
Lipid Droplets, Phospholipase A2, Arachidonic Acid, and Atherosclerosis




Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
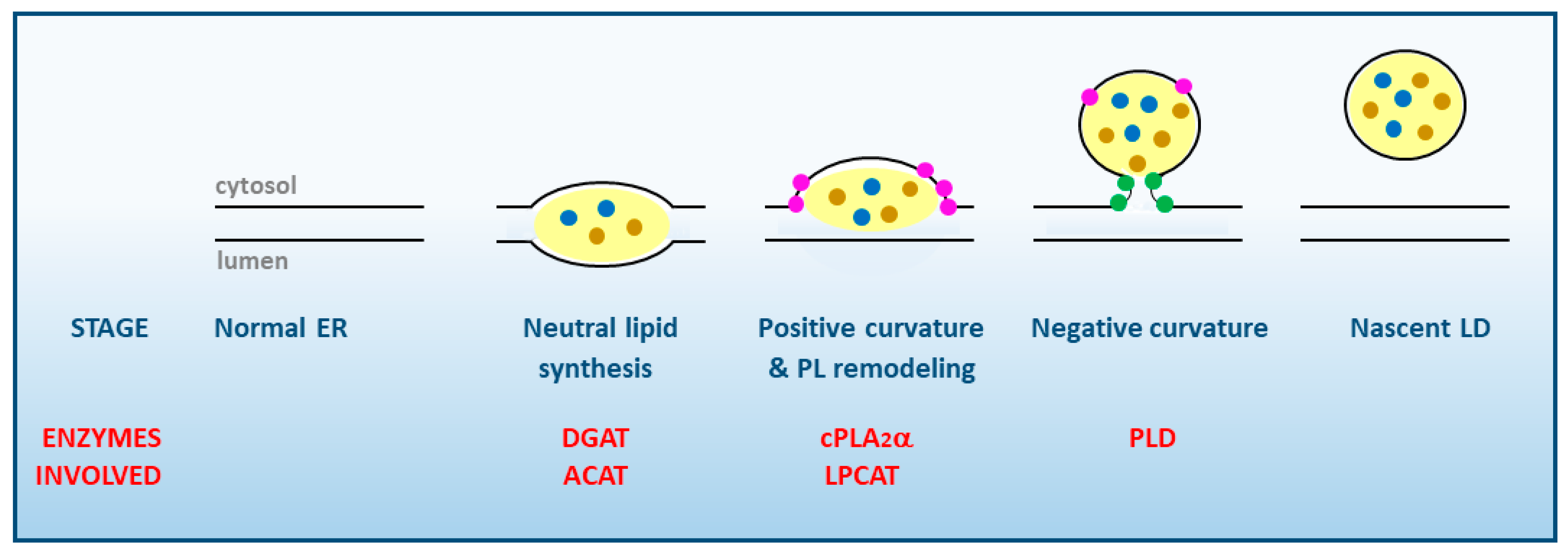
1. Lipid Droplet Biogenesis. General Aspects
2. Functional Diversity of Lipid Droplets
3. Lipid Droplets and Atherosclerosis
4. Circulating Foamy Monocytes
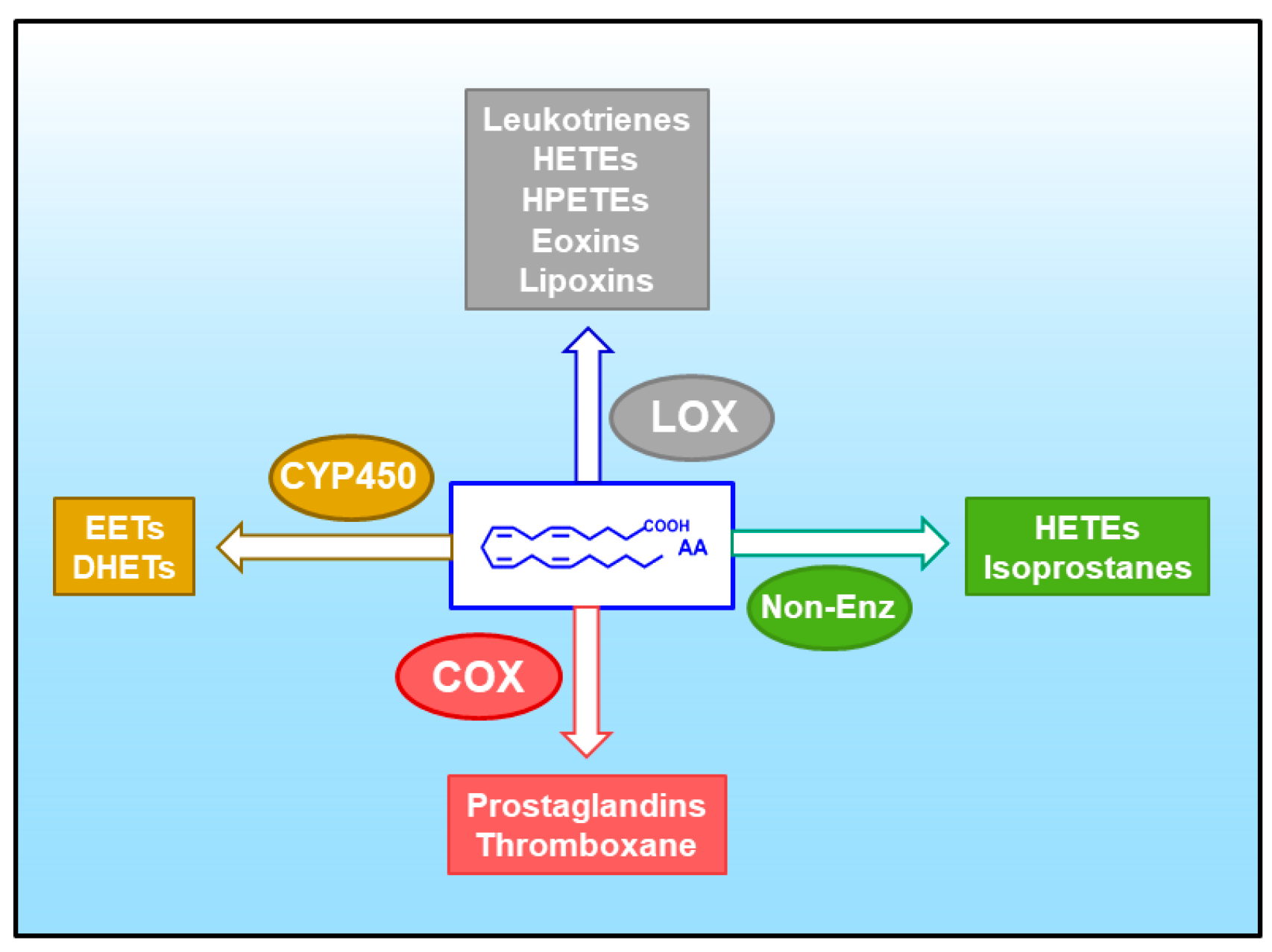
5. Arachidonic Acid, a Compound Released in Atherosclerotic Lesions
6. Glycerophospholipid Hydrolysis as a Major Pathway for the Mobilization of AA
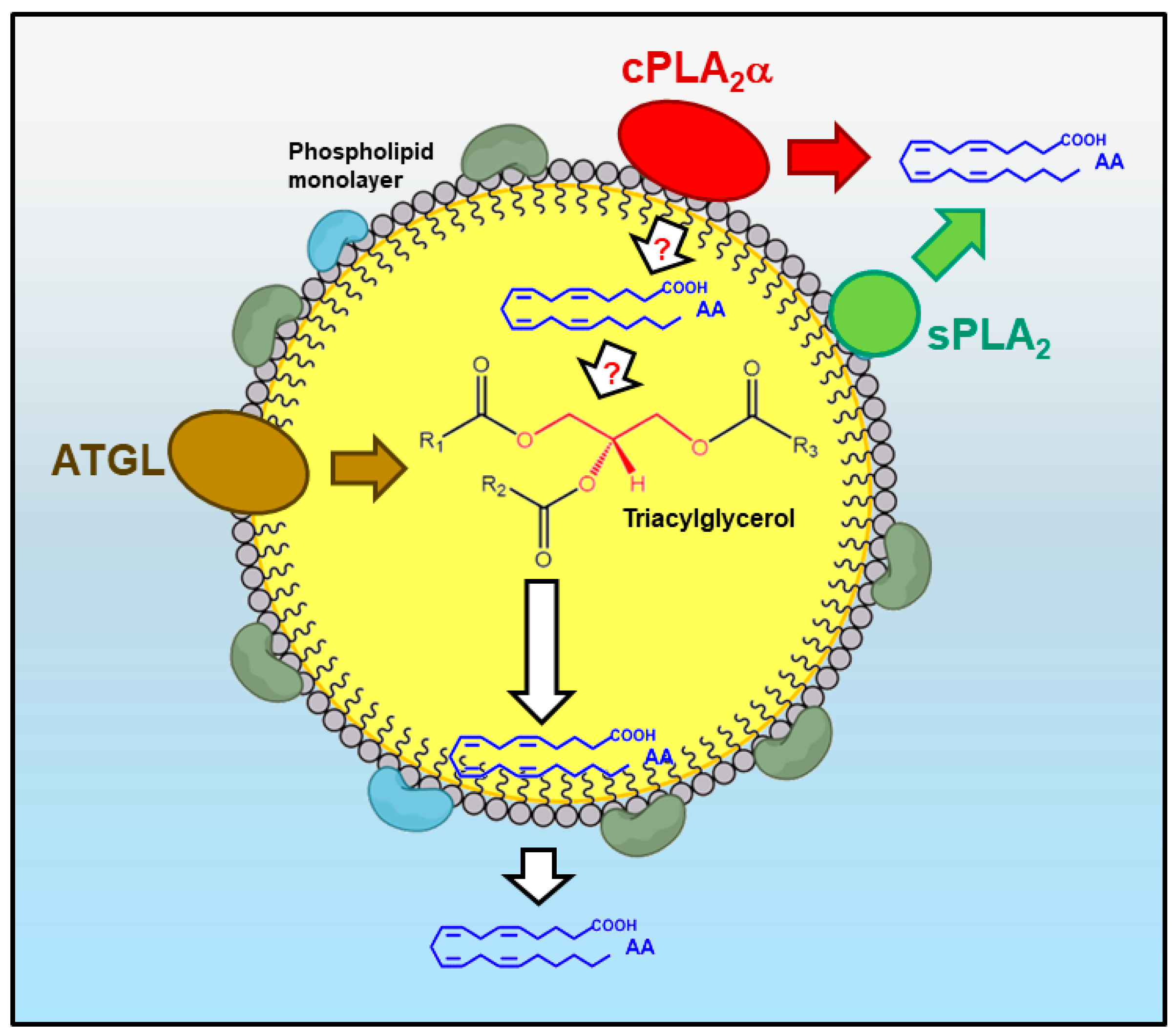
7. Lipid Droplets as a Source of Free AA for Lipid Mediator Synthesis
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AA | arachidonic acid |
| ATGL | triacylglycerol lipase |
| CE | cholesterol esters |
| ER | endoplasmic reticulum |
| LD | lipid droplet |
| LDL | low density lipoprotein |
| PA | phosphatidic acid |
| PC | choline-containing glycerophospholipids |
| PE | ethanolamine-containing glycerophospholipids |
| PI | phosphatidylinositol |
| PS | phosphatidylserine |
| PUFA | polyunsaturated fatty acid |
| PLA2 | phospholipase A2 |
| cPLA2α | group IVA cytosolic phospholipase A2α |
| cPLA2γ | group IVC cytosolic phospholipase A2γ |
| iPLA2β | group VIA calcium-independent phospholipase A2β |
| sPLA2 | secreted phospholipase A2 |
| TG | triacylglycerol |
References
- Olzmann, J.A.; Carvalho, P. Dynamics and Functions of Lipid Droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Grillitsch, K.; Connerth, M.; Köfeler, H.; Arrey, T.N.; Rietschel, B.; Wagner, B.; Karas, M.; Daum, G. Lipid Particles/Droplets of the Yeast Saccharomyces Cerevisiae Revisited: Lipidome Meets Proteome. Biochim. Biophys. Acta 2011, 1811, 1165–1176. [Google Scholar] [CrossRef] [Green Version]
- Thiam, A.R.; Farese, R.V., Jr.; Walther, T.C. The Biophysics and Cell Biology of Lipid Droplets. Nat. Rev. Mol. Cell Biol. 2013, 14, 775–786. [Google Scholar] [CrossRef] [Green Version]
