kiener, H.P.; Baghestanian, M.; Dominkus, M.; Walchshofer, S.; Ghannadan, M.; Willheim, M.; Sillaber, C.; Graninger, W.B.; Smolen, J.S.; Valent, P. Expression of the C5a receptor (CD88) on synovial mast cells in patients with rheumatoid arthritis. Arthritis Rheum. 1998, 41, 233–245. [Google Scholar] [CrossRef]
- Theiner, G.; Gessner, A.; Lutz, M.B. The mast cell mediator PGD2 suppresses IL-12 release by dendritic cells leading to Th2 polarized immune responses in vivo. Immunobiology 2006, 211, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Vanbervliet, B.; Akdis, M.; Vocanson, M.; Rozieres, A.; Benetiere, J.; Rouzaire, P.; Akdis, C.A.; Nicolas, J.F.; Hennino, A. Histamine receptor H1 signaling on dendritic cells plays a key role in the IFN-gamma/IL-17 balance in T cell-mediated skin inflammation. J. Allergy Clin. Immunol. 2011, 127, 943–953.e10. [Google Scholar] [CrossRef]
- Eissmann, M.F.; Dijkstra, C.; Jarnicki, A.; Phesse, T.; Brunnberg, J.; Poh, A.R.; Etemadi, N.; Tsantikos, E.; Thiem, S.; Huntington, N.D.; et al. IL-33-mediated mast cell activation promotes gastric cancer through macrophage mobilization. Nat. Commun. 2019, 10, 2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammendola, M.; Gadaleta, C.D.; Frampton, A.E.; Piardi, T.; Memeo, R.; Zuccala, V.; Luposella, M.; Patruno, R.; Zizzo, N.; Gadaleta, P.; et al. The density of mast cells c-Kit(+) and tryptase(+) correlates with each other and with angiogenesis in pancreatic cancer patients. Oncotarget 2017, 8, 70463–70471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellstrand, K.; Asea, A.; Hermodsson, S. Histaminergic regulation of antibody-dependent cellular cytotoxicity of granulocytes, monocytes, and natural killer cells. J. Leukoc. Biol. 1994, 55, 392–397. [Google Scholar] [CrossRef] [Green Version]
- Hellstrand, K.; Hermodsson, S. Cell-to-cell mediated inhibition of natural killer cell proliferation by monocytes and its regulation by histamine H2-receptors. Scand. J. Immunol. 1991, 34, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Vosskuhl, K.; Greten, T.F.; Manns, M.P.; Korangy, F.; Wedemeyer, J. Lipopolysaccharide-mediated mast cell activation induces IFN-gamma secretion by NK cells. J. Immunol. 2010, 185, 119–125. [Google Scholar] [CrossRef]
- Panagi, M.; Voutouri, C.; Mpekris, F.; Papageorgis, P.; Martin, M.R.; Martin, J.D.; Demetriou, P.; Pierides, C.; Polydorou, C.; Stylianou, A.; et al. TGF-beta inhibition combined with cytotoxic nanomedicine normalizes triple negative breast cancer microenvironment towards anti-tumor immunity. Theranostics 2020, 10, 1910–1922. [Google Scholar] [CrossRef]
- Uwagawa, T.; Misawa, T.; Sakamoto, T.; Ito, R.; Gocho, T.; Shiba, H.; Wakiyama, S.; Hirohara, S.; Sadaoka, S.; Yanaga, K. A phase I study of full-dose gemcitabine and regional arterial infusion of nafamostat mesilate for advanced pancreatic cancer. Ann. Oncol. 2009, 20, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Uwagawa, T.; Misawa, T.; Tsutsui, N.; Ito, R.; Gocho, T.; Hirohara, S.; Sadaoka, S.; Yanaga, K. Phase II study of gemcitabine in combination with regional arterial infusion of nafamostat mesilate for advanced pancreatic cancer. Am. J. Clin. Oncol. 2013, 36, 44–48. [Google Scholar] [CrossRef]
- Ammendola, M.; Leporini, C.; Marech, I.; Gadaleta, C.D.; Scognamillo, G.; Sacco, R.; Sammarco, G.; De Sarro, G.; Russo, E.; Ranieri, G. Targeting mast cells tryptase in tumor microenvironment: A potential antiangiogenetic strategy. Biomed. Res. Int. 2014, 2014, 154702. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-beta and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, M.; Zhang, Y.; Rosenblatt, J.D. B cell regulation of the anti-tumor response and role in carcinogenesis. J. Immunother. Cancer 2016, 4, 40. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Seo, H.K. Emerging treatments for bacillus Calmette-Guerin-unresponsive non-muscle-invasive bladder cancer. Investig. Clin. Urol. 2021, 62, 361–377. [Google Scholar] [CrossRef]
