
Polyglutamine Ataxias: Our Current Molecular Understanding and What the Future Holds for Antisense Therapies


Abstract
:1. Introduction
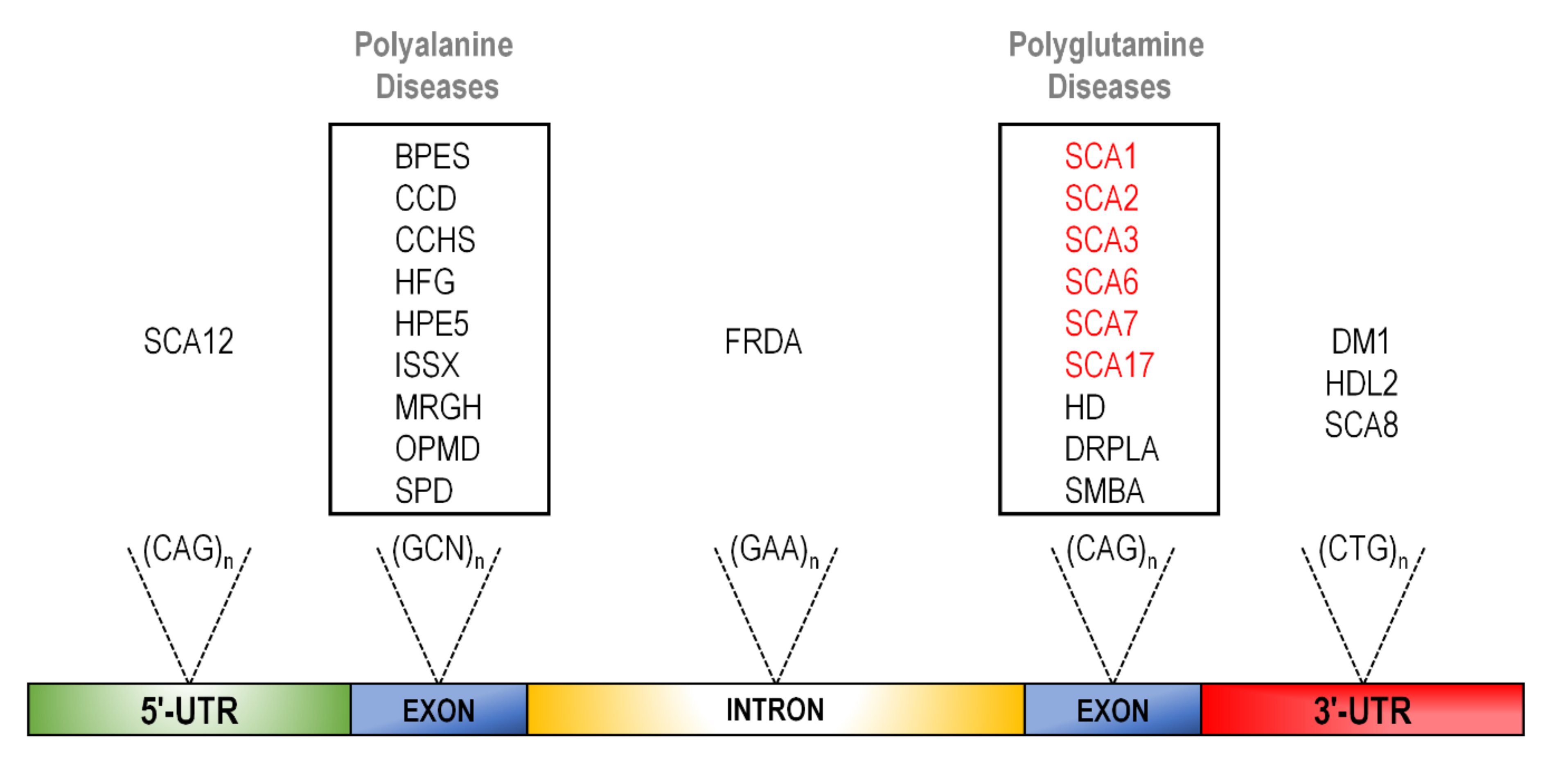
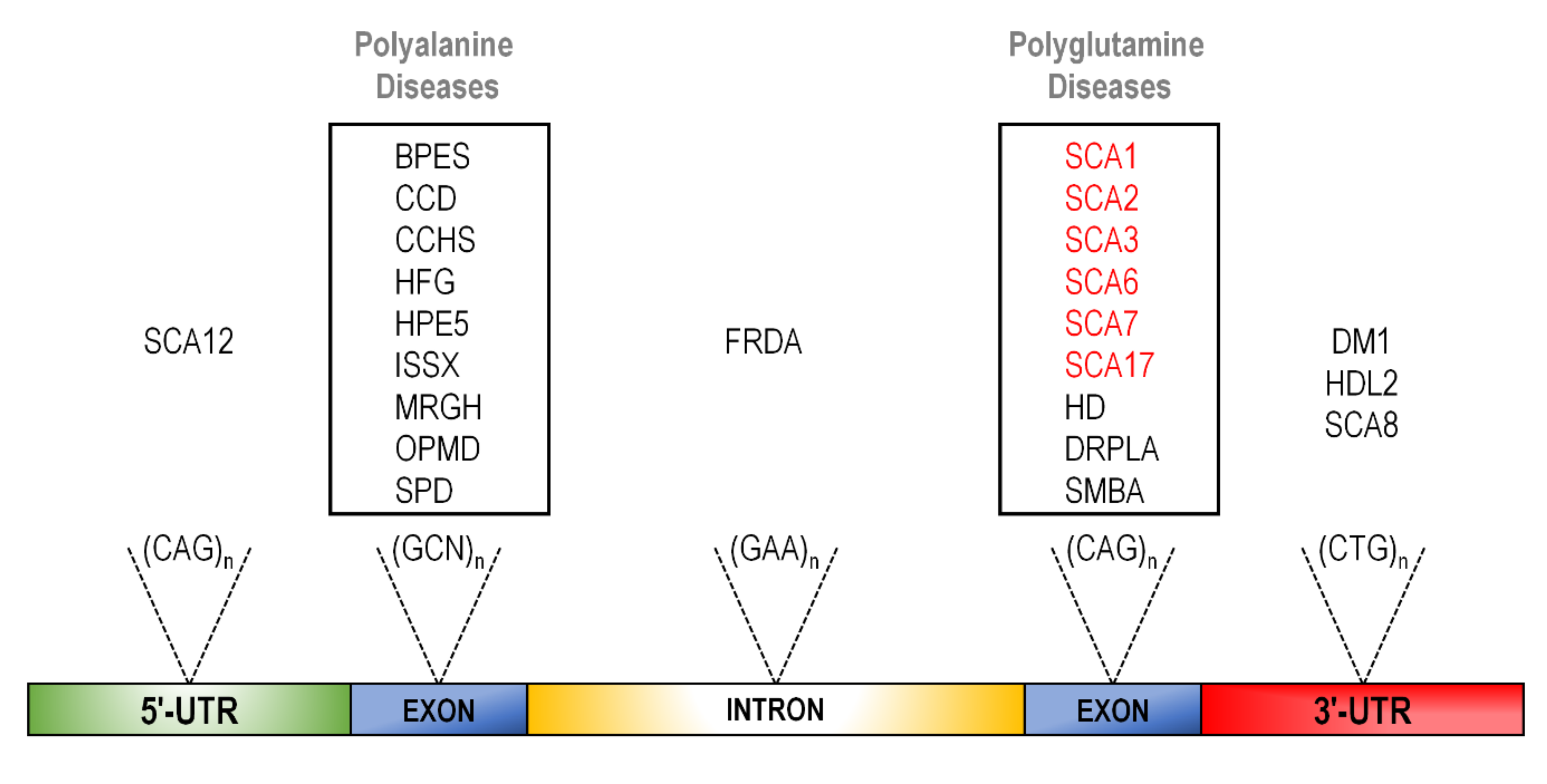
2. The CAG Expansion
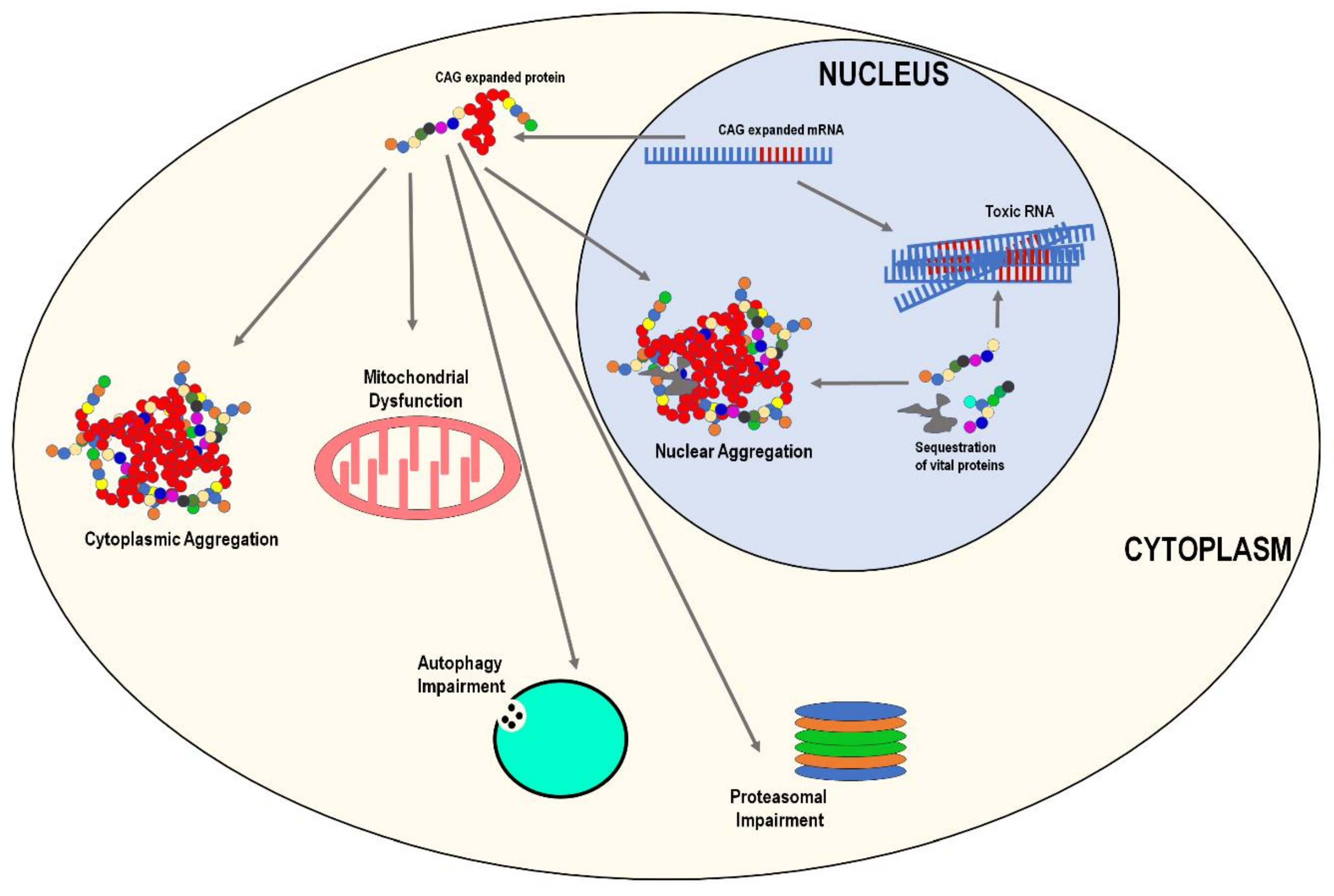
3. Common Pathological Features
3.1. Protein Aggregation
3.2. Mitochondrial Dysfunction and Oxidative Stress
3.3. Proteasomal and Autophagy Impairment
3.4. RNA Toxicity
3.5. Neuroinflammation
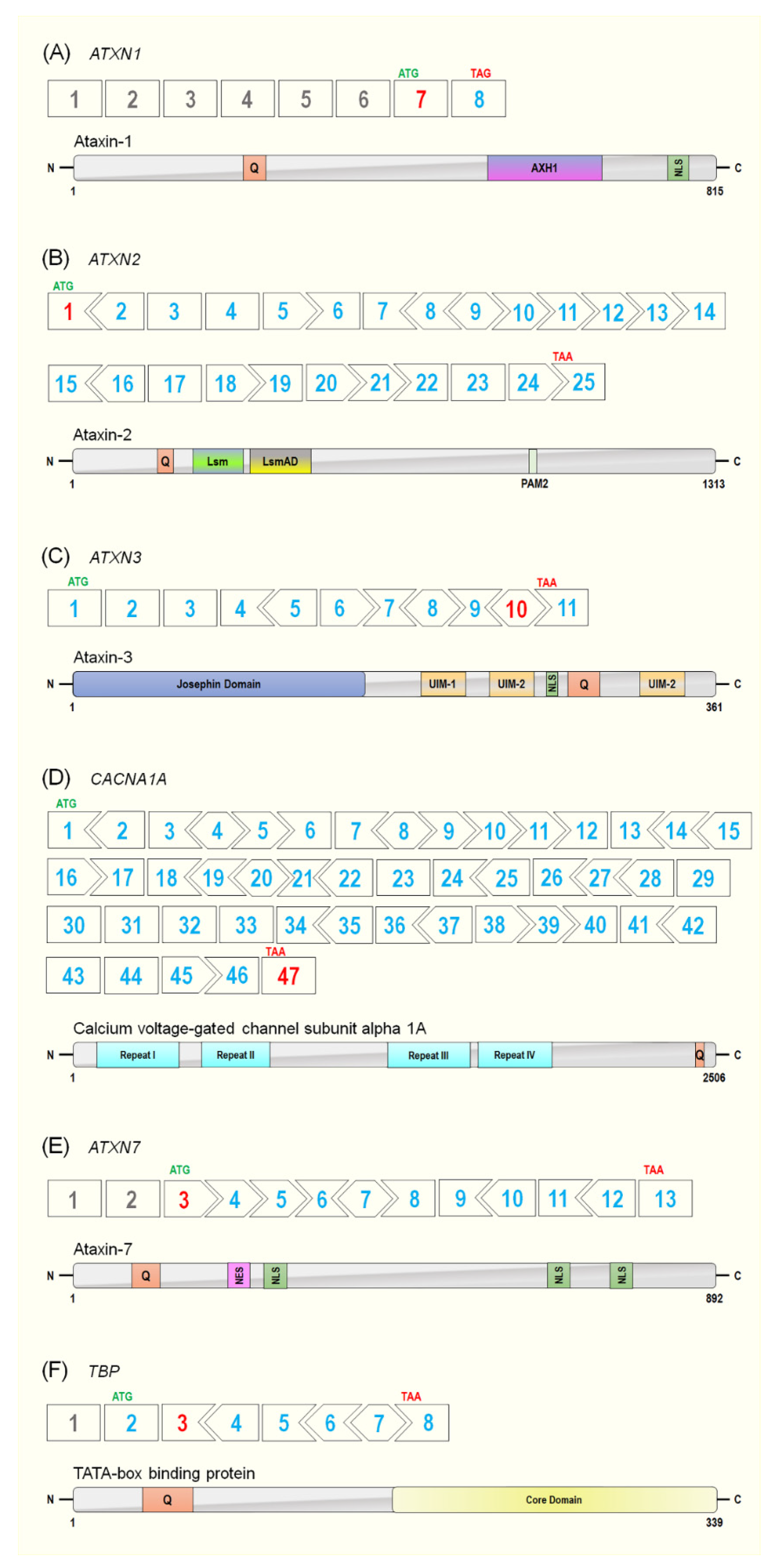
4. Spinocerebellar Ataxia Type 1 (SCA1)
5. Spinocerebellar Ataxia Type 2 (SCA2)
6. Spinocerebellar Ataxia Type 3 (SCA3)
7. Spinocerebellar Ataxia Type 6 (SCA6)
8. Spinocerebellar Ataxia Type 7 (SCA7)
9. Spinocerebellar Ataxia Type 17 (SCA17)
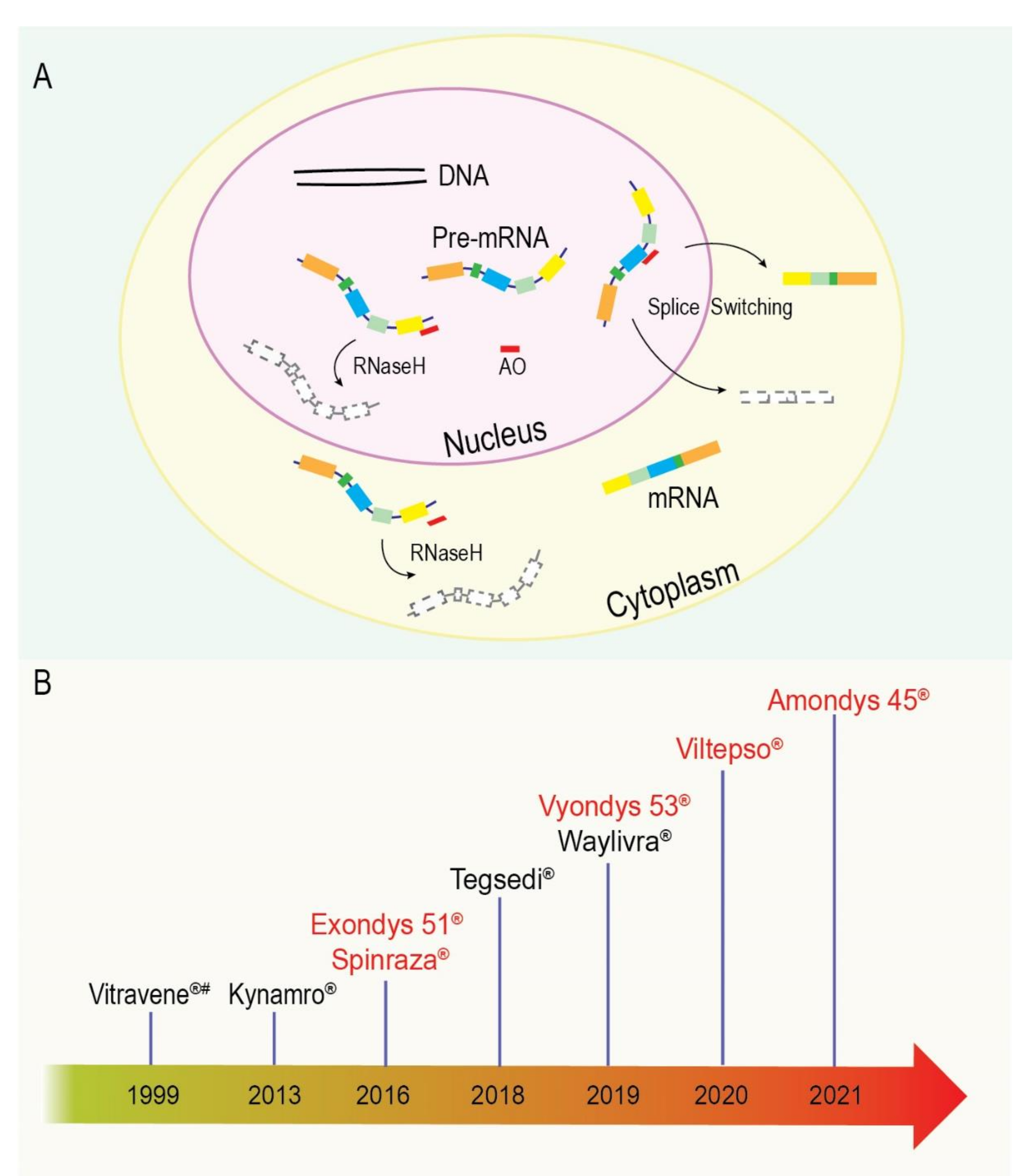
10. Brief Background into Antisense Therapeutics
10.1. RNaseH Degradation of a Targeted RNA Transcript
10.2. Splice Switching
11. Antisense Therapeutics for SCAs
11.1. SCA1
11.2. SCA2
11.3. SCA3
11.4. SCA7
12. A History and Outcome of Recent Huntington’s Disease Clinical Trial
13. Conclusions and Future Potential
Author Contributions
Funding
Conflicts of Interest
References
- Buijsen, R.A.; Toonen, L.J.; Gardiner, S.L.; van Roon-Mom, W.M. Genetics, Mechanisms, and Therapeutic Progress in Polyglutamine Spinocerebellar Ataxias. Neurotherapeutics 2019, 16, 263–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntosh, C.; Aung-Htut, M.; Fletcher, S.; Wilton, S. Polyglutamine ataxias: From Clinical and Molecular Features to Current Therapeutic Strategies. J. Genet. Syndr. Gene Ther. 2017, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Jeong, S.-Y.; Uchihara, T.; Anno, M.; Nagashima, K.; Nagashima, T.; Ikeda, S.-I.; Tsuji, S.; Kanazawa, I. SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum. Mol. Genet. 2001, 10, 1441–1448. [Google Scholar] [CrossRef] [Green Version]
- Paulson, H.L.; Shakkottai, V.G.; Clark, H.B.; Orr, H.T. Polyglutamine spinocerebellar ataxias—From genes to potential treatments. Nat. Rev. Neurosci. 2017, 18, 613. [Google Scholar] [CrossRef] [PubMed]
- Trottier, Y.; Lutz, Y.; Stevanin, G.; Imbert, G. Polyglutamine expansion as a pathological epitope in Huntington’s disease and four dominant cerebellar ataxias. Nature 1995, 378, 403. [Google Scholar] [CrossRef]
- Zhuchenko, O.; Bailey, J.; Bonnen, P.; Ashizawa, T.; Stockton, D.W.; Amos, C.; Dobyns, W.B.; Subramony, S.; Zoghbi, H.Y.; Lee, C.C. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the α1A-voltage-dependent calcium channel. Nat. Genet. 1997, 15, 62–69. [Google Scholar] [CrossRef]
- Durr, A. Autosomal dominant cerebellar ataxias: Polyglutamine expansions and beyond. Lancet Neurol. 2010, 9, 885–894. [Google Scholar] [CrossRef]
- Bushart, D.D.; Murphy, G.G.; Shakkottai, V.G. Precision medicine in spinocerebellar ataxias: Treatment based on common mechanisms of disease. Ann. Transl. Med. 2016, 4, 25. [Google Scholar]
- Du, Y.-C.; Ma, Y.; Shao, Y.-R.; Gan, S.-R.; Dong, Y.; Wu, Z.-Y. Factors Associated with Intergenerational Instability of ATXN3 CAG Repeat and Genetic Anticipation in Chinese Patients with Spinocerebellar Ataxia Type 3. Cerebellum 2020, 19, 902–906. [Google Scholar] [CrossRef] [PubMed]
- Duenas, A.M.; Goold, R.; Giunti, P. Molecular pathogenesis of spinocerebellar ataxias. Brain 2006, 129, 1357–1370. [Google Scholar] [CrossRef] [Green Version]
- Soong, B.-W.; Lu, Y.-C.; Choo, K.-B.; Lee, H.-Y. Frequency analysis of autosomal dominant cerebellar ataxias in Taiwanese patients and clinical and molecular characterization of spinocerebellar ataxia type 6. Arch. Neurol. 2001, 58, 1105–1109. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.-C.; Ho, L.-I.; Chi, C.-S.; Chen, S.-J.; Peng, G.-S.; Chan, T.-M.; Lin, S.-Z.; Harn, H.-J. Polyglutamine (PolyQ) diseases: Genetics to treatments. Cell Transplant. 2014, 23, 441–458. [Google Scholar] [CrossRef] [Green Version]
- Ellerby, L.M. Repeat Expansion Disorders: Mechanisms and Therapeutics; Springer: Cham, Switzerland, 2020. [Google Scholar]
- Paulson, H. Repeat expansion diseases. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 147, pp. 105–123. [Google Scholar]
- Lee, D.-Y.; McMurray, C.T. Trinucleotide expansion in disease: Why is there a length threshold? Curr. Opin. Genet. Dev. 2014, 26, 131–140. [Google Scholar] [CrossRef] [Green Version]
- La Spada, A.R.; Taylor, J.P. Repeat expansion disease: Progress and puzzles in disease pathogenesis. Nat. Rev. Genet. 2010, 11, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Gatchel, J.R.; Zoghbi, H.Y. Diseases of unstable repeat expansion: Mechanisms and common principles. Nat. Rev. Genet. 2005, 6, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Usdin, K. The biological effects of simple tandem repeats: Lessons from the repeat expansion diseases. Genome Res. 2008, 18, 1011–1019. [Google Scholar] [CrossRef] [Green Version]
- Orr, H.T.; Zoghbi, H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007, 30, 575–621. [Google Scholar] [CrossRef]
- Mirkin, S.M. Expandable DNA repeats and human disease. Nature 2007, 447, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Petruska, J.; Hartenstine, M.J.; Goodman, M.F. Analysis of strand slippage in DNA polymerase expansions of CAG/CTG triplet repeats associated with neurodegenerative disease. J. Biol. Chem. 1998, 273, 5204–5210. [Google Scholar] [CrossRef] [Green Version]
- Levinson, G.; Gutman, G.A. Slipped-strand mispairing: A major mechanism for DNA sequence evolution. Mol. Biol. Evol. 1987, 4, 203–221. [Google Scholar]
- Takeuchi, T.; Nagai, Y. Protein misfolding and aggregation as a therapeutic target for polyglutamine diseases. Brain Sci. 2017, 7, 128. [Google Scholar] [CrossRef] [Green Version]
- Raj, K.; Chanu, S.I.; Sarkar, S. Protein misfolding and aggregation in neurodegenerative disorders: Focus on chaperone-mediated protein folding machinery. Int. J. Neurol. Res. 2015, 1, 72–78. [Google Scholar] [CrossRef]