- Brasaemle, D.L. The Perilipin Family of Structural Lipid Droplet Proteins: Stabilization of Lipid Droplets and Control of Lipolysis. J. Lipid Res. 2007, 48, 2547–2559. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Gong, J.; Wu, H.; Xu, W.; Wu, L.; Xu, D.; Gao, J.; Wu, J.W.; Yang, H.; Yang, M.; et al. Perilipin1 Promotes Unilocular Lipid Droplet Formation through the Activation of Fsp27 in Adipocytes. Nat. Commun. 2013, 4, 1594. [Google Scholar] [CrossRef] [Green Version]
- Mardani, I.; Dalen, K.I.; Drevinge, C.; Miljanovic, A.; Ståhlman, M.; Klevstig, M.; Täng, M.S.; Fogelstrand, P.; Levin, M.; Ekstrand, M.; et al. Plin2-Deficiency Reduces Lipophagy and Results in Increased Lipid Accumulation in the Heart. Sci. Rep. 2019, 9, 6909. [Google Scholar] [CrossRef] [Green Version]
- MacPherson, R.E.K.; Vandenboom, R.; Roy, B.D.; Peters, S.J. Skeletal Muscle PLIN3 and PLIN5 Are Serine Phosphorylated at Rest and Following Lipolysis during Adrenergic or Contractile Stimulation. Physiol. Rep. 2013, 1, e00084. [Google Scholar] [CrossRef]
- Čopič, A.; Antoine-Bally, S.; Giménez-Andrés, M.; La Torre Garay, C.; Antonny, B.; Manni, M.M.; Pagnotta, S.; Guihot, J.; Jackson, C.L. A Giant Amphipathic Helix from a Perilipin That Is Adapted for Coating Lipid Droplets. Nat. Commun. 2018, 9, 1332. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, P.B.M.; Krizanac, M.; Weiskirchen, R.; Asimakopoulos, A. Understanding the Role of Perilipin 5 in Non-Alcoholic Fatty Liver Disease and Its Role in Hepatocellular Carcinoma: A Review of Novel Insights. Int. J. Mol. Sci. 2021, 22, 5284. [Google Scholar] [CrossRef]
- Lass, A.; Zimmermann, R.; Oberer, M.; Zechner, R. Lipolysis-A Highly Regulated Multi-Enzyme Complex Mediates the Catabolism of Cellular Fat Stores. Prog. Lipid Res. 2011, 50, 14–27. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.J.; Patel, S.; Miyoshi, H.; Greenberg, A.S.; Kraemer, F.B. Functional Interaction of Hormone-Sensitive Lipase and Perilipin in Lipolysis. J. Lipid Res. 2009, 50, 2306–2313. [Google Scholar] [CrossRef] [Green Version]
- Krahmer, N.; Guo, Y.; Wilfling, F.; Hilger, M.; Lingrell, S.; Heger, K.; Newman, H.W.; Schmidt-Supprian, M.; Vance, D.E.; Mann, M.; et al. Phosphatidylcholine Synthesis for Lipid Droplet Expansion Is Mediated by Localized Activation of CTP:Phosphocholine Cytidylyltransferase. Cell Metab. 2011, 14, 504–515. [Google Scholar] [CrossRef] [Green Version]