- Bohle, A.; Jocham, D.; Bock, P.R. Intravesical bacillus Calmette-Guerin versus mitomycin C for superficial bladder cancer: A formal meta-analysis of comparative studies on recurrence and toxicity. J. Urol. 2003, 169, 90–95. [Google Scholar] [CrossRef]
- Malmstrom, P.U.; Sylvester, R.J.; Crawford, D.E.; Friedrich, M.; Krege, S.; Rintala, E.; Solsona, E.; Di Stasi, S.M.; Witjes, J.A. An individual patient data meta-analysis of the long-term outcome of randomised studies comparing intravesical mitomycin C versus bacillus Calmette-Guerin for non-muscle-invasive bladder cancer. Eur. Urol. 2009, 56, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Sfakianos, J.P.; Salome, B.; Daza, J.; Farkas, A.; Bhardwaj, N.; Horowitz, A. Bacillus Calmette-Guerin (BCG): Its fight against pathogens and cancer. Urol. Oncol. 2021, 39, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Babjuk, M.; Burger, M.; Comperat, E.M.; Gontero, P.; Mostafid, A.H.; Palou, J.; van Rhijn, B.W.G.; Roupret, M.; Shariat, S.F.; Sylvester, R.; et al. European Association of Urology Guidelines on Non-muscle-invasive Bladder Cancer (TaT1 and Carcinoma In Situ)—2019 Update. Eur. Urol. 2019, 76, 639–657. [Google Scholar] [CrossRef]
- Chang, S.S.; Boorjian, S.A.; Chou, R.; Clark, P.E.; Daneshmand, S.; Konety, B.R.; Pruthi, R.; Quale, D.Z.; Ritch, C.R.; Seigne, J.D.; et al. Diagnosis and Treatment of Non-Muscle Invasive Bladder Cancer: AUA/SUO Guideline. J. Urol. 2016, 196, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Flaig, T.W.; Spiess, P.E.; Agarwal, N.; Bangs, R.; Boorjian, S.A.; Buyyounouski, M.K.; Chang, S.; Downs, T.M.; Efstathiou, J.A.; Friedlander, T.; et al. Bladder Cancer, Version 3.2020, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2020, 18, 329–354. [Google Scholar] [CrossRef] [Green Version]
- Pak, S.; Kim, S.Y.; Kim, S.H.; Joung, J.Y.; Park, W.S.; Chung, J.; Lee, K.H.; Seo, H.K. Association Between Antibiotic Treatment and the Efficacy of Intravesical BCG Therapy in Patients With High-Risk Non-Muscle Invasive Bladder Cancer. Front. Oncol. 2021, 11, 570077. [Google Scholar] [CrossRef]
- Convit, J.; Montesinos, H.; Oviedo, H.; Romero, G.; Maccarone, B.; Essenfeld, E.; Convit, A.; Palacios, L.E. Autologous tumor lysate/Bacillus Calmette-Guerin immunotherapy as an adjuvant to conventional breast cancer therapy. Clin. Transl. Oncol. 2015, 17, 884–887. [Google Scholar] [CrossRef] [Green Version]
- Ayari, C.; LaRue, H.; Hovington, H.; Decobert, M.; Harel, F.; Bergeron, A.; Tetu, B.; Lacombe, L.; Fradet, Y. Bladder tumor infiltrating mature dendritic cells and macrophages as predictors of response to bacillus Calmette-Guerin immunotherapy. Eur. Urol. 2009, 55, 1386–1395. [Google Scholar] [CrossRef] [PubMed]
- Takayama, H.; Nishimura, K.; Tsujimura, A.; Nakai, Y.; Nakayama, M.; Aozasa, K.; Okuyama, A.; Nonomura, N. Increased infiltration of tumor associated macrophages is associated with poor prognosis of bladder carcinoma in situ after intravesical bacillus Calmette-Guerin instillation. J. Urol. 2009, 181, 1894–1900. [Google Scholar] [CrossRef]
- Brandau, S.; Riemensberger, J.; Jacobsen, M.; Kemp, D.; Zhao, W.; Zhao, X.; Jocham, D.; Ratliff, T.L.; Bohle, A. NK cells are essential for effective BCG immunotherapy. Int. J. Cancer 2001, 92, 697–702. [Google Scholar] [CrossRef]
