
Mitochondrial Electron Transport Chain Protein Abnormalities Detected in Plasma Extracellular Vesicles in Alzheimer’s Disease


Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Isolation of Neuron-Derived EVs (NDEVs)
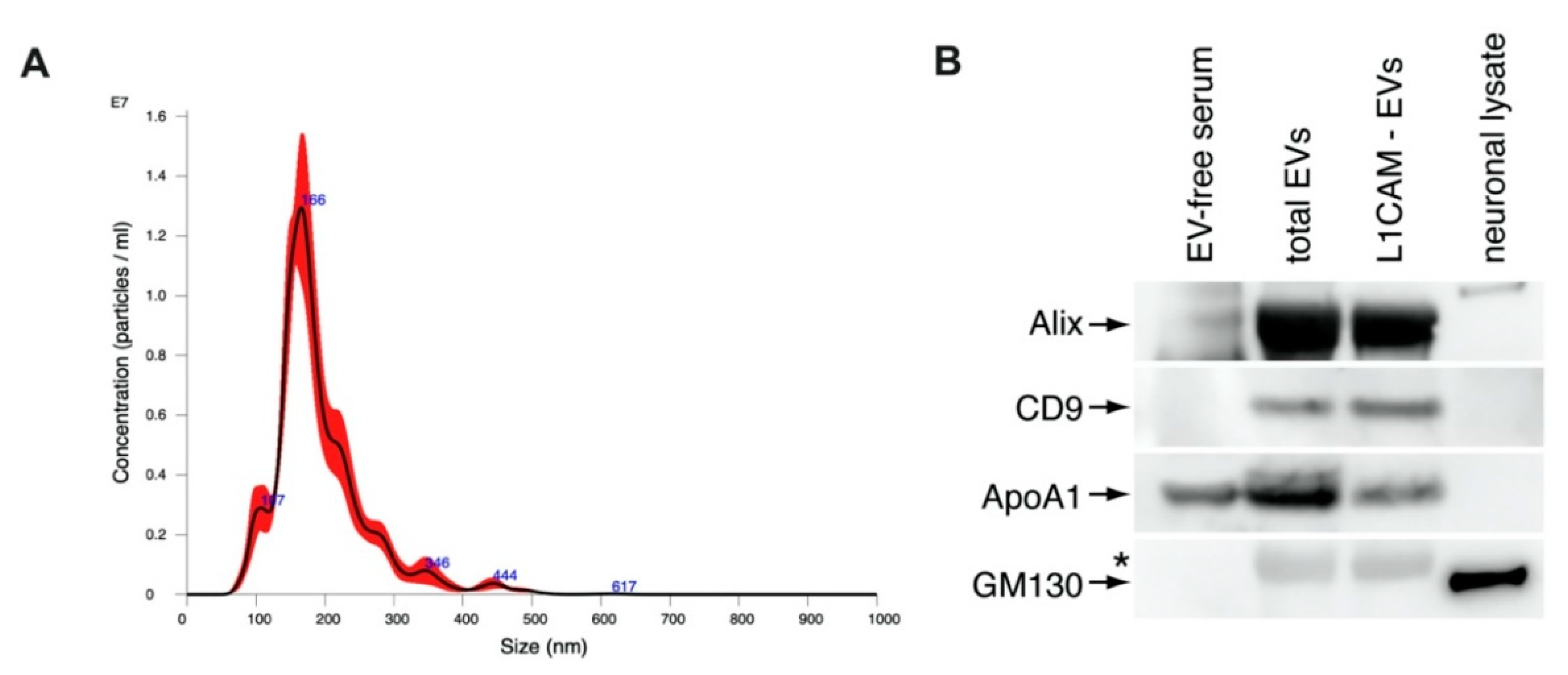
2.3. NDEV Characterization
2.4. Quantification of NDEV Proteins
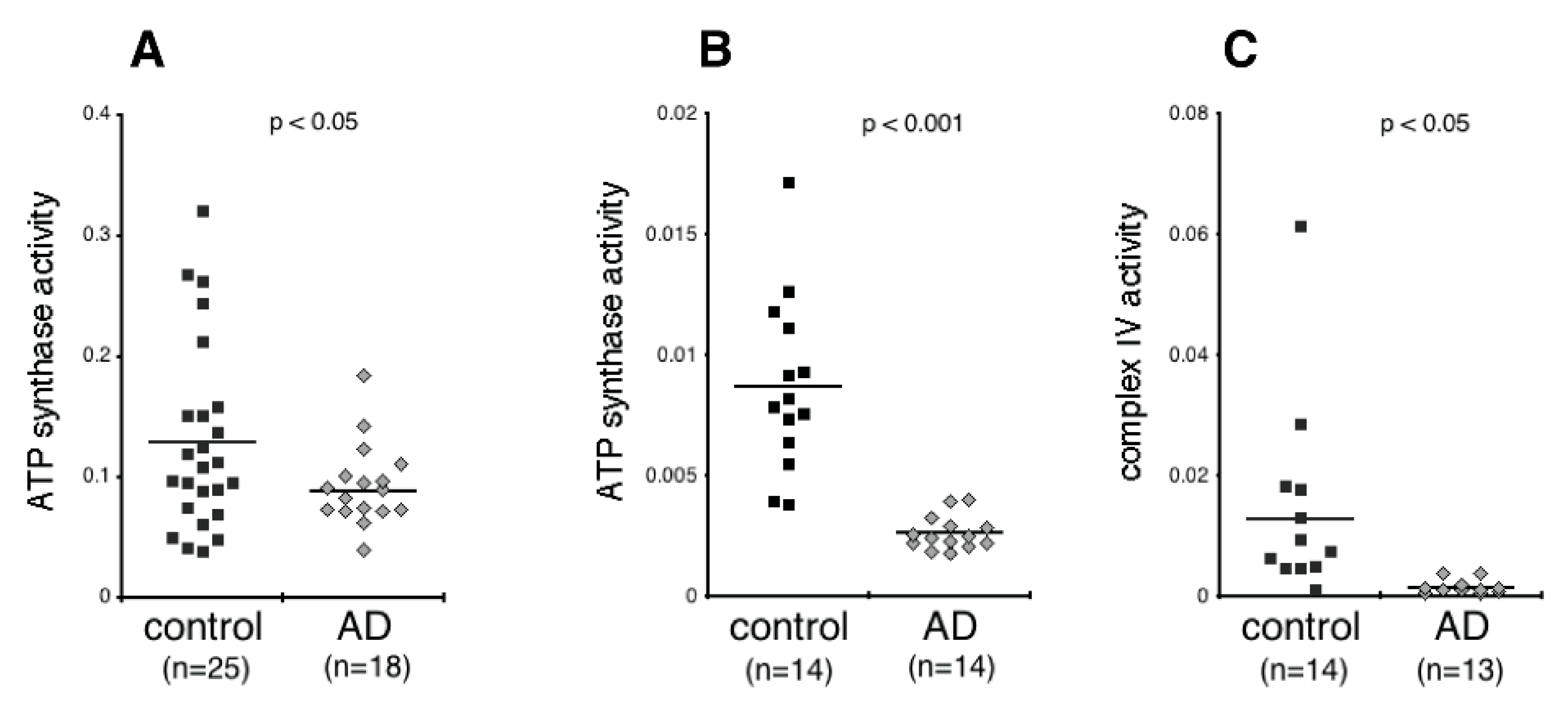
2.5. Measurement of ATP Synthase and Complex IV Activity
3. Results
3.1. NDEV Characterization
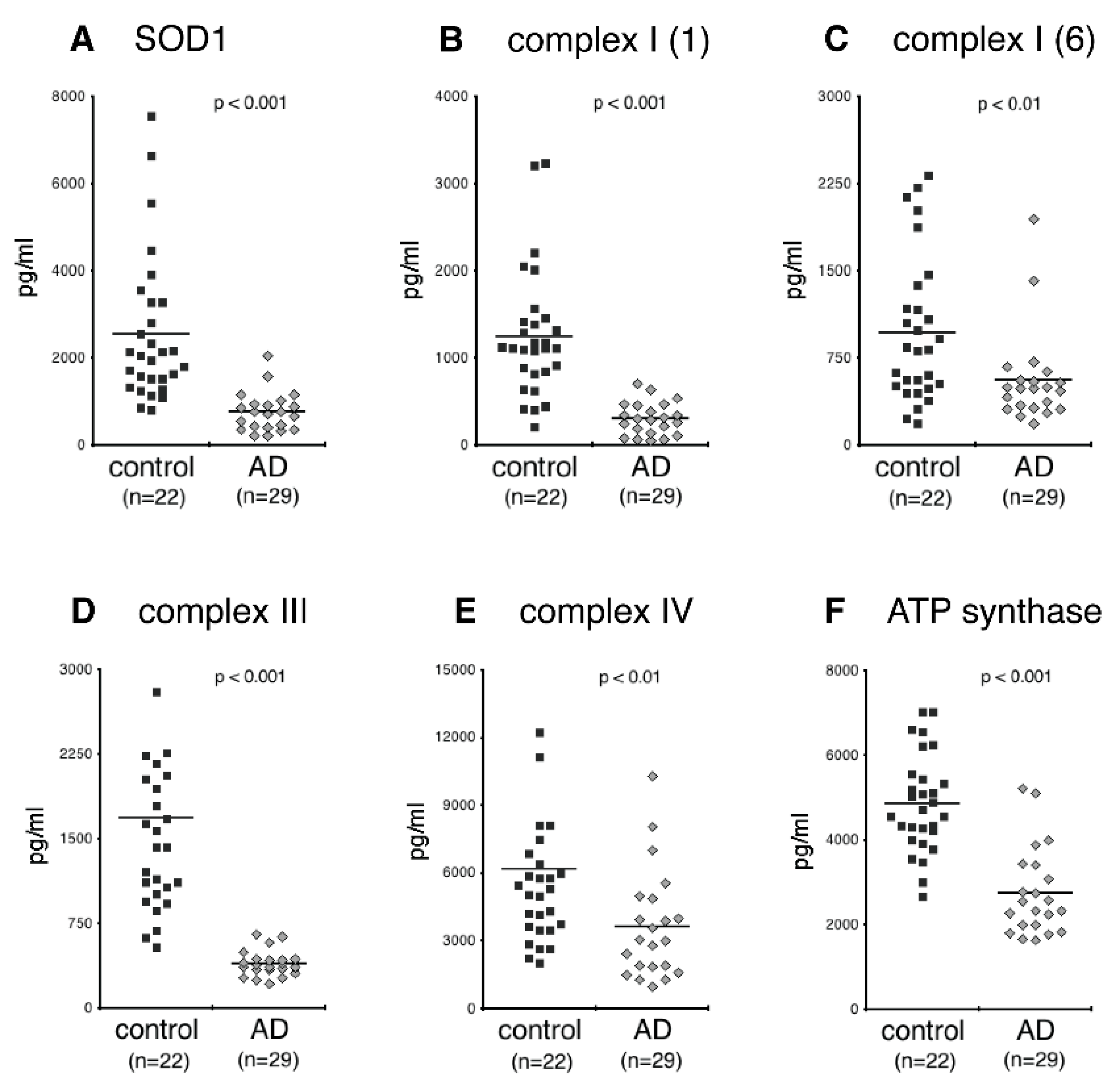
3.2. NDEV Mitochondrial Proteins
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [Green Version]
- Massaad, C.A.; Pautler, R.G.; Klann, E. Mitochondrial superoxide: A key player in Alzheimer’s disease. Aging 2009, 1, 758–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, K.; Dong, Y.T.; Xiang, J.; Xu, Y.; Hong, W.; Song, H.; Guan, Z.Z. Reduced expression of SIRT1 and SOD-1 and the correlation between these levels in various regions of the brains of patients with Alzheimer’s disease. J. Clin. Pathol. 2018, 71, 1090–1099. [Google Scholar] [CrossRef]
- Youssef, P.; Chami, B.; Lim, J.; Middleton, T.; Sutherland, G.T.; Witting, P.K. Evidence supporting oxidative stress in a moderately affected area of the brain in Alzheimer’s disease. Sci. Rep. 2018, 8, 11553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement. 2015, 11, 600–607.e1. [Google Scholar] [CrossRef] [Green Version]
- Mustapic, M.; Eitan, E.; Werner, J.K., Jr.; Berkowitz, S.T.; Lazaropoulos, M.P.; Tran, J.; Goetzl, E.J.; Kapogiannis, D. Plasma Extracellular Vesicles Enriched for Neuronal Origin: A Potential Window into Brain Pathologic Processes. Front. Neurosci. 2017, 11, 278. [Google Scholar] [CrossRef] [Green Version]