- Aitchison, A.J.; Arsenault, D.J.; Ridgway, N.D. Nuclear-Localized CTP: Phosphocholine Cytidylyltransferase α Regulates Phosphatidylcholine Synthesis Required for Lipid Droplet Biogenesis. Mol. Biol. Cell 2015, 26, 2927–2938. [Google Scholar] [CrossRef]
- Moessinger, C.; Kuerschner, L.; Spandl, J.; Shevchenko, A.; Thiele, C. Human Lysophosphatidylcholine Acyltransferases 1 and 2 Are Located in Lipid Droplets Where They Catalyze the Formation of Phosphatidylcholine. J. Biol. Chem. 2011, 286, 21330–21339. [Google Scholar] [CrossRef] [Green Version]
- Poppelreuther, M.; Rudolph, B.; Du, C.; Grossmann, R.; Becker, M.; Thiele, C.; Ehehalt, R.; Füllekrug, J. The N-Terminal Region of Acyl-CoA Synthetase 3 Is Essential for Both the Localization on Lipid Droplets and the Function in Fatty Acid Uptake. J. Lipid Res. 2012, 53, 888–900. [Google Scholar] [CrossRef] [Green Version]
- Valdearcos, M.; Esquinas, E.; Meana, C.; Gil-de-Gómez, L.; Guijas, C.; Balsinde, J.; Balboa, M.A. Subcellular Localization and Role of Lipin-1 in Human Macrophages. J. Immunol. 2011, 186, 6004–6013. [Google Scholar] [CrossRef] [Green Version]
- Sembongi, H.; Miranda, M.; Han, G.S.; Fakas, S.; Grimsey, N.; Vendrell, J.; Carman, G.M.; Siniossoglou, S. Distinct Roles of the Phosphatidate Phosphatases Lipin 1 and 2 during Adipogenesis and Lipid Droplet Biogenesis in 3T3-L1 Cells. J. Biol. Chem. 2013, 288, 34502–34513. [Google Scholar] [CrossRef] [Green Version]
- Melo, R.C.N.; Weller, P.F. Unraveling the Complexity of Lipid Body Organelles in Human Eosinophils. J. Leukoc. Biol. 2014, 96, 703–712. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Bozza, P.T.; Tzizik, D.M.; Gray, J.P.; Cassara, J.; Dvorak, A.M.; Weller, P.F. Co-Compartmentalization of MAP Kinases and Cytosolic Phospholipase A2 at Cytoplasmic Arachidonate-Rich Lipid Bodies. Am. J. Pathol. 1998, 152, 759–769. [Google Scholar]