- Pichler, R.; Fritz, J.; Zavadil, C.; Schafer, G.; Culig, Z.; Brunner, A. Tumor-infiltrating immune cell subpopulations influence the oncologic outcome after intravesical Bacillus Calmette-Guerin therapy in bladder cancer. Oncotarget 2016, 7, 39916–39930. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, T.; Shimizu, M.; Owaki, A.; Takahashi, M.; Shinya, E.; Nishimura, T.; Takahashi, H. A possible mechanism of intravesical BCG therapy for human bladder carcinoma: Involvement of innate effector cells for the inhibition of tumor growth. Cancer Immunol. Immunother. 2009, 58, 1245–1255. [Google Scholar] [CrossRef] [Green Version]
- Naoe, M.; Ogawa, Y.; Takeshita, K.; Morita, J.; Iwamoto, S.; Miyazaki, A.; Yoshida, H. Bacillus Calmette-Guerin-pulsed dendritic cells stimulate natural killer T cells and gammadeltaT cells. Int. J. Urol. 2007, 14, 532–538; discussion 538. [Google Scholar] [CrossRef]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef]
- Dowell, A.C.; Cobby, E.; Wen, K.; Devall, A.J.; During, V.; Anderson, J.; James, N.D.; Cheng, K.K.; Zeegers, M.P.; Bryan, R.T.; et al. Interleukin-17-positive mast cells influence outcomes from BCG for patients with CIS: Data from a comprehensive characterisation of the immune microenvironment of urothelial bladder cancer. PLoS ONE 2017, 12, e0184841. [Google Scholar] [CrossRef] [Green Version]
- Miranda-Filho, A.; Bray, F.; Charvat, H.; Rajaraman, S.; Soerjomataram, I. The world cancer patient population (WCPP): An updated standard for international comparisons of population-based survival. Cancer Epidemiol. 2020, 69, 101802. [Google Scholar] [CrossRef]
- Somasundaram, R.; Connelly, T.; Choi, R.; Choi, H.; Samarkina, A.; Li, L.; Gregorio, E.; Chen, Y.; Thakur, R.; Abdel-Mohsen, M.; et al. Tumor-infiltrating mast cells are associated with resistance to anti-PD-1 therapy. Nat. Commun. 2021, 12, 346. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhu, Y.; Xu, L.; Zhang, J.; Xie, H.; Fu, H.; Zhou, Q.; Chang, Y.; Dai, B.; Xu, J. Tumor stroma-infiltrating mast cells predict prognosis and adjuvant chemotherapeutic benefits in patients with muscle invasive bladder cancer. Oncoimmunology 2018, 7, e1474317. [Google Scholar] [CrossRef] [Green Version]
- Lyons, J.J.; Metcalfe, D.D. Targeting Mast Cells with Biologics. Immunol. Allergy Clin. N. Am. 2020, 40, 667–685. [Google Scholar] [CrossRef]
- Soucek, L.; Lawlor, E.R.; Soto, D.; Shchors, K.; Swigart, L.B.; Evan, G.I. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat. Med. 2007, 13, 1211–1218. [Google Scholar] [CrossRef]
- Wroblewski, M.; Bauer, R.; Cubas Cordova, M.; Udonta, F.; Ben-Batalla, I.; Legler, K.; Hauser, C.; Egberts, J.; Janning, M.; Velthaus, J.; et al. Mast cells decrease efficacy of anti-angiogenic therapy by secreting matrix-degrading granzyme B. Nat. Commun. 2017, 8, 269. [Google Scholar] [CrossRef] [PubMed]
- Finke, J.H.; Rini, B.; Ireland, J.; Rayman, P.; Richmond, A.; Golshayan, A.; Wood, L.; Elson, P.; Garcia, J.; Dreicer, R.; et al. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin. Cancer Res. 2008, 14, 6674–6682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, J.S.; Zea, A.H.; Rini, B.I.; Ireland, J.L.; Elson, P.; Cohen, P.; Golshayan, A.; Rayman, P.A.; Wood, L.; Garcia, J.; et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin. Cancer Res. 2009, 15, 2148–2157. [Google Scholar] [CrossRef] [Green Version]
- Andre, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Horak, F.; Puri, K.D.; Steiner, B.H.; Holes, L.; Xing, G.; Zieglmayer, P.; Zieglmayer, R.; Lemell, P.; Yu, A. Randomized phase 1 study of the phosphatidylinositol 3-kinase delta inhibitor idelalisib in patients with allergic rhinitis. J. Allergy Clin. Immunol. 2016, 137, 1733–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siraganian, R.P.; de Castro, R.O.; Barbu, E.A.; Zhang, J. Mast cell signaling: The role of protein tyrosine kinase Syk, its activation and screening methods for new pathway participants. FEBS Lett. 2010, 584, 4933–4940. [Google Scholar] [CrossRef] [Green Version]