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoghbi, H.Y.; Orr, H.T. Polyglutamine diseases: Protein cleavage and aggregation. Curr. Opin. Neurobiol. 1999, 9, 566–570. [Google Scholar] [CrossRef]
- Ross, O.A.; Rutherford, N.J.; Baker, M.; Soto-Ortolaza, A.I.; Carrasquillo, M.M.; DeJesus-Hernandez, M.; Adamson, J.; Li, M.; Volkening, K.; Finger, E. Ataxin-2 repeat-length variation and neurodegeneration. Hum. Mol. Genet. 2011, 20, 3207–3212. [Google Scholar] [CrossRef]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- Kuiper, E.; de Mattos, E.P.; Jardim, L.B.; Kampinga, H.H.; Bergink, S. Chaperones in polyglutamine aggregation: Beyond the Q-stretch. Front. Neurosci. 2017, 11, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, S.R.; Lieberman, A.P. The Ubiquitination, Disaggregation and Proteasomal Degradation Machineries in Polyglutamine Disease. Front. Mol. Neurosci. 2017, 10, 78. [Google Scholar] [CrossRef]
- Seidel, K.; Siswanto, S.; Fredrich, M.; Bouzrou, M.; Brunt, E.; Leeuwen, F.; Kampinga, H.; Korf, H.W.; Rüb, U.; Dunnen, W. Polyglutamine aggregation in Huntington’s disease and spinocerebellar ataxia type 3: Similar mechanisms in aggregate formation. Neuropathol. Appl. Neurobiol. 2015, 42, 153–166. [Google Scholar] [CrossRef]
- Burright, E.N.; Clark, H.B.; Servadio, A.; Matilla, T.; Feddersen, R.M.; Yunis, W.S.; Duvick, L.A.; Zoghbi, H.Y.; Orr, H.T. SCA1 transgenic mice: A model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell 1995, 82, 937–948. [Google Scholar] [CrossRef] [Green Version]
- Meierhofer, D.; Halbach, M.; Şen, N.E.; Gispert, S.; Auburger, G. Ataxin-2 (Atxn2)-knock-out mice show branched chain amino acids and fatty acids pathway alterations. Mol. Cell. Proteom. 2016, 15, 1728–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bichelmeier, U.; Schmidt, T.; Hübener, J.; Boy, J.; Rüttiger, L.; Häbig, K.; Poths, S.; Bonin, M.; Knipper, M.; Schmidt, W.J. Nuclear localization of ataxin-3 is required for the manifestation of symptoms in SCA3: In vivo evidence. J. Neurosci. 2007, 27, 7418–7428. [Google Scholar] [CrossRef] [Green Version]
- Watase, K. Spinocerebellar ataxia type 6: Lessons from faithful knock-in mouse models. Neurol. Clin. Neurosci. 2015, 3, 14–17. [Google Scholar] [CrossRef] [Green Version]
- Watase, K.; Barrett, C.F.; Miyazaki, T.; Ishiguro, T.; Ishikawa, K.; Hu, Y.; Unno, T.; Sun, Y.; Kasai, S.; Watanabe, M. Spinocerebellar ataxia type 6 knockin mice develop a progressive neuronal dysfunction with age-dependent accumulation of mutant CaV2. 1 channels. Proc. Natl. Acad. Sci. USA 2008, 105, 11987–11992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breuer, P.; Haacke, A.; Evert, B.O.; Wüllner, U. Nuclear aggregation of polyglutamine-expanded Ataxin-3 fragments escape the cytoplasmic quality control. J. Biol. Chem. 2010, 285, 6532–6537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansorge, O.; Giunti, P.; Michalik, A.; Van Broeckhoven, C.; Harding, B.; Wood, N.; Scaravilli, F. Ataxin-7 aggregation and ubiquitination in infantile SCA7 with 180 CAG repeats. Ann. Neurol. 2004, 56, 448–452. [Google Scholar] [CrossRef]
- Cornelius, N.; Wardman, J.H.; Hargreaves, I.P.; Neergheen, V.; Bie, A.S.; Tümer, Z.; Nielsen, J.E.; Nielsen, T.T. Evidence of oxidative stress and mitochondrial dysfunction in spinocerebellar ataxia type 2 (SCA2) patient fibroblasts: Effect of coenzyme Q10 supplementation on these parameters. Mitochondrion 2017, 34, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.-Y.; Jhang, Y.-L.; Cheng, P.-H.; Chang, Y.-F.; Mao, S.-H.; Yang, H.-I.; Lin, C.-W.; Chen, C.-M.; Yang, S.-H. The truncated C-terminal fragment of mutant ATXN3 disrupts mitochondria dynamics in spinocerebellar ataxia type 3 models. Front. Mol. Neurosci. 2017, 10, 196. [Google Scholar] [CrossRef] [Green Version]
- Laço, M.N.; Oliveira, C.R.; Paulson, H.L.; Rego, A.C. Compromised mitochondrial complex II in models of Machado–Joseph disease. BBA Mol. Basis Dis. 2012, 1822, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Furuta, N.; Tsukagoshi, S.; Hirayanagi, K.; Ikeda, Y. Suppression of the yeast elongation factor Spt4 ortholog reduces expanded SCA36 GGCCUG repeat aggregation and cytotoxicity. Brain Res. 2019, 1711, 29–40. [Google Scholar] [CrossRef]
- Ito, H.; Fujita, K.; Tagawa, K.; Chen, X.; Homma, H.; Sasabe, T.; Shimizu, J.; Shimizu, S.; Tamura, T.; Muramatsu, S.I. HMGB 1 facilitates repair of mitochondrial DNA damage and extends the lifespan of mutant ataxin-1 knock-in mice. EMBO Mol. Med. 2015, 7, 78–101. [Google Scholar] [CrossRef] [PubMed]
- Kazachkova, N.; Raposo, M.; Montiel, R.; Cymbron, T.; Bettencourt, C.; Silva-Fernandes, A.; Silva, S.; Maciel, P.; Lima, M. Patterns of mitochondrial DNA damage in blood and brain tissues of a transgenic mouse model of Machado-Joseph disease. Neurodegener. Dis. 2013, 11, 206–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ripolone, M.; Lucchini, V.; Ronchi, D.; Fagiolari, G.; Bordoni, A.; Fortunato, F.; Mondello, S.; Bonato, S.; Meregalli, M.; Torrente, Y. Purkinje cell COX deficiency and mtDNA depletion in an animal model of spinocerebellar ataxia type 1. J. Neurosci. Res. 2018, 96, 1576–1585. [Google Scholar] [CrossRef]
- Bettencourt, C.; Hensman-Moss, D.; Flower, M.; Wiethoff, S.; Brice, A.; Goizet, C.; Stevanin, G.; Koutsis, G.; Karadima, G.; Panas, M. DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases. Ann. Neurol. 2016, 79, 983–990. [Google Scholar] [CrossRef]
- Mantle, D.; Hargreaves, I.P. Ataxia and coenzyme Q10: An overview. Br. J. Neurosci. Nurs. 2018, 14, 108–114. [Google Scholar] [CrossRef]
- Fan, L.; Feng, Y.; Chen, G.-C.; Qin, L.-Q.; Fu, C.-L.; Chen, L.-H. Effects of coenzyme Q10 supplementation on inflammatory markers: A systematic review and meta-analysis of randomized controlled trials. Pharmacol. Res. 2017, 119, 128–136. [Google Scholar] [CrossRef]
- Bates, C.; Baxter, P.; Bonney, H.; Bremner, F.; Bunn, L.; Perez-Tome, M.C.; Chung, M.; Cipolotti, L.; de Silva, R.; Duberley, K. Management of the Ataxias towards Best Clinical Practice; Ataxia UK: London, UK, 2016. [Google Scholar]