- Wooten, R.E.; Willingham, M.C.; Daniel, L.W.; Leslie, C.C.; Rogers, L.C.; Sergeant, S.; O’Flaherty, J.T. Novel Translocation Responses of Cytosolic Phospholipase A2α Fluorescent Proteins. Biochim. Biophys. Acta 2008, 1783, 1544–1550. [Google Scholar] [CrossRef] [Green Version]
- Jarc, E.; Petan, T. A Twist of FATe: Lipid Droplets and Inflammatory Lipid Mediators. Biochimie 2020, 169, 69–87. [Google Scholar] [CrossRef]
- Pérez-Chacón, G.; Astudillo, A.M.; Ruipérez, V.; Balboa, M.A.; Balsinde, J. Signaling Role for Lysophosphatidylcholine Acyltransferase 3 in Receptor-Regulated Arachidonic Acid Reacylation Reactions in Human Monocytes. J. Immunol. 2010, 184, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Arrese, E.L.; Saudale, F.Z.; Soulages, J.L. Lipid Droplets as Signaling Platforms Linking Metabolic and Cellular Functions. Lipid Insights 2014, 7, 7–16. [Google Scholar] [CrossRef]
- Guijas, C.; Rodríguez, J.P.; Rubio, J.M.; Balboa, M.A.; Balsinde, J. Phospholipase A2 Regulation of Lipid Droplet Formation. Biochim. Biophys. Acta 2014, 1841, 1661–1671. [Google Scholar] [CrossRef] [Green Version]
- Wilfling, F.; Wang, H.; Haas, J.T.; Krahmer, N.; Gould, T.J.; Uchida, A.; Cheng, J.X.; Graham, M.; Christiano, R.; Fröhlich, F.; et al. Triacylglycerol Synthesis Enzymes Mediate Lipid Droplet Growth by Relocalizing from the ER to Lipid Droplets. Dev. Cell 2013, 24, 384–399. [Google Scholar] [CrossRef] [Green Version]
- Jackson, C.L. Lipid Droplet Biogenesis. Curr. Opin. Cell Biol. 2018, 59, 88–96. [Google Scholar] [CrossRef]
- Coleman, R.A.; Lee, D.P. Enzymes of Triacylglycerol Synthesis and Their Regulation. Prog. Lipid Res. 2004, 43, 134–176. [Google Scholar] [CrossRef]
- Thiam, A.R.; Ikonen, E. Lipid Droplet Nucleation. Trends Cell Biol. 2021, 31, 108–118. [Google Scholar] [CrossRef]
- Pol, A.; Gross, S.P.; Parton, R.G. Biogenesis of the Multifunctional Lipid Droplet: Lipids, Proteins, and Sites. J. Cell Biol. 2014, 204, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Guijas, C.; Pérez-Chacón, G.; Astudillo, A.M.; Rubio, J.M.; Gil-de-Gómez, L.; Balboa, M.A.; Balsinde, J. Simultaneous Activation of p38 and JNK by Arachidonic Acid Stimulates the Cytosolic Phospholipase A2-Dependent Synthesis of Lipid Droplets in Human Monocytes. J. Lipid Res. 2012, 53, 2343–2354. [Google Scholar] [CrossRef] [Green Version]