- Teo, P.Z.; Utz, P.J.; Mollick, J.A. Using the allergic immune system to target cancer: Activity of IgE antibodies specific for human CD20 and MUC1. Cancer Immunol. Immunother. 2012, 61, 2295–2309. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, P.; Singer, J.; Hunt, J.; Gan, S.K.; Rudman, S.M.; Mechtcheriakova, D.; Knittelfelder, R.; Daniels, T.R.; Hobson, P.S.; Beavil, A.J.; et al. Characterisation of an engineered trastuzumab IgE antibody and effector cell mechanisms targeting HER2/neu-positive tumour cells. Cancer Immunol. Immunother. 2009, 58, 915–930. [Google Scholar] [CrossRef] [Green Version]
- Busse, W.; Corren, J.; Lanier, B.Q.; McAlary, M.; Fowler-Taylor, A.; Cioppa, G.D.; van As, A.; Gupta, N. Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J. Allergy Clin. Immunol. 2001, 108, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, B.A.; Brock, E.C.; Leung, J.; Falahati, R.; Bryce, P.J.; Bright, J.; Williams, J.; Shultz, L.D.; Greiner, D.L.; Brehm, M.A.; et al. AK002, a Humanized Sialic Acid-Binding Immunoglobulin-Like Lectin-8 Antibody that Induces Antibody-Dependent Cell-Mediated Cytotoxicity against Human Eosinophils and Inhibits Mast Cell-Mediated Anaphylaxis in Mice. Int. Arch. Allergy Immunol. 2019, 180, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, B.A.; Brock, E.C.; Leung, J.; Falahati, R.; Bochner, B.S.; Rasmussen, H.S.; Peterson, K.; Bebbington, C.; Tomasevic, N. Siglec-8 antibody reduces eosinophils and mast cells in a transgenic mouse model of eosinophilic gastroenteritis. JCI Insight 2019, 4, e126219. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Lei, Z.; Zhang, G.M.; Li, D.; Song, C.; Li, B.; Liu, Y.; Yuan, Y.; Unkeless, J.; Xiong, H.; et al. SCF-mediated mast cell infiltration and activation exacerbate the inflammation and immunosuppression in tumor microenvironment. Blood 2008, 112, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Gounaris, E.; Erdman, S.E.; Restaino, C.; Gurish, M.F.; Friend, D.S.; Gounari, F.; Lee, D.M.; Zhang, G.; Glickman, J.N.; Shin, K.; et al. Mast cells are an essential hematopoietic component for polyp development. Proc. Natl. Acad. Sci. USA 2007, 104, 19977–19982. [Google Scholar] [CrossRef] [Green Version]
- Shi, G.; Yang, Q.; Zhang, Y.; Jiang, Q.; Lin, Y.; Yang, S.; Wang, H.; Cheng, L.; Zhang, X.; Li, Y.; et al. Modulating the Tumor Microenvironment via Oncolytic Viruses and CSF-1R Inhibition Synergistically Enhances Anti-PD-1 Immunotherapy. Mol. Ther. 2019, 27, 244–260. [Google Scholar] [CrossRef] [Green Version]
- Bedognetti, D.; Ceccarelli, M.; Galluzzi, L.; Lu, R.; Palucka, K.; Samayoa, J.; Spranger, S.; Warren, S.; Wong, K.K.; Ziv, E.; et al. Toward a comprehensive view of cancer immune responsiveness: A synopsis from the SITC workshop. J. Immunother. Cancer 2019, 7, 131. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, H.W.; Naskar, M.; Seo, H.K.; Lee, H.W. Tumor-Associated Mast Cells in Urothelial Bladder Cancer: Optimizing Immuno-Oncology. Biomedicines 2021, 9, 1500. https://doi.org/10.3390/biomedicines9111500
Choi HW, Naskar M, Seo HK, Lee HW. Tumor-Associated Mast Cells in Urothelial Bladder Cancer: Optimizing Immuno-Oncology. Biomedicines. 2021; 9(11):1500. https://doi.org/10.3390/biomedicines9111500
Chicago/Turabian StyleChoi, Hae Woong, Manisha Naskar, Ho Kyung Seo, and Hye Won Lee. 2021. "Tumor-Associated Mast Cells in Urothelial Bladder Cancer: Optimizing Immuno-Oncology" Biomedicines 9, no. 11: 1500. https://doi.org/10.3390/biomedicines9111500