- Eren, E.; Hunt, J.F.V.; Shardell, M.; Chawla, S.; Tran, J.; Gu, J.; Vogt, N.M.; Johnson, S.C.; Bendlin, B.B.; Kapogiannis, D. Extracellular vesicle biomarkers of Alzheimer’s disease associated with sub-clinical cognitive decline in late middle age. Alzheimers Dement. 2020, 16, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Kapogiannis, D.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Biragyn, A.; Masharani, U.; Frassetto, L.; Petersen, R.C.; Miller, B.L.; Goetzl, E.J. Dysfunctionally phosphorylated type 1 insulin receptor substrate in neural-derived blood exosomes of preclinical Alzheimer’s disease. FASEB J. 2015, 29, 589–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapogiannis, D.; Mustapic, M.; Shardell, M.D.; Berkowitz, S.T.; Diehl, T.C.; Spangler, R.D.; Tran, J.; Lazaropoulos, M.P.; Chawla, S.; Gulyani, S.; et al. Association of Extracellular Vesicle Biomarkers With Alzheimer Disease in the Baltimore Longitudinal Study of Aging. JAMA Neurol. 2019, 76, 1340–1351. [Google Scholar] [CrossRef]
- Walker, K.A.; Chawla, S.; Nogueras-Ortiz, C.; Coresh, J.; Sharrett, A.R.; Wong, D.F.; Jack, C.R., Jr.; Spychalla, A.J.; Gottesman, R.F.; Kapogiannis, D. Neuronal insulin signaling and brain structure in nondemented older adults: The Atherosclerosis Risk in Communities Study. Neurobiol. Aging. 2021, 97, 65–72. [Google Scholar] [CrossRef]
- Kim, K.M.; Meng, Q.; Perez de Acha, O.; Mustapic, M.; Cheng, A.; Eren, E.; Kundu, G.; Piao, Y.; Munk, R.; Wood, W.H., 3rd; et al. Mitochondrial RNA in Alzheimer’s Disease Circulating Extracellular Vesicles. Front. Cell Dev. Biol. 2020, 8, 581882. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Srihari, V.H.; Guloksuz, S.; Ferrara, M.; Tek, C.; Heninger, G.R. Decreased mitochondrial electron transport proteins and increased complement mediators in plasma neural-derived exosomes of early psychosis. Transl. Psychiatry 2020, 10, 361. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Srihari, V.H.; Guloksuz, S.; Ferrara, M.; Tek, C.; Heninger, G.R. Neural cell-derived plasma exosome protein abnormalities implicate mitochondrial impairment in first episodes of psychosis. FASEB J. 2021, 35, e21339. [Google Scholar] [CrossRef]