- Gubern, A.; Casas, J.; Barceló-Torns, M.; Barneda, D.; de la Rosa, X.; Masgrau, R.; Picatoste, F.; Balsinde, J.; Balboa, M.A.; Claro, E. Group IVA Phospholipase A2 Is Necessary for the Biogenesis of Lipid Droplets. J. Biol. Chem. 2008, 283, 27369–27382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubern, A.; Barceló-Torns, M.; Casas, J.; Barneda, D.; Masgrau, R.; Picatoste, F.; Balsinde, J.; Balboa, M.A.; Claro, E. Lipid Droplet Biogenesis Induced by Stress Involves Triacylglycerol Synthesis that Depends on Group VIA Phospholipase A2. J. Biol. Chem. 2009, 284, 5697–5708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubern, A.; Barceló-Torns, M.; Barneda, D.; López, J.M.; Masgrau, R.; Picatoste, F.; Chalfant, C.E.; Balsinde, J.; Balboa, M.A.; Claro, E. JNK and Ceramide Kinase Govern the Biogenesis of Lipid Droplets through Activation of Group IVA Phospholipase A2. J. Biol. Chem. 2009, 284, 32359–32369. [Google Scholar] [CrossRef] [Green Version]
- Leslie, C.C. Cytosolic Phospholipase A2: Physiological Function and Role in Disease. J. Lipid Res. 2015, 56, 1386–1402. [Google Scholar] [CrossRef] [Green Version]
- Balsinde, J.; Winstead, M.V.; Dennis, E.A. Phospholipase A2 Regulation of Arachidonic Acid Mobilization. FEBS Lett. 2002, 531, 2–6. [Google Scholar] [CrossRef]
- Dennis, E.A.; Norris, P.C. Eicosanoid Storm in Infection and Inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Astudillo, A.M.; Balboa, M.A.; Balsinde, J. Selectivity of Phospholipid Hydrolysis by Phospholipase A2 Enzymes in Activated Cells Leading to Polyunsaturated Fatty Acid Mobilization. Biochim. Biophys. Acta 2019, 1864, 772–783. [Google Scholar] [CrossRef]
- Melo, R.C.N.; Weller, P. Lipid Droplets in Leukocytes: Organelles Linked to Inflammatory Responses. Exp. Cell. Res. 2016, 340, 193–197. [Google Scholar] [CrossRef] [Green Version]
- Bozza, P.T.; Bakker-Abreu, I.; Navarro-Xavier, R.A.; Bandeira-Melo, C. Lipid Body Function in Eicosanoid Synthesis: An Update. Prostaglandins Leukot. Essent. Fatty Acids 2011, 85, 205–213. [Google Scholar] [CrossRef]