- Torres-Ramos, Y.; Montoya-Estrada, A.; Cisneros, B.; Tercero-Pérez, K.; León-Reyes, G.; Leyva-García, N.; Hernández-Hernández, O.; Magaña, J.J. Oxidative stress in spinocerebellar ataxia type 7 is associated with disease severity. Cerebellum 2018, 17, 601–609. [Google Scholar] [CrossRef]
- de Assis, A.M.; Saute, J.A.M.; Longoni, A.; Haas, C.B.; Torrez, V.R.; Brochier, A.W.; Souza, G.N.; Furtado, G.V.; Gheno, T.C.; Russo, A. Peripheral oxidative stress biomarkers in spinocerebellar ataxia type 3/Machado–Joseph disease. Front. Neurol. 2017, 8, 485. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Sanchez, M.; Thomson, F.; Zavodszky, E.; Rubinsztein, D.C. Autophagy and polyglutamine diseases. Prog. Neurobiol. 2012, 97, 67–82. [Google Scholar] [CrossRef] [Green Version]
- Sittler, A.; Muriel, M.P.; Marinello, M.; Brice, A.; den Dunnen, W.; Alves, S. Deregulation of autophagy in postmortem brains of Machado-Joseph disease patients. Neuropathology 2018, 38, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Nascimento-Ferreira, I.; Santos-Ferreira, T.; Sousa-Ferreira, L.; Auregan, G.; Onofre, I.; Alves, S.; Dufour, N.; Gould, V.F.C.; Koeppen, A.; Déglon, N. Overexpression of the autophagic beclin-1 protein clears mutant ataxin-3 and alleviates Machado–Joseph disease. Brain 2011, 134, 1400–1415. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Bento, C.F.; Ricketts, T.; Vicinanza, M.; Siddiqi, F.; Pavel, M.; Squitieri, F.; Hardenberg, M.C.; Imarisio, S.; Menzies, F.M. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 2017, 545, 108. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, A.; Bento, C.F.; Ricketts, T.; Vicinanza, M.; Siddiqi, F.; Pavel, M.; Squitieri, F.; Hardenberg, M.C.; Imarisio, S.; Menzies, F.M. Polyglutamine tracts regulate autophagy. Autophagy 2017, 13, 1613–1614. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Lee, Y.-I.; Lee, Y.-S.; Lee, S.B. The mechanisms of nuclear proteotoxicity in polyglutamine spinocerebellar ataxias. Front. Neurosci. 2020, 14, 489. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.; Chang, K.-H.; Chen, Y.-J.; Chen, Y.-C.; Lee-Chen, G.-J.; Chen, C.-M. Pueraria lobata and daidzein reduce cytotoxicity by enhancing ubiquitin-proteasome system function in SCA3-iPSC-derived neurons. Oxid. Med. Cell. Longev. 2019, 2019, 8130481. [Google Scholar] [CrossRef]
- Chen, C.-M.; Chen, W.-L.; Hung, C.-T.; Lin, T.-H.; Lee, M.-C.; Chen, I.-C.; Lin, C.-H.; Chao, C.-Y.; Wu, Y.-R.; Chang, K.-H. Shaoyao Gancao Tang (SG-Tang), a formulated Chinese medicine, reduces aggregation and exerts neuroprotection in spinocerebellar ataxia type 17 (SCA17) cell and mouse models. Aging 2019, 11, 986. [Google Scholar] [CrossRef]
- Mykowska, A.; Sobczak, K.; Wojciechowska, M.; Kozlowski, P.; Krzyzosiak, W.J. CAG repeats mimic CUG repeats in the misregulation of alternative splicing. Nucleic Acids Res. 2011, 39, 8938–8951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scoles, D.R.; Ho, M.H.; Dansithong, W.; Pflieger, L.T.; Petersen, L.W.; Thai, K.K.; Pulst, S.M. Repeat associated non-AUG translation (RAN translation) dependent on sequence downstream of the ATXN2 CAG repeat. PLoS ONE 2015, 10, e0128769. [Google Scholar] [CrossRef] [Green Version]
- Martí, E. RNA toxicity induced by expanded CAG repeats in H untington’s disease. Brain Pathol. 2016, 26, 779–786. [Google Scholar] [CrossRef]
- De Mezer, M.; Wojciechowska, M.; Napierala, M.; Sobczak, K.; Krzyzosiak, W.J. Mutant CAG repeats of Huntingtin transcript fold into hairpins, form nuclear foci and are targets for RNA interference. Nucleic Acids Res. 2011, 39, 3852–3863. [Google Scholar] [CrossRef] [Green Version]
- Jasinska, A.; Michlewski, G.; De Mezer, M.; Sobczak, K.; Kozlowski, P.; Napierala, M.; Krzyzosiak, W.J. Structures of trinucleotide repeats in human transcripts and their functional implications. Nucleic Acids Res. 2003, 31, 5463–5468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.B.; Yu, Z.; Teng, X.; Bonini, N.M. RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature 2008, 453, 1107–1111. [Google Scholar] [CrossRef] [Green Version]
- Bhambri, A.; Pinto, A.; Pillai, B. Interferon mediated neuroinflammation in polyglutamine disease is not caused by RNA toxicity. Cell Death Dis. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Chiu, Y.-J.; Lin, S.-A.; Chen, W.-L.; Lin, T.-H.; Lin, C.-H.; Yao, C.-F.; Lin, W.; Wu, Y.-R.; Chang, K.-H.; Lee-Chen, G.-J. Pathomechanism characterization and potential therapeutics identification for SCA3 targeting neuroinflammation. Aging 2020, 12, 23619. [Google Scholar] [CrossRef] [PubMed]
- Rekatsina, M.; Paladini, A.; Piroli, A.; Zis, P.; Pergolizzi, J.V.; Varrassi, G. Pathophysiology and therapeutic perspectives of oxidative stress and neurodegenerative diseases: A narrative review. Adv. Ther. 2020, 37, 113–139. [Google Scholar] [CrossRef] [Green Version]
- Evert, B.O.; Vogt, I.R.; Kindermann, C.; Ozimek, L.; De Vos, R.A.; Brunt, E.R.; Schmitt, I.; Klockgether, T.; Wüllner, U. Inflammatory genes are upregulated in expanded ataxin-3-expressing cell lines and spinocerebellar ataxia type 3 brains. J. Neurosci. 2001, 21, 5389–5396. [Google Scholar] [CrossRef] [Green Version]
- Tong, X.; Gui, H.; Jin, F.; Heck, B.W.; Lin, P.; Ma, J.; Fondell, J.D.; Tsai, C.C. Ataxin-1 and Brother of ataxin-1 are components of the Notch signalling pathway. EMBO Rep. 2011, 12, 428–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, C.-C.; Kao, H.-Y.; Mitzutani, A.; Banayo, E.; Rajan, H.; McKeown, M.; Evans, R.M. Ataxin 1, a SCA1 neurodegenerative disorder protein, is functionally linked to the silencing mediator of retinoid and thyroid hormone receptors. Proc. Natl. Acad. Sci. USA 2004, 101, 4047–4052. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, A.; Wang, L.; Rajan, H.; Vig, P.J.; Alaynick, W.A.; Thaler, J.P.; Tsai, C.C. Boat, an AXH domain protein, suppresses the cytotoxicity of mutant ataxin-1. EMBO J. 2005, 24, 3339–3351. [Google Scholar] [CrossRef] [Green Version]
- Bowman, A.B.; Lam, Y.C.; Jafar-Nejad, P.; Chen, H.-K.; Richman, R.; Samaco, R.C.; Fryer, J.D.; Kahle, J.J.; Orr, H.T.; Zoghbi, H.Y. Duplication of Atxn1l suppresses SCA1 neuropathology by decreasing incorporation of polyglutamine-expanded ataxin-1 into native complexes. Nat. Genet. 2007, 39, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.S.; Geoghegan, J.C.; Boudreau, R.L.; Lennox, K.A.; Davidson, B.L. RNAi or overexpression: Alternative therapies for Spinocerebellar Ataxia Type 1. Neurobiol. Dis. 2013, 56, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Seidel, K.; Siswanto, S.; Brunt, E.R.; Den Dunnen, W.; Korf, H.-W.; Rüb, U. Brain pathology of spinocerebellar ataxias. Acta Neuropathol. 2012, 124, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Genís, D.; Matilla, T.; Volpini, V.; Rosell, J.; Dávalos, A.; Ferrer, I.; Molins, A.; Estivill, X. Clinical, neuropathologic, and genetic studies of a large spinocerebellar ataxia type 1 (SCA1) kindred:(CAG) n expansion and early premonitory signs and symptoms. Neurology 1995, 45, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Stoyas, C.A.; La Spada, A.R. The CAG–polyglutamine repeat diseases: A clinical, molecular, genetic, and pathophysiologic nosology. Handb. Clin. Neurol. 2018, 147, 143–170. [Google Scholar]
- Pérez, L.V.; Cruz, G.S.; Falcón, N.S.; Mederos, L.E.A.; Batallan, K.E.; Labrada, R.R.; Herrera, M.P.; Mesa, J.M.L.; Díaz, J.C.R.; Rodríguez, R.A. Molecular epidemiology of spinocerebellar ataxias in Cuba: Insights into SCA2 founder effect in Holguin. Neurosci. Lett. 2009, 454, 157–160. [Google Scholar] [CrossRef]
- Velázquez-Pérez, L.; Medrano-Montero, J.; Rodríguez-Labrada, R.; Canales-Ochoa, N.; Alí, J.C.; Rodes, F.J.C.; Graña, T.R.; Oliver, M.O.H.; Rodríguez, R.A.; Barrios, Y.D. Hereditary ataxias in Cuba: A Nationwide epidemiological and clinical study in 1001 patients. Cerebellum 2020, 19, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Labrada, R.; Martins, A.C.; Magaña, J.J.; Vazquez-Mojena, Y.; Medrano-Montero, J.; Fernandez-Ruíz, J.; Cisneros, B.; Teive, H.; McFarland, K.N.; Saraiva-Pereira, M.L. Founder Effects of Spinocerebellar Ataxias in the American Continents and the Caribbean. Cerebellum 2020, 19, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Carmo-Silva, S.; Nobrega, C.; de Almeida, L.P.; Cavadas, C. Unraveling the Role of Ataxin-2 in Metabolism. Trends Endocrinol. Metab. 2017, 28, 309–318. [Google Scholar] [CrossRef]
- Pfeffer, M.; Gispert, S.; Auburger, G.; Wicht, H.; Korf, H.-W. Impact of Ataxin-2 knock out on circadian locomotor behavior and PER immunoreaction in the SCN of mice. Chronobiol. Int. 2017, 34, 129–137. [Google Scholar] [CrossRef]
- Ostrowski, L.A.; Hall, A.C.; Mekhail, K. Ataxin-2: From RNA control to human health and disease. Genes 2017, 8, 157. [Google Scholar] [CrossRef]
- Lee, J.; Yoo, E.; Lee, H.; Park, K.; Hur, J.-H.; Lim, C. LSM12 and ME31B/DDX6 define distinct modes of posttranscriptional regulation by ATAXIN-2 protein complex in Drosophila circadian pacemaker neurons. Mol. Cell 2017, 66, 129–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inagaki, H.; Hosoda, N.; Tsuiji, H.; Hoshino, S.-I. Direct evidence that Ataxin-2 is a translational activator mediating cytoplasmic polyadenylation. J. Biol. Chem. 2020, 295, 15810–15825. [Google Scholar] [CrossRef] [PubMed]
- Nezhad, H.G.; Franklin, J.P.; Alix, J.J.; Moll, T.; Pattrick, M.; Cooper-Knock, J.; Shanmugarajah, P.; Beauchamp, N.J.; Hadjivissiliou, M.; Paling, D. Simultaneous ALS and SCA2 associated with an intermediate-length ATXN2 CAG-repeat expansion. Amyotroph. Lateral Scler. Front. Degener. 2020, 1–3. [Google Scholar] [CrossRef]
- Elden, A.C.; Kim, H.-J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Becker, L.A.; Huang, B.; Bieri, G.; Ma, R.; Knowles, D.A.; Jafar-Nejad, P.; Messing, J.; Kim, H.J.; Soriano, A.; Auburger, G. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 2017, 544, 367–371. [Google Scholar] [CrossRef] [Green Version]
- Yomono, H.S.; Kurisaki, H.; Hebisawa, A.; Sakiyama, Y.; Saito, Y.; Murayama, S. Autopsy case of SCA2 with Parkinsonian phenotype. Rinsho Shinkeigaku Clin. Neurol. 2010, 50, 156–162. [Google Scholar] [CrossRef]
- Socal, M.; Emmel, V.; Rieder, C.; Hilbig, A.; Saraiva-Pereira, M.; Jardim, L. Intrafamilial variability of Parkinson phenotype in SCAs: Novel cases due to SCA2 and SCA3 expansions. Parkinsonism Relat. Disord. 2009, 15, 374–378. [Google Scholar] [CrossRef]
- Furtado, S.; Payami, H.; Lockhart, P.J.; Hanson, M.; Nutt, J.G.; Singleton, A.A.; Singleton, A.; Bower, J.; Utti, R.J.; Bird, T.D. Profile of families with parkinsonism-predominant spinocerebellar ataxia type 2 (SCA2). Mov. Disord. 2004, 19, 622–629. [Google Scholar] [CrossRef]
- Ying, S.; Choi, S.; Lee, M.; Perlman, S.; Baloh, R.; Toga, A.; Zee, D. Relative atrophy of the flocculus and ocular motor dysfunction in SCA2 and SCA6. Ann. N. Y. Acad. Sci. 2005, 1039, 430–435. [Google Scholar] [CrossRef]
- Sasaki, H.; Fukazawa, T.; Wakisaka, A.; Hamada, K.; Hamada, T.; Koyama, T.; Tsuji, S.; Tashiro, K. Central phenotype and related varieties of spinocerebellar ataxia 2 (SCA2): A clinical and genetic study with a pedigree in the Japanese. J. Neurol. Sci. 1996, 144, 176–181. [Google Scholar] [CrossRef]
- Tang, B.; Liu, C.; Shen, L.; Dai, H.; Pan, Q.; Jing, L.; Ouyang, S.; Xia, J. Frequency of SCA1, SCA2, SCA3/MJD, SCA6, SCA7, and DRPLA CAG trinucleotide repeat expansion in patients with hereditary spinocerebellar ataxia from Chinese kindreds. Arch. Neurol. 2000, 57, 540–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudarsky, L.; Coutinho, P. Machado-Joseph disease. Clin. Neurosci. 1995, 3, 17–22. [Google Scholar]
- Nakano, K.K.; Dawson, D.M.; Spence, A. Machado disease A hereditary ataxia in Portuguese emigrants to Massachusetts. Neurology 1972, 22, 49. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.M.; Toonen, L.J.; van Roon-Mom, W.M. Ataxin-3 protein and RNA toxicity in spinocerebellar ataxia type 3: Current insights and emerging therapeutic strategies. Mol. Neurobiol. 2014, 49, 1513–1531. [Google Scholar] [CrossRef] [Green Version]
- Harris, G.M.; Dodelzon, K.; Gong, L.; Gonzalez-Alegre, P.; Paulson, H.L. Splice isoforms of the polyglutamine disease protein ataxin-3 exhibit similar enzymatic yet different aggregation properties. PLoS ONE 2010, 5, e13695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettencourt, C.; Santos, C.; Montiel, R.; do Carmo Costa, M.; Cruz-Morales, P.; Santos, L.R.; Simões, N.; Kay, T.; Vasconcelos, J.; Maciel, P. Increased transcript diversity: Novel splicing variants of Machado–Joseph Disease gene (ATXN3). Neurogenetics 2010, 11, 193–202. [Google Scholar] [CrossRef]
- Bettencourt, C.; Santos, C.; Montiel, R.; Kay, T.; Vasconcelos, J.; Maciel, P.; Lima, M. The (CAG) n tract of Machado–Joseph Disease gene (ATXN3): A comparison between DNA and mRNA in patients and controls. Eur. J. Hum. Genet. 2010, 18, 621–623. [Google Scholar] [CrossRef] [Green Version]
- Ashizawa, T.; Öz, G.; Paulson, H.L. Spinocerebellar ataxias: Prospects and challenges for therapy development. Nat. Rev. Neurol. 2018, 14, 590–605. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Landwehrmeyer, G.B.; Schmitt, I.; Trottier, Y.; Auburger, G.; Laccone, F.; Klockgether, T.; Völpel, M.; Epplen, J.T.; Schöls, L. An isoform of ataxin—3 accumulates in the nucleus of neuronal cells in affected brain regions of SCA3 patients. Brain Pathol. 1998, 8, 669–679. [Google Scholar] [CrossRef]
- Faber, J.; Schaprian, T.; Berkan, K.; Reetz, K.; França, M.C., Jr.; de Rezende, T.J.R.; Hong, J.; Liao, W.; van de Warrenburg, B.; van Gaalen, J. Regional Brain and Spinal Cord Volume Loss in Spinocerebellar Ataxia Type 3. Mov. Disord. 2021. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.; Mayer, A.K.; Bakula, D.; Freude, J.; Weber, J.J.; Weiss, A.; Riess, O.; Schmidt, T. Vulnerability of frontal brain neurons for the toxicity of expanded ataxin-3. Hum. Mol. Genet. 2019, 28, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Mendonça, N.; França, M.C., Jr.; Goncalves, A.F.; Januario, C. Clinical features of Machado-Joseph disease. Adv. Exp. Med. Biol. 2018, 1049, 255–273. [Google Scholar]
- McLoughlin, H.S.; Moore, L.R.; Paulson, H.L. Pathogenesis of SCA3 and implications for other polyglutamine diseases. Neurobiol. Dis. 2020, 134, 104635. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Kojima, H.; Yabe, I.; Tashiro, K.; Hamada, T.; Sawa, H.; Hiraga, H.; Nagashima, K. Neuropathological and molecular studies of spinocerebellar ataxia type 6 (SCA6). Acta Neuropathol. 1998, 95, 199–204. [Google Scholar] [CrossRef]
- Matsuyama, Z.; Kawakami, H.; Maruyama, H.; Maruyama, H.; Izumi, Y.; Komure, O.; Udaka, F.; Kameyama, M.; Nishio, T.; Kuroda, Y. Molecular features of the CAG repeats of spinocerebellar ataxia 6 (SCA6). Hum. Mol. Genet. 1997, 6, 1283–1287. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Gomez, C.M. Spinocerebellar ataxia type 6: Molecular mechanisms and calcium channel genetics. In Polyglutamine Disorders; Springer: Cham, Switzerland, 2018; pp. 147–173. [Google Scholar]
- Pradotto, L.; Mencarelli, M.; Bigoni, M.; Milesi, A.; Di Blasio, A.; Mauro, A. Episodic ataxia and SCA6 within the same family due to the D302N CACNA1A gene mutation. J. Neurol. Sci. 2016, 371, 81–84. [Google Scholar] [CrossRef]
- Bürk, K.; Kaiser, F.J.; Tennstedt, S.; Schöls, L.; Kreuz, F.R.; Wieland, T.; Strom, T.M.; Büttner, T.; Hollstein, R.; Braunholz, D. A novel missense mutation in CACNA1A evaluated by in silico protein modeling is associated with non-episodic spinocerebellar ataxia with slow progression. Eur. J. Med. Genet. 2014, 57, 207–211. [Google Scholar] [CrossRef]
- Barros, J.; Damásio, J.; Tuna, A.; Alves, I.; Silveira, I.; Pereira-Monteiro, J.; Sequeiros, J.; Alonso, I.; Sousa, A.; Coutinho, P. Cerebellar Ataxia, Hemiplegic Migraine, and Related Phenotypes Due to a CACNA1A Missense Mutation: 12-year follow-up of a large Portuguese family. JAMA Neurol. 2013, 70, 235–240. [Google Scholar] [CrossRef] [Green Version]
- Indelicato, E.; Boesch, S. From Genotype to Phenotype: Expanding the Clinical Spectrum of CACNA1A Variants in the Era of Next Generation Sequencing. Front. Neurol. 2021, 12, 263. [Google Scholar] [CrossRef]
- Wiethoff, S.; O’Connor, E.; Haridy, N.A.; Nethisinghe, S.; Wood, N.; Giunti, P.; Bettencourt, C.; Houlden, H. Sequencing analysis of the SCA6 CAG expansion excludes an influence of repeat interruptions on disease onset. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1226–1227. [Google Scholar] [CrossRef] [Green Version]
- Giunti, P.; Mantuano, E.; Frontali, M.; Veneziano, L. Molecular mechanism of Spinocerebellar Ataxia type 6: Glutamine repeat disorder, channelopathy and transcriptional dysregulation. The multifaceted aspects of a single mutation. Front. Cell. Neurosci. 2015, 9, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipscombe, D.; Andrade, A.; Allen, S.E. Alternative splicing: Functional diversity among voltage-gated calcium channels and behavioral consequences. BBA Biomembr. 2013, 1828, 1522–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrobon, D. Calcium channels and migraine. BBA Biomembr. 2013, 1828, 1655–1665. [Google Scholar] [CrossRef]
- Rajakulendran, S.; Kaski, D.; Hanna, M.G. Neuronal P/Q-type calcium channel dysfunction in inherited disorders of the CNS. Nat. Rev. Neurol. 2012, 8, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Pietrobon, D. Calcium channels and channelopathies of the central nervous system. Mol. Neurobiol. 2002, 25, 31–50. [Google Scholar] [CrossRef]
- Toru, S.; Murakoshi, T.; Ishikawa, K.; Saegusa, H.; Fujigasaki, H.; Uchihara, T.; Nagayama, S.; Osanai, M.; Mizusawa, H.; Tanabe, T. Spinocerebellar ataxia type 6 mutation alters P-type calcium channel function. J. Biol. Chem. 2000, 275, 10893–10898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordasiewicz, H.B.; Thompson, R.M.; Clark, H.B.; Gomez, C.M. C-termini of P/Q-type Ca2+ channel α1A subunits translocate to nuclei and promote polyglutamine-mediated toxicity. Hum. Mol. Genet. 2006, 15, 1587–1599. [Google Scholar] [CrossRef] [Green Version]
- Matsuyama, Z.; Wakamori, M.; Mori, Y.; Kawakami, H.; Nakamura, S.; Imoto, K. Direct alteration of the P/Q-type Ca2+ channel property by polyglutamine expansion in spinocerebellar ataxia 6. J. Neurosci 1999, 19, RC14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froment, J.; Bonnet, P.; Colrat, A. Heredo-degenerations retinienne et spino-cerebelleuse: Variantes ophtalmoscopiques et neurologiques presentees par trois generations successives. J. Med. Lyon 1937, 22, 153–163. [Google Scholar]
- David, G.; Giunti, P.; Abbas, N.; Coullin, P.; Stevanin, G.; Horta, W.; Gemmill, R.; Weissenbach, J.; Wood, N.; Cunha, S. The gene for autosomal dominant cerebellar ataxia type II is located in a 5-cM region in 3p12-p13: Genetic and physical mapping of the SCA7 locus. Am. J. Hum. Genet. 1996, 59, 1328. [Google Scholar]
- Gouw, L.G.; Kaplan, C.D.; Haines, J.H.; Digre, K.B.; Rutledge, S.L.; Matilla, A.; Leppert, M.; Zoghbi, H.Y.; Ptácek, L.J. Retinal degeneration characterizes a spinocerebellar ataxia mapping to chromosome 3p. Nat. Genet. 1995, 10, 89–93. [Google Scholar] [CrossRef]
- Holmberg, M.; Johansson, J.; Forsgren, L.; Heijbel, J.; Sandgren, O.; Holmgren, G. Localization of autosomal dominant cerebellar ataxia associated with retinal degeneration and anticipation to chromosome 3p12-p21. 1. Hum. Mol. Genet. 1995, 4, 1441–1445. [Google Scholar] [CrossRef] [PubMed]
- David, G.; Abbas, N.; Stevanin, G.; Dürr, A.; Yvert, G.; Cancel, G.; Weber, C.; Imbert, G.; Saudou, F.; Antoniou, E. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat. Genet. 1997, 17, 65–70. [Google Scholar] [CrossRef]
- Smith, D.; Bryer, A.; Watson, L.; Greenberg, L. Inherited polyglutamine spinocerebellar ataxias in South Africa. S. Afr. Med. J. 2012, 102, 683–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, L.; Wood, M.; Smith, D.; Scholefield, J.; Ballo, R.; Kidson, S.; Greenberg, L. Spinocerebellar ataxia type 7 in South Africa: Epidemiology, pathogenesis and therapy: The new millennium. S. Afr. Med. J. 2016, 106, 107–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmlinger, D.; Hardy, S.; Sasorith, S.; Klein, F.; Robert, F.; Weber, C.; Miguet, L.; Potier, N.; Van-Dorsselaer, A.; Wurtz, J.-M. Ataxin-7 is a subunit of GCN5 histone acetyltransferase-containing complexes. Hum. Mol. Genet. 2004, 13, 1257–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaytor, M.D.; Duvick, L.A.; Skinner, P.J.; Koob, M.D.; Ranum, L.P.; Orr, H.T. Nuclear localization of the spinocerebellar ataxia type 7 protein, ataxin-7. Hum. Mol. Genet. 1999, 8, 1657–1664. [Google Scholar] [CrossRef] [Green Version]
- La Spada, A.R.; Fu, Y.-H.; Sopher, B.L.; Libby, R.T.; Wang, X.; Li, L.Y.; Einum, D.D.; Huang, J.; Possin, D.E.; Smith, A.C. Polyglutamine-expanded ataxin-7 antagonizes CRX function and induces cone-rod dystrophy in a mouse model of SCA7. Neuron 2001, 31, 913–927. [Google Scholar] [CrossRef] [Green Version]
- Mohan, R.D.; Dialynas, G.; Weake, V.M.; Liu, J.; Martin-Brown, S.; Florens, L.; Washburn, M.P.; Workman, J.L.; Abmayr, S.M. Loss of Drosophila Ataxin-7, a SAGA subunit, reduces H2B ubiquitination and leads to neural and retinal degeneration. Genes Dev. 2014, 28, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Palhan, V.B.; Chen, S.; Peng, G.-H.; Tjernberg, A.; Gamper, A.M.; Fan, Y.; Chait, B.T.; La Spada, A.R.; Roeder, R.G. Polyglutamine-expanded ataxin-7 inhibits STAGA histone acetyltransferase activity to produce retinal degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 8472–8477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niewiadomska-Cimicka, A.; Doussau, F.; Perot, J.-B.; Roux, M.J.; Keime, C.; Hache, A.; Piguet, F.; Novati, A.; Weber, C.; Yalcin, B. SCA7 mouse cerebellar pathology reveals preferential downregulation of key Purkinje cell-identity genes and shared disease signature with SCA1 and SCA2. J. Neurosci. 2021, 41, 4910–4936. [Google Scholar] [CrossRef]
- Koide, R.; Kobayashi, S.; Shimohata, T.; Ikeuchi, T.; Maruyama, M.; Saito, M.; Yamada, M.; Takahashi, H.; Tsuji, S. A neurological disease caused by an expanded CAG trinucleotide repeat in the TATA-binding protein gene: A new polyglutamine disease? Hum. Mol. Genet. 1999, 8, 2047–2053. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Pan, Y.; Li, X.-J.; Li, S. Molecular mechanisms and therapeutics for SCA17. Neurotherapeutics 2019, 16, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Nikolov, D.B.; Hu, S.-H.; Lin, J.; Gasch, A.; Hoffmann, A.; Horikoshi, M.; Chua, N.-H.; Roeder, R.G.; Burley, S.K. Crystal structure of TFIID TATA-box binding protein. Nature 1992, 360, 40–46. [Google Scholar] [CrossRef]
- Nikolov, D.B.; Chen, H.; Halay, E.D.; Hoffman, A.; Roeder, R.G.; Burley, S.K. Crystal structure of a human TATA box-binding protein/TATA element complex. Proc. Natl. Acad. Sci. USA 1996, 93, 4862–4867. [Google Scholar] [CrossRef] [Green Version]
- Burley, S.; Roeder, R. Biochemistry and structural biology of transcription factor IID (TFIID). Annu. Rev. Biochem. 1996, 65, 769–799. [Google Scholar] [CrossRef]
- Reid, S.J.; Rees, M.I.; van Roon-Mom, W.M.; Jones, A.L.; MacDonald, M.E.; Sutherland, G.; During, M.J.; Faull, R.L.; Owen, M.J.; Dragunow, M. Molecular investigation of TBP allele length:: A SCA17 cellular model and population study. Neurobiol. Dis. 2003, 13, 37–45. [Google Scholar] [CrossRef]
- Yang, S.; Li, X.-J.; Li, S. Molecular mechanisms underlying Spinocerebellar Ataxia 17 (SCA17) pathogenesis. Rare Dis. 2016, 4, 349–365. [Google Scholar] [CrossRef] [Green Version]
- Rolfs, A.; Koeppen, A.H.; Bauer, I.; Bauer, P.; Buhlmann, S.; Topka, H.; Schöls, L.; Riess, O. Clinical features and neuropathology of autosomal dominant spinocerebellar ataxia (SCA17). Ann. Neurol. 2003, 54, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, Y.; Takahashi, H. Spinocerebellar ataxia type 17 (SCA17). In Polyglutamine Disorders; Springer: Cham, Switzerland, 2018; pp. 219–231. [Google Scholar]
- Huang, M.; Verbeek, D.S. Why do so many genetic insults lead to Purkinje Cell degeneration and spinocerebellar ataxia? Neurosci. Lett. 2019, 688, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-C.; Yao, N.-W.; Lin, C.-W.; Su, W.-S.; Wu, C.-T.; Chang, C.; Hsieh-Li, H.M. Neuroimaging Spectrum at Pre-, Early, and Late Symptomatic Stages of SCA17 Mice. Cerebellum 2020, 19, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Yang, S.; Li, X.J.; Li, S. Genetically modified rodent models of SCA17. J. Neurosci. Res. 2017, 95, 1540–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, N.C.; Crum, W.R.; Scahill, R.I.; Stevens, J.M.; Janssen, J.C.; Rossor, M.N. Imaging of onset and progression of Alzheimer’s disease with voxel-compression mapping of serial magnetic resonance images. Lancet 2001, 358, 201–205. [Google Scholar] [CrossRef]
- Koscik, T.R.; Sloat, L.; van der Plas, E.; Joers, J.M.; Deelchand, D.K.; Lenglet, C.; Öz, G.; Nopoulos, P.C. Brainstem and striatal volume changes are detectable in under 1 year and predict motor decline in spinocerebellar ataxia type 1. Brain Commun. 2020, 2, fcaa184. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef] [Green Version]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef] [Green Version]
- Watts, J.K.; Brown, R.H.; Khvorova, A. Nucleic Acid Therapeutics for Neurological Diseases. Neurotherapeutics 2019, 16, 245–247. [Google Scholar] [CrossRef] [Green Version]
- Khvorova, A.; Watts, J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef]
- Zaw, K.; Greer, K.; Aung-Htut, M.T.; Veedu, R.N.; Fletcher, S.; Wilton, S.D. Making the Inactive Active through Changes in Antisense Oligonucleotide Chemistries. Front. Genet. 2019, 10, 1249. [Google Scholar] [CrossRef]
- Bennett, C.F. Therapeutic antisense oligonucleotides are coming of age. Annu. Rev. Med. 2019, 70, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Aung-Htut, M.T.; McIntosh, C.S.; Ham, K.A.; Pitout, I.L.; Flynn, L.L.; Greer, K.; Fletcher, S.; Wilton, S.D. Systematic approach to developing splice modulating antisense oligonucleotides. Int. J. Mol. Sci. 2019, 20, 5030. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, C.; Wood, M.J. Antisense oligonucleotides: The next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 2018, 14, 9. [Google Scholar] [CrossRef]
- Crooke, S.T.; Baker, B.F.; Crooke, R.M.; Liang, X.-h. Antisense technology: An overview and prospectus. Nat. Rev. Drug Discov. 2021, 20, 427–453. [Google Scholar] [CrossRef]
- Lim, K.H.; Han, Z.; Jeon, H.Y.; Kach, J.; Jing, E.; Weyn-Vanhentenryck, S.; Downs, M.; Corrionero, A.; Oh, R.; Scharner, J. Antisense oligonucleotide modulation of non-productive alternative splicing upregulates gene expression. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Parra, M.; Zhang, W.; Vu, J.; DeWitt, M.; Conboy, J.G. Antisense targeting of decoy exons can reduce intron retention and increase protein expression in human erythroblasts. RNA 2020, 26, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.N.; Luo, D.; Singh, R.N. Pre-mRNA splicing modulation by antisense oligonucleotides. In Exon Skipping and Inclusion Therapies; Springer: Cham, Switzerland, 2018; pp. 415–437. [Google Scholar]
- Chan, J.H.; Lim, S.; Wong, W.F. Antisense oligonucleotides: From design to therapeutic application. Clin. Exp. Pharmacol. Physiol. 2006, 33, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Pitout, I.; Flynn, L.L.; Wilton, S.D.; Fletcher, S. Antisense-mediated splice intervention to treat human disease: The odyssey continues. F1000Research 2019, 8, 710. [Google Scholar] [CrossRef] [Green Version]
- Doxakis, E. Therapeutic antisense oligonucleotides for movement disorders. Med. Res. Rev. 2020, 41, 2656–2688. [Google Scholar] [CrossRef]
- Agrawal, S. The Evolution of Antisense Oligonucleotide Chemistry—A Personal Journey. Biomedicines 2021, 9, 503. [Google Scholar] [CrossRef]
- Adams, A.M.; Harding, P.L.; Iversen, P.L.; Coleman, C.; Fletcher, S.; Wilton, S.D. Antisense oligonucleotide induced exon skipping and the dystrophin gene transcript: Cocktails and chemistries. BMC Mol. Biol. 2007, 8, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClorey, G.; Moulton, H.; Iversen, P.; Fletcher, S.; Wilton, S. Antisense oligonucleotide-induced exon skipping restores dystrophin expression in vitro in a canine model of DMD. Gene Ther. 2006, 13, 1373. [Google Scholar] [CrossRef] [PubMed]
- Mann, C.J.; Honeyman, K.; McClorey, G.; Fletcher, S.; Wilton, S.D. Improved antisense oligonucleotide induced exon skipping in the mdx mouse model of muscular dystrophy. J. Gene Med. 2002, 4, 644–654. [Google Scholar] [CrossRef]
- Ham, K.A.; Aung-Htut, M.T.; Fletcher, S.; Wilton, S.D. Nonsequential Splicing Events Alter Antisense-Mediated Exon Skipping Outcome in COL7A1. Int. J. Mol. Sci. 2020, 21, 7705. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, C.S.; Aung-Htut, M.T.; Fletcher, S.; Wilton, S.D. Removal of the Polyglutamine Repeat of Ataxin-3 by Redirecting pre-mRNA Processing. Int. J. Mol. Sci. 2019, 20, 5434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aung-Htut, M.T.; Comerford, I.; Johnsen, R.; Foyle, K.; Fletcher, S.; Wilton, S.D. Reduction of integrin alpha 4 activity through splice modulating antisense oligonucleotides. Sci. Rep. 2019, 9, 12994. [Google Scholar] [CrossRef] [PubMed]
- Flynn, L.L.; Mitrpant, C.; Pitout, I.L.; Fletcher, S.; Wilton, S.D. Antisense Oligonucleotide-Mediated Terminal Intron Retention of the SMN2 Transcript. Mol. Ther. Nucleic Acids 2018, 11, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, S. Viltolarsen: First approval. Drugs 2020, 80, 1027–1031. [Google Scholar] [CrossRef]
- Afonso-Reis, R.; Afonso, I.T.; Nóbrega, C. Current Status of Gene Therapy Research in Polyglutamine Spinocerebellar Ataxias. Int. J. Mol. Sci. 2021, 22, 4249. [Google Scholar] [CrossRef]
- Friedrich, J.; Kordasiewicz, H.B.; O’Callaghan, B.; Handler, H.P.; Wagener, C.; Duvick, L.; Swayze, E.E.; Rainwater, O.; Hofstra, B.; Benneyworth, M. Antisense oligonucleotide–mediated ataxin-1 reduction prolongs survival in SCA1 mice and reveals disease-associated transcriptome profiles. JCI Insight 2018, 3, e123193. [Google Scholar] [CrossRef] [PubMed]
- Matilla, A.; Roberson, E.D.; Banfi, S.; Morales, J.; Armstrong, D.L.; Burright, E.N.; Orr, H.T.; Sweatt, J.D.; Zoghbi, H.Y.; Matzuk, M.M. Mice lacking ataxin-1 display learning deficits and decreased hippocampal paired-pulse facilitation. J. Neurosci. 1998, 18, 5508–5516. [Google Scholar] [CrossRef]
- Kourkouta, E.; Weij, R.; González-Barriga, A.; Mulder, M.; Verheul, R.; Bosgra, S.; Groenendaal, B.; Puoliväli, J.; Toivanen, J.; van Deutekom, J.C. Suppression of mutant protein expression in SCA3 and SCA1 mice using a CAG repeat-targeting antisense oligonucleotide. Mol. Ther. Nucleic Acids 2019, 17, 601–614. [Google Scholar] [CrossRef] [Green Version]
- Huynh, D.P.; Maalouf, M.; Silva, A.J.; Schweizer, F.E.; Pulst, S.M. Dissociated fear and spatial learning in mice with deficiency of ataxin-2. PLoS ONE 2009, 4, e6235. [Google Scholar] [CrossRef] [Green Version]
- Kiehl, T.-R.; Nechiporuk, A.; Figueroa, K.P.; Keating, M.T.; Huynh, D.P.; Pulst, S.-M. Generation and characterization of Sca2 (ataxin-2) knockout mice. Biochem. Biophys. Res. Commun. 2006, 339, 17–24. [Google Scholar] [CrossRef]
- Scoles, D.R.; Meera, P.; Schneider, M.D.; Paul, S.; Dansithong, W.; Figueroa, K.P.; Hung, G.; Rigo, F.; Bennett, C.F.; Otis, T.S. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature 2017, 544, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Toonen, L.J.; Rigo, F.; van Attikum, H.; van Roon-Mom, W.M. Antisense oligonucleotide-mediated removal of the polyglutamine repeat in spinocerebellar ataxia type 3 mice. Mol. Ther. Nucleic Acids 2017, 8, 232–242. [Google Scholar] [CrossRef] [Green Version]
- Evers, M.M.; Tran, H.-D.; Zalachoras, I.; Pepers, B.A.; Meijer, O.C.; den Dunnen, J.T.; van Ommen, G.-J.B.; Aartsma-Rus, A.; van Roon-Mom, W.M. Ataxin-3 protein modification as a treatment strategy for spinocerebellar ataxia type 3: Removal of the CAG containing exon. Neurobiol. Dis. 2013, 58, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLoughlin, H.S.; Moore, L.R.; Chopra, R.; Komlo, R.; McKenzie, M.; Blumenstein, K.G.; Zhao, H.; Kordasiewicz, H.B.; Shakkottai, V.G.; Paulson, H.L. Oligonucleotide therapy mitigates disease in spinocerebellar ataxia type 3 mice. Ann. Neurol. 2018, 84, 64–77. [Google Scholar] [CrossRef]
- Rodríguez-Lebrón, E.; Costa, M.D.; Luna-Cancalon, K.; Peron, T.M.; Fischer, S.; Boudreau, R.L.; Davidson, B.L.; Paulson, H.L. Silencing mutant ATXN3 expression resolves molecular phenotypes in SCA3 transgenic mice. Mol. Ther. 2013, 21, 1909–1918. [Google Scholar] [CrossRef] [Green Version]
- Niu, C.; Prakash, T.P.; Kim, A.; Quach, J.L.; Huryn, L.A.; Yang, Y.; Lopez, E.; Jazayeri, A.; Hung, G.; Sopher, B.L. Antisense oligonucleotides targeting mutant Ataxin-7 restore visual function in a mouse model of spinocerebellar ataxia type 7. Sci. Transl. Med. 2018, 10, eaap8677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, S.-Y.; Pennesi, M.E.; Weeber, E.J.; Xu, B.; Atkinson, R.; Chen, S.; Armstrong, D.L.; Wu, S.M.; Sweatt, J.D.; Zoghbi, H.Y. SCA7 knockin mice model human SCA7 and reveal gradual accumulation of mutant ataxin-7 in neurons and abnormalities in short-term plasticity. Neuron 2003, 37, 383–401. [Google Scholar] [CrossRef] [Green Version]
- Schobel, S.; Palermo, G.; Trundell, D.; Kremer, T.; Sanwald-Ducray, P.; Smith, A.; Boak, L.; Doody, R. A Global Development Program Testing RG6042, an Antisense Oligonucleotide, for the Treatment of Early Manifest Huntington’s Disease. In Proceedings of the European Huntington’s Disease Network 2018 Plenary Meeting, Vienna, Austria, 14–16 September 2018; pp. 14–16. [Google Scholar]
- Kordasiewicz, H.B.; Stanek, L.M.; Wancewicz, E.V.; Mazur, C.; McAlonis, M.M.; Pytel, K.A.; Artates, J.W.; Weiss, A.; Cheng, S.H.; Shihabuddin, L.S. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 2012, 74, 1031–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, D. Failure of genetic therapies for Huntington’s devastates community. Nature 2021, 593, 180. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Causative Gene | Normal Gene Function * | PolyQ Location | Healthy Repeat Range | Pre-Mutation Repeat Range | Pathogenetic Repeat Range |
|---|---|---|---|---|---|---|
| SCA1 MIM #164400 | ATXN1 MIM #601556 | RNA metabolism and transcriptional repression | Exon 7 | 6–35 | 36–40 | 41–89 |
| SCA2 MIM #183090 | ATXN2 MIM #601517 | Transcriptional repression and RNA metabolism | Exon 1 | 17–29 | 30–36 | 37–100+ |
| SCA3 MIM #109150 | ATXN3 MIM #607047 | Deubiquitination and proteasomal protein degradation | Exon 10 | 7–44 | 45–54 | 55–89 |
| SCA6 MIM #183086 | CACNA1A MIM #601011 | Gives rise to P/Q calcium channels and neurotransmitter release from presynaptic terminals | Exon 47 | 4–18 | 19–20 | 21–30 |
| SCA7 MIM #164500 | ATXN7 MIM #607640 | Mediates the interaction between the CRX and STAGA complex | Exon 3 | 7–19 | 20–35 | 36–400+ |
| SCA17 MIM #607136 | TBP MIM #600075 | General transcription factor and mediates the initiation of transcription through TFIID binding to the TAT box | Exon 3 | 25–42 | 43–46 | 47–66 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McIntosh, C.S.; Li, D.; Wilton, S.D.; Aung-Htut, M.T. Polyglutamine Ataxias: Our Current Molecular Understanding and What the Future Holds for Antisense Therapies. Biomedicines 2021, 9, 1499. https://doi.org/10.3390/biomedicines9111499
McIntosh CS, Li D, Wilton SD, Aung-Htut MT. Polyglutamine Ataxias: Our Current Molecular Understanding and What the Future Holds for Antisense Therapies. Biomedicines. 2021; 9(11):1499. https://doi.org/10.3390/biomedicines9111499
Chicago/Turabian StyleMcIntosh, Craig S., Dunhui Li, Steve D. Wilton, and May T. Aung-Htut. 2021. "Polyglutamine Ataxias: Our Current Molecular Understanding and What the Future Holds for Antisense Therapies" Biomedicines 9, no. 11: 1499. https://doi.org/10.3390/biomedicines9111499