- Adav, S.S.; Park, J.E.; Sze, S.K. Quantitative profiling brain proteomes revealed mitochondrial dysfunction in Alzheimer’s disease. Mol. Brain. 2019, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, L.M.; Vanderstichele, H.; Knapik-Czajka, M.; Clark, C.M.; Aisen, P.S.; Petersen, R.C.; Blennow, K.; Soares, H.; Simon, A.; Lewczuk, P.; et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann. Neurol. 2009, 65, 403–413. [Google Scholar] [CrossRef] [Green Version]
- Goetzl, E.J.; Kapogiannis, D.; Schwartz, J.B.; Lobach, I.V.; Goetzl, L.; Abner, E.L.; Jicha, G.A.; Karydas, A.M.; Boxer, A.; Miller, B.L. Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer’s disease. FASEB J. 2016, 30, 4141–4148. [Google Scholar] [CrossRef] [Green Version]
- D’Acunzo, P.; Perez-Gonzalez, R.; Kim, Y.; Hargash, T.; Miller, C.; Alldred, M.J.; Erdjument-Bromage, H.; Penikalapati, S.C.; Pawlik, M.; Saito, M.; et al. Mitovesicles are a novel population of extracellular vesicles of mitochondrial origin altered in Down syndrome. Sci. Adv. 2021, 7, eabe5085. [Google Scholar] [CrossRef]
- Vilcaes, A.A.; Chanaday, N.L.; Kavalali, E.T. Interneuronal exchange and functional integration of synaptobrevin via extracellular vesicles. Neuron 2021, 109, 971–983.e5. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Weidling, I.; Koppel, S.; Menta, B.; Perez Ortiz, J.; Kalani, A.; Wilkins, H.M.; Swerdlow, R.H. Detection of mitochondria-pertinent components in exosomes. Mitochondrion 2020, 55, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef] [Green Version]