- Kooijman, E.E.; Chupin, V.; Fuller, N.L.; Kozlov, M.M.; de Kruijff, B.; Burger, K.N.; Rand, P.R. Spontaneous Curvature of Phosphatidic Acid and Lysophosphatidic Acid. Biochemistry 2005, 44, 2097–2102. [Google Scholar] [CrossRef]
- Andersson, L.; Boström, P.; Ericson, J.; Rutberg, M.; Magnusson, B.; Marchesan, D.; Ruiz, M.; Asp, L.; Huang, P.; Frohman, M.A.; et al. PLD1 and ERK2 Regulate Cytosolic Lipid Droplet Formation. J. Cell Sci. 2006, 119, 2246–2257. [Google Scholar] [CrossRef] [Green Version]
- Balsinde, J.; Diez, E.; Fernández, B.; Mollinedo, F. Biochemical Characterization of Phospholipase D Activity from Human Neutrophils. Eur. J. Biochem. 1989, 186, 717–724. [Google Scholar] [CrossRef]
- Tan, J.S.; Seow, C.J.; Goh, V.J.; Silver, D.L. Recent Advances in Understanding Proteins Involved in Lipid Droplet Formation, Growth and Fusion. J. Genet. Genom. 2014, 41, 251–259. [Google Scholar] [CrossRef]
- Penno, A.; Hackenbroich, G.; Thiele, C. Phospholipids and Lipid Droplets. Biochim. Biophys. Acta 2013, 1831, 589–594. [Google Scholar] [CrossRef]
- Henne, M.; Goodman, J.M.; Hariri, H. Spatial Compartmentalization of Lipid Droplet Biogenesis. Biochim. Biophys. Acta 2020, 1865, 158499. [Google Scholar] [CrossRef]
- Nettebrock, N.T.; Bohnert, M. Born This Way-Biogenesis of Lipid Droplets from Specialized ER Subdomains. Biochim. Biophys. Acta 2020, 1865, 158448. [Google Scholar] [CrossRef]
- Cabodevilla, A.G.; Sánchez-Caballero, L.; Nintou, E.; Boiadjieva, V.G.; Picatoste, F.; Gubern, A.; Claro, E. Cell Survival during Complete Nutrient Deprivation Depends on Lipid Droplet-Fueled β-Oxidation of Fatty Acids. J. Biol. Chem. 2013, 288, 27777–27788. [Google Scholar] [CrossRef] [Green Version]
- Bosch, M.; Sánchez-Álvarez, M.; Fajardo, A.; Kapetanovic, R.; Steiner, B.; Dutra, F.; Moreira, L.; López, J.A.; Campo, R.; Marí, M.; et al. Mammalian Lipid Droplets Are Innate Immune Hubs Integrating Cell Metabolism and Host Defense. Science 2020, 370, eaay8085. [Google Scholar] [CrossRef]
- Vallochi, A.L.; Teixeira, L.; Oliveira, K.D.S.; Maya-Monteiro, C.M.; Bozza, P.T. Lipid Droplet, a Key Player in Host-Parasite Interactions. Front. Immunol. 2018, 9, 1022. [Google Scholar] [CrossRef]
- Monson, E.A.; Crosse, K.M.; Duan, M.; Chen, W.; O’Shea, R.D.; Wakim, L.M.; Carr, J.M.; Whelan, D.R.; Helbig, K.J. Intracellular Lipid Droplet Accumulation Occurs Early Following Viral Infection and Is Required for an Efficient Interferon Response. Nat. Commun. 2021, 12, 4303. [Google Scholar] [CrossRef]
- Wolins, N.E.; Quaynor, B.K.; Skinner, J.R.; Schoenfish, M.J.; Tzekov, A.; Bickel, P.E. S3–12, Adipophilin, and TIP47 Package Lipid in Adipocytes. J. Biol. Chem. 2005, 280, 19146–19155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiam, A.R.; Beller, M. The Why, When and How of Lipid Droplet Diversity. J. Cell Sci. 2017, 130, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Yavuz, A.; Wang, M.C. Dissecting Lipid Droplet Biology with Coherent Raman Scattering Microscopy. J. Cell Sci. 2022, 135, jcs252353. [Google Scholar] [CrossRef] [PubMed]
- Hodges, B.D.; Wu, C.C. Proteomic Insights into an Expanded Cellular Role for Cytoplasmic Lipid Droplets. J. Lipid Res. 2010, 51, 262–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herker, E.; Vieyres, G.; Beller, M.; Krahmer, N.; Bohnert, M. Lipid Droplet Contact Sites in Health and Disease. Trends Cell Biol. 2021, 31, 345–358. [Google Scholar] [CrossRef]
- Benador, I.Y.; Veliova, M.; Liesa, M.; Shirihai, O.S. Mitochondria Bound to Lipid Droplets: Where Mitochondrial Dynamics Regulate Lipid Storage and Utilization. Cell Metab. 2019, 29, 827–835. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, I.J.; Reue, K.; Abumrad, N.A.; Bickel, P.E.; Cohen, S.; Fisher, E.A.; Galis, Z.S.; Granneman, G.J.; Lewandowski, D.E.; Murphy, R.; et al. Deciphering the Role of Lipid Droplets in Cardiovascular Disease. Circulation 2018, 138, 305–315. [Google Scholar] [CrossRef]
- Pagidipati, N.J.; Gaziano, T.A. Estimating Deaths from Cardiovascular Disease: A Review of Global Methodologies of Mortality Measurement. Circulation 2013, 127, 749–756. [Google Scholar] [CrossRef]
- Tabas, I.; Williams, K.J.; Borén, J. Subendothelial Lipoprotