
CCR6 Deficiency Increases Infarct Size after Murine Acute Myocardial Infarction

 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Coronary Ischemia-Reperfusion Surgery
2.2. Bone Marrow Transplantation
2.3. Echocardiography
2.4. Histology and Immunohistochemistry
2.5. Statistical Analysis
3. Results
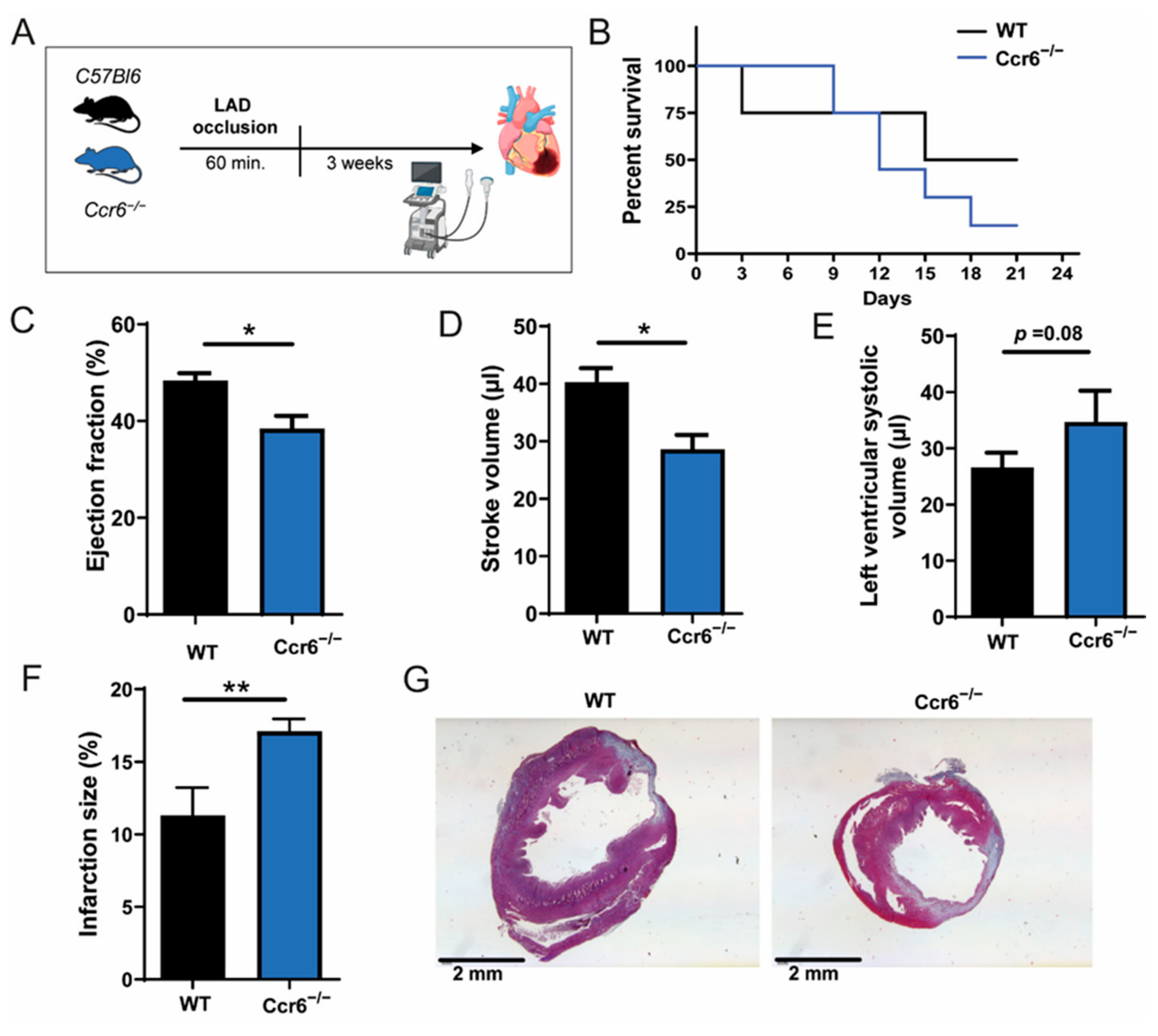
3.1. CCR6 Deficiency Worsens Cardiac Function and Increases Infarction Size after Ischemia-Reperfusion Injury
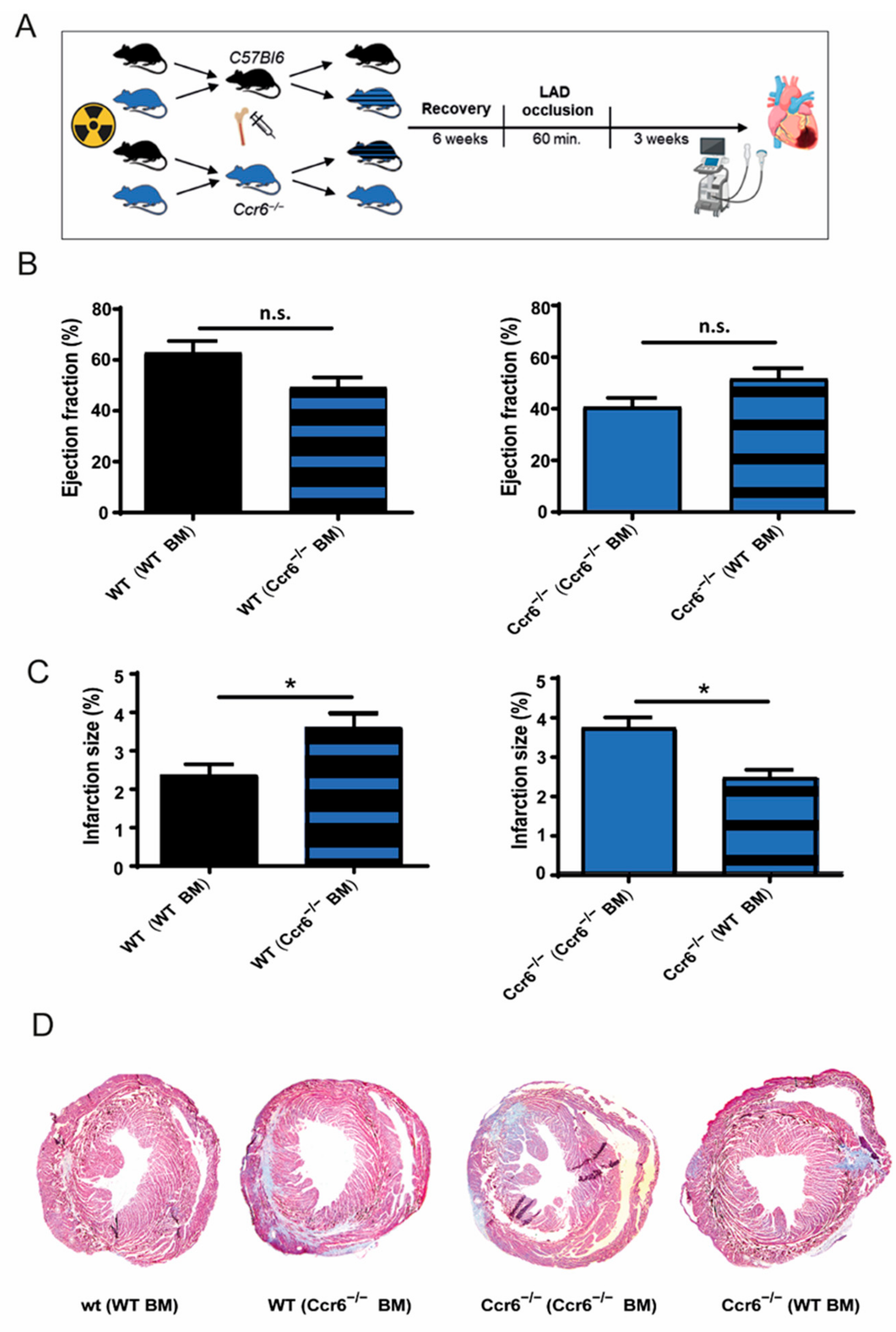
3.2. Bone Marrow Derived Immune Cells Are Responsible for the Effects of Ccr6−/− after Coronary Ischemia-Reperfusion Injury
3.3. Increased Cardiac Infiltration of Inflammatory Immune Cells in CCR6−/− Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burke, A.P.; Virmani, R. Pathophysiology of Acute Myocardial Infarction. Med Clin. North Am. 2007, 91, 553–572. [Google Scholar] [CrossRef]
- O’Gara, P.T.; Kushner, F.G.; Ascheim, D.D.; Casey, D.E.; Chung, M.K.; de Lemos, J.A.; Ettinger, S.M.; Fang, J.C.; Fesmire, F.M.; Franklin, B.A.; et al. 2013 ACCF/AHA guideline for the management of st-elevation myocardial infarction: A report of the American college of cardiology foundation/american heart association task force on practice guidelines. J. Am. Coll. Cardiol. 2013, 61, 78–140. [Google Scholar] [CrossRef] [Green Version]
- Pagliaro, P.; Moro, F.; Tullio, F.; Perrelli, M.-G.; Penna, C. Cardioprotective Pathways During Reperfusion: Focus on Redox Signaling and Other Modalities of Cell Signaling. Antioxidants Redox Signal. 2011, 14, 833–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulati, A.; Jabbour, A.; Ismail, T.F.; Guha, K.; Khwaja, J.; Raza, S.; Morarji, K.; Brown, T.D.H.; Ismail, N.A.; Dweck, M.R.; et al. Association of Fibrosis With Mortality and Sudden Cardiac Death in Patients With Nonischemic Dilated Cardiomyopathy. JAMA 2013, 309, 896–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahrendorf, M. Myeloid cell contributions to cardiovascular health and disease. Nat. Med. 2018, 24, 711–720. [Google Scholar] [CrossRef]
- Puhl, S.-L.; Steffens, S. Neutrophils in Post-myocardial Infarction Inflammation: Damage vs. Resolution? Front. Cardiovasc. Med. 2019, 6, 25. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Frangogiannis, N.G. Chemokines in cardiac fibrosis. Curr. Opin. Physiol. 2021, 19, 80–91. [Google Scholar] [CrossRef]
- Noels, H.; Weber, C.; Koenen, R.R. Chemokines as Therapeutic Targets in Cardiovascular Disease. Arter. Thromb. Vasc. Biol. 2019, 39, 583–592. [Google Scholar] [CrossRef] [Green Version]
- Koenen, R.; Weber, C. Chemokines: Established and novel targets in atherosclerosis. EMBO Mol. Med. 2011, 3, 713–725. [Google Scholar] [CrossRef]
- Majmudar, M.D.; Keliher, E.J.; Heidt, T.; Leuschner, F.; Truelove, J.; Sena, B.F.; Gorbatov, R.; Iwamoto, Y.; Dutta, P.; Wojtkiewicz, G.; et al. Monocyte-Directed RNAi Targeting CCR2 Improves Infarct Healing in Atherosclerosis-Prone Mice. Circ. 2013, 127, 2038–2046. [Google Scholar] [CrossRef] [Green Version]
- Hao, H.; Hu, S.; Chen, H.; Bu, D.; Zhu, L.; Xu, C.; Chu, F.; Huo, X.; Tang, Y.; Sun, X.; et al. Loss of Endothelial CXCR7 Impairs Vascular Homeostasis and Cardiac Remodeling After Myocardial Infarction. Circ. 2017, 135, 1253–1264. [Google Scholar] [CrossRef]
- Vajen, T.; Koenen, R.R.; Werner, I.; Staudt, M.; Projahn, D.; Curaj, A.; Sönmez, T.T.; Simsekyilmaz, S.; Schumacher, D.; Möllmann, J.; et al. Blocking CCL5-CXCL4 heteromerization preserves heart function after myocardial infarction by attenuating leukocyte recruitment and NETosis. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Projahn, D.; Simsekyilmaz, S.; Singh, S.; Kanzler, I.; Kramp, B.K.; Langer, M.; Burlacu, A.; Bernhagen, J.; Klee, D.; Zernecke, A.; et al. Controlled intramyocardial release of engineered chemokines by biodegradable hydrogels as a treatment approach of myocardial infarction. J. Cell. Mol. Med. 2014, 18, 790–800. [Google Scholar] [CrossRef]
- Braunersreuther, V.; Pelli, G.; Galan, K.; Proudfoot, A.E.; Belin, A.; Vuilleumier, N.; Burger, F.; Lenglet, S.; Caffa, I.; Soncini, D.; et al. Treatment with the CC chemokine-binding protein Evasin-4 improves post-infarction myocardial injury and survival in mice. Thromb. Haemost. 2013, 110, 807–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safa, A.; Rashidinejad, H.; Khalili, M.; Dabiri, S.; Nemati, M.; Mohammadi, M.; Jafarzadeh, A. Higher circulating levels of chemokines CXCL10, CCL20 and CCL22 in patients with ischemic heart disease. Cytokine 2016, 83, 147–157. [Google Scholar] [CrossRef]
- Lin, C.-F.; Su, C.-J.; Liu, J.-H.; Chen, S.-T.; Huang, H.-L.; Pan, S.-L. Potential Effects of CXCL9 and CCL20 on Cardiac Fibrosis in Patients with Myocardial Infarction and Isoproterenol-Treated Rats. J. Clin. Med. 2019, 8, 659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arunachalam, P.; Ludewig, P.; Melich, P.; Arumugam, T.V.; Gerloff, C.; Prinz, I.; Magnus, T.; Gelderblom, M. CCR6 (CC Chemokine Receptor 6) Is Essential for the Migration of Detrimental Natural Interleukin-17–Producing γδ T Cells in Stroke. Stroke 2017, 48, 1957–1965. [Google Scholar] [CrossRef]
- Hofmann, U.; Frantz, S. Role of Lymphocytes in Myocardial Injury, Healing, and Remodeling After Myocardial Infarction. Circ. Res. 2015, 116, 354–367. [Google Scholar] [CrossRef] [Green Version]
- Santos-Zas, I.; Lemarié, J.; Tedgui, A.; Ait-Oufella, H. Adaptive Immune Responses Contribute to Post-ischemic Cardiac Remodeling. Front. Cardiovasc. Med. 2019, 5, 198. [Google Scholar] [CrossRef]
- Cook, D.; Prosser, D.M.; Forster, R.; Zhang, J.; A Kuklin, N.; Abbondanzo, S.J.; Niu, X.-D.; Chen, S.-C.; Manfra, D.J.; Wiekowski, M.T.; et al. CCR6 Mediates Dendritic Cell Localization, Lymphocyte Homeostasis, and Immune Responses in Mucosal Tissue. Immun. 2000, 12, 495–503. [Google Scholar] [CrossRef] [Green Version]
- Curaj, A.; Simsekyilmaz, S.; Staudt, M.; Liehn, E.A. Minimal Invasive Surgical Procedure of Inducing Myocardial Infarction in Mice. J. Vis. Exp. 2015, e52197, e52197. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, D.; Curaj, A.; Staudt, M.; Cordes, F.; Dumitraşcu, A.; Rolles, B.; Beckers, C.; Soppert, J.; Rusu, M.; Simsekyilmaz, S.; et al. Phosphatidylserine Supplementation as a Novel Strategy for Reducing Myocardial Infarct Size and Preventing Adverse Left Ventricular Remodeling. Int. J. Mol. Sci. 2021, 22, 4401. [Google Scholar] [CrossRef]
- Liehn, E.A.; Kanzler, I.; Konschalla, S.; Kroh, A.; Simsekyilmaz, S.; Sönmez, T.T.; Bucala, R.; Bernhagen, J.; Weber, C. Compartmentalized Protective and Detrimental Effects of Endogenous Macrophage Migration-Inhibitory Factor Mediated by CXCR2 in a Mouse Model of Myocardial Ischemia/Reperfusion. Arter. Thromb. Vasc. Biol. 2013, 33, 2180–2186. [Google Scholar] [CrossRef] [Green Version]
- Curaj, A.; Pop-Fele, L.; Jovanovic, M.; Jonas, S.M.; Moellmann, J.; Ghertescu, D.; Novac, O.C.; Rusu, M.; Liehn, E.A. Advanced modular automated calculation of the morpho-histological parameters in myocardial infarction. Discov. 2016, 4, e66. [Google Scholar] [CrossRef]
- Ortega, S.B.; Torres, V.O.; Latchney, S.E.; Whoolery, C.W.; Noorbhai, I.Z.; Poinsatte, K.; Selvaraj, U.M.; Benson, M.A.; Meeuwissen, A.J.M.; Plautz, E.J.; et al. B cells migrate into remote brain areas and support neurogenesis and functional recovery after focal stroke in mice. Proc. Natl. Acad. Sci. USA 2020, 117, 4983–4993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, T.; Yang, X.O.; Chung, Y.; Fukunaga, A.; Nurieva, R.; Pappu, B.; Martin-Orozco, N.; Kang, H.S.; Ma, L.; Panopoulos, A.; et al. CCR6 Regulates the Migration of Inflammatory and Regulatory T Cells. J. Immunol. 2008, 181, 8391–8401. [Google Scholar] [CrossRef] [Green Version]
- Zouggari, Y.; Ait-Oufella, H.; Bonnin, P.; Simon, T.; Sage, A.; Guérin, C.; Vilar, J.; Caligiuri, G.; Tsiantoulas, D.; Laurans, L.; et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat. Med. 2013, 19, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Reimer, D.; Lee, A.Y.S.; Bannan, J.; Fromm, P.; E Kara, E.; Comerford, I.; McColl, S.; Wiede, F.; Mielenz, D.; Körner, H. Early CCR6 expression on B cells modulates germinal centre kinetics and efficient antibody responses. Immunol. Cell Biol. 2016, 95, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Wiede, F.; Fromm, P.D.; Comerford, I.; Kara, E.; Bannan, J.; Schuh, W.; Ranasinghe, C.; Tarlinton, D.; Winkler, T.; McColl, S.R.; et al. CCR6 is transiently upregulated on B cells after activation and modulates the germinal center reaction in the mouse. Immunol. Cell Biol. 2013, 91, 335–339. [Google Scholar] [CrossRef]
- Elgueta, R.; Marks, E.; Nowak, E.; Menezes, S.; Benson, M.; Raman, V.S.; Ortiz, C.; O’Connell, S.; Hess, H.; Lord, G.; et al. CCR6-Dependent Positioning of Memory B Cells Is Essential for Their Ability To Mount a Recall Response to Antigen. J. Immunol. 2015, 194, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Suan, D.; Kräutler, N.J.; Maag, J.; Butt, D.; Bourne, K.; Hermes, J.R.; Avery, D.T.; Young, C.; Statham, A.; Elliott, M.; et al. CCR6 Defines Memory B Cell Precursors in Mouse and Human Germinal Centers, Revealing Light-Zone Location and Predominant Low Antigen Affinity. Immun. 2017, 47, 1142–1153. [Google Scholar] [CrossRef] [Green Version]
- Katayama, H. Development of psoriasis by continuous neutrophil infiltration into the epidermis. Exp. Dermatol. 2018, 27, 1084–1091. [Google Scholar] [CrossRef] [Green Version]
- González-Guerrero, C.; Morgado-Pascual, J.L.; Cannata-Ortiz, P.; Ramos-Barron, M.A.; Gómez-Alamillo, C.; Arias, M.; Mezzano, S.; Egido, J.; Ruiz-Ortega, M.; Ortiz, A.; et al. CCL20 blockade increases the severity of nephrotoxic folic acid-induced acute kidney injury. J. Pathol. 2018, 246, 191–204. [Google Scholar] [CrossRef]
- Yeh, C.-Y.; Shun, C.-T.; Kuo, Y.-M.; Jung, C.-J.; Hsieh, S.-C.; Chiu, Y.-L.; Chen, J.-W.; Hsu, R.-B.; Yang, C.-J.; Chia, J.-S. Activated Human Valvular Interstitial Cells Sustain Interleukin-17 Production To Recruit Neutrophils in Infective Endocarditis. Infect. Immun. 2015, 83, 2202–2212. [Google Scholar] [CrossRef] [Green Version]
- Wojkowska, D.; Szpakowski, P.; Ksiazek-Winiarek, D.; Leszczynski, M.; Glabinski, A. Interactions between Neutrophils, Th17 Cells, and Chemokines during the Initiation of Experimental Model of Multiple Sclerosis. Mediat. Inflamm. 2014, 2014, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Meitei, H.T.; Jadhav, N.; Lal, G. CCR6-CCL20 axis as a therapeutic target for autoimmune diseases. Autoimmun. Rev. 2021, 20, 102846. [Google Scholar] [CrossRef]
- Hammerich, L.; Bangen, J.M.; Govaere, O.; Zimmermann, H.W.; Gassler, N.; Huss, S.; Liedtke, C.; Prinz, I.; Lira, S.A.; Luedde, T.; et al. Chemokine receptor CCR6-dependent accumulation of γδ T cells in injured liver restricts hepatic inflammation and fibrosis. Hepatol. 2014, 59, 630–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liehn, E.A.; Postea, O.; Curaj, A.; Marx, N. Repair After Myocardial Infarction, Between Fantasy and Reality: The Role of Chemokines. J. Am. Coll. Cardiol. 2011, 58, 2357–2362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frangogiannis, N.G. Targeting the inflammatory response in healing myocardial infarcts. Curr. Med. Chem. 2006, 13, 1877–1893. [Google Scholar] [CrossRef]
- Yan, X.; Shichita, T.; Katsumata, Y.; Matsuhashi, T.; Ito, H.; Ito, K.; Anzai, A.; Endo, J.; Tamura, Y.; Kimura, K.; et al. Deleterious Effect of the IL-23/IL-17A Axis and γδT Cells on Left Ventricular Remodeling After Myocardial Infarction. J. Am. Hear. Assoc. 2012, 1, e004408. [Google Scholar] [CrossRef] [Green Version]
- Dobaczewski, M.; Xia, Y.; Bujak, M.; Gonzalez-Quesada, C.; Frangogiannis, N. CCR5 Signaling Suppresses Inflammation and Reduces Adverse Remodeling of the Infarcted Heart, Mediating Recruitment of Regulatory T Cells. Am. J. Pathol. 2010, 176, 2177–2187. [Google Scholar] [CrossRef]
- Saxena, A.; Dobaczewski, M.; Rai, V.; Haque, Z.; Chen, W.; Li, N.; Frangogiannis, N.G. Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function. Am. J. Physiol. Circ. Physiol. 2014, 307, H1233–H1242. [Google Scholar] [CrossRef] [PubMed]
- Weirather, J.; Hofmann, U.D.; Beyersdorf, N.; Ramos, G.; Vogel, B.; Frey, A.; Ertl, G.; Kerkau, T.; Frantz, S. Foxp3 + CD4 + T Cells Improve Healing After Myocardial Infarction by Modulating Monocyte/Macrophage Differentiation. Circ. Res. 2014, 115, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Lai, X.; Huang, Z.; Gua, Z. Elevated circulating level of CCR6 is related to acute myocardial infarction and severity of coronary artery stenosis. Int. J. Clin. Exp. Med. 2018, 11, 2292–2298. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schumacher, D.; Liehn, E.A.; Singh, A.; Curaj, A.; Wijnands, E.; Lira, S.A.; Tacke, F.; Jankowski, J.; Biessen, E.A.L.; van der Vorst, E.P.C. CCR6 Deficiency Increases Infarct Size after Murine Acute Myocardial Infarction. Biomedicines 2021, 9, 1532. https://doi.org/10.3390/biomedicines9111532
Schumacher D, Liehn EA, Singh A, Curaj A, Wijnands E, Lira SA, Tacke F, Jankowski J, Biessen EAL, van der Vorst EPC. CCR6 Deficiency Increases Infarct Size after Murine Acute Myocardial Infarction. Biomedicines. 2021; 9(11):1532. https://doi.org/10.3390/biomedicines9111532
Chicago/Turabian StyleSchumacher, David, Elisa A. Liehn, Anjana Singh, Adelina Curaj, Erwin Wijnands, Sergio A. Lira, Frank Tacke, Joachim Jankowski, Erik A.L. Biessen, and Emiel P.C. van der Vorst. 2021. "CCR6 Deficiency Increases Infarct Size after Murine Acute Myocardial Infarction" Biomedicines 9, no. 11: 1532. https://doi.org/10.3390/biomedicines9111532
APA StyleSchumacher, D., Liehn, E. A., Singh, A., Curaj, A., Wijnands, E., Lira, S. A., Tacke, F., Jankowski, J., Biessen, E. A. L., & van der Vorst, E. P. C. (2021). CCR6 Deficiency Increases Infarct Size after Murine Acute Myocardial Infarction. Biomedicines, 9(11), 1532. https://doi.org/10.3390/biomedicines9111532