- Kriebel, M.; Ebel, J.; Battke, F.; Griesbach, S.; Volkmer, H. Interference With Complex IV as a Model of Age-Related Decline in Synaptic Connectivity. Front. Mol. Neurosci. 2020, 13, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prokopenko, D.; Morgan, S.L.; Mullin, K.; Hofmann, O.; Chapman, B.; Kirchner, R.; Alzheimer’s Disease Neuroimaging, I.; Amberkar, S.; Wohlers, I.; Lange, C.; et al. Whole-genome sequencing reveals new Alzheimer’s disease-associated rare variants in loci related to synaptic function and neuronal development. Alzheimers Dement. 2021, 17, 1509–1527. [Google Scholar] [CrossRef] [PubMed]
- Mota, S.I.; Costa, R.O.; Ferreira, I.L.; Santana, I.; Caldeira, G.L.; Padovano, C.; Fonseca, A.C.; Baldeiras, I.; Cunha, C.; Letra, L.; et al. Oxidative stress involving changes in Nrf2 and ER stress in early stages of Alzheimer’s disease. Biochim. Biophys. Acta 2015, 1852, 1428–1441. [Google Scholar] [CrossRef] [Green Version]
- Omar, R.A.; Chyan, Y.J.; Andorn, A.C.; Poeggeler, B.; Robakis, N.K.; Pappolla, M.A. Increased Expression but Reduced Activity of Antioxidant Enzymes in Alzheimer’s Disease. J. Alzheimers Dis. 1999, 1, 139–145. [Google Scholar] [CrossRef]
- Butterfield, D.A. Perspectives on Oxidative Stress in Alzheimer’s Disease and Predictions of Future Research Emphases. J. Alzheimers Dis. 2018, 64, S469–S479. [Google Scholar] [CrossRef] [PubMed]
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging. Cell 2019, 18, e13031. [Google Scholar] [CrossRef] [Green Version]
- Leuner, K.; Schutt, T.; Kurz, C.; Eckert, S.H.; Schiller, C.; Occhipinti, A.; Mai, S.; Jendrach, M.; Eckert, G.P.; Kruse, S.E.; et al. Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid. Redox Signal. 2012, 16, 1421–1433. [Google Scholar] [CrossRef] [Green Version]
- Llanos-Gonzalez, E.; Henares-Chavarino, A.A.; Pedrero-Prieto, C.M.; Garcia-Carpintero, S.; Frontinan-Rubio, J.; Sancho-Bielsa, F.J.; Alcain, F.J.; Peinado, J.R.; Rabanal-Ruiz, Y.; Duran-Prado, M. Interplay Between Mitochondrial Oxidative Disorders and Proteostasis in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 1444. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Fujii, S. Extracellular ATP modulates synaptic plasticity induced by activation of metabotropic glutamate receptors in the hippocampus. Biomed. Res. 2015, 36, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Gabuzda, D.; Busciglio, J.; Chen, L.B.; Matsudaira, P.; Yankner, B.A. Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J. Biol. Chem. 1994, 269, 13623–13628. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Yoon, E.J.; Park, H.J.; Kim, G.Y.; Cho, H.M.; Choi, J.H.; Park, H.Y.; Jang, J.Y.; Rhim, H.S.; Kang, S.M. Intracellular amyloid beta interacts with SOD1 and impairs the enzymatic activity of SOD1: Implications for the pathogenesis of amyotrophic lateral sclerosis. Exp. Mol. Med. 2009, 41, 611–617. [Google Scholar] [CrossRef]
- Cavallaro, R.A.; Nicolia, V.; Fiorenza, M.T.; Scarpa, S.; Fuso, A. S-Adenosylmethionine and Superoxide Dismutase 1 Synergistically Counteract Alzheimer’s Disease Features Progression in TgCRND8 Mice. Antioxidants 2017, 6, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, K.; Murata, N.; Noda, Y.; Tahara, S.; Kaneko, T.; Kinoshita, N.; Hatsuta, H.; Murayama, S.; Barnham, K.J.; Irie, K.; et al. SOD1 (copper/zinc superoxide dismutase) deficiency drives amyloid beta protein oligomerization and memory loss in mouse model of Alzheimer disease. J. Biol. Chem. 2011, 286, 44557–44568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopeikina, K.J.; Carlson, G.A.; Pitstick, R.; Ludvigson, A.E.; Peters, A.; Luebke, J.I.; Koffie, R.M.; Frosch, M.P.; Hyman, B.T.; Spires-Jones, T.L. Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer’s disease brain. Am. J. Pathol. 2011, 179, 2071–2082. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef] [Green Version]
- Yao, P.J.; Eren, E.; Petralia, R.S.; Gu, J.W.; Wang, Y.X.; Kapogiannis, D. Mitochondrial Protrusions in Neuronal Cells. iScience 2020, 23, 101514. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Sun, J.; Zhang, W.; Yu, J.; Zhuang, C. Transcription factor NRF2 as a promising therapeutic target for Alzheimer’s disease. Free. Radic. Biol. Med. 2020, 159, 87–102. [Google Scholar] [CrossRef]
- Delaidelli, A.; Richner, M.; Jiang, L.; van der Laan, A.; Bergholdt Jul Christiansen, I.; Ferreira, N.; Nyengaard, J.R.; Vaegter, C.B.; Jensen, P.H.; Mackenzie, I.R.; et al. alpha-Synuclein pathology in Parkinson disease activates homeostatic NRF2 anti-oxidant response. Acta Neuropathol. Commun. 2021, 9, 105. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| First Cohort | Second Cohort | |||
|---|---|---|---|---|
| Control | AD | Control | AD | |
| Age | 73.4 ± 2.11 | 73.2 ± 1.65 | 73.1 ± 2.11 | 73.1 ± 2.36 |
| Sex (F/M) | 13/9 | 17/12 | 7/7 | 7/7 |
| MMSE | 28–30 | 23–26 | - | 18–30 |
| ADAS-Cog (21−23) | 0–5 | 10–16 | - | 5–25 |
| CDR | 0.0 | 0.5–1.0 | - | 0.5–1.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, P.J.; Eren, E.; Goetzl, E.J.; Kapogiannis, D. Mitochondrial Electron Transport Chain Protein Abnormalities Detected in Plasma Extracellular Vesicles in Alzheimer’s Disease. Biomedicines 2021, 9, 1587. https://doi.org/10.3390/biomedicines9111587
Yao PJ, Eren E, Goetzl EJ, Kapogiannis D. Mitochondrial Electron Transport Chain Protein Abnormalities Detected in Plasma Extracellular Vesicles in Alzheimer’s Disease. Biomedicines. 2021; 9(11):1587. https://doi.org/10.3390/biomedicines9111587
Chicago/Turabian StyleYao, Pamela J., Erden Eren, Edward J. Goetzl, and Dimitrios Kapogiannis. 2021. "Mitochondrial Electron Transport Chain Protein Abnormalities Detected in Plasma Extracellular Vesicles in Alzheimer’s Disease" Biomedicines 9, no. 11: 1587. https://doi.org/10.3390/biomedicines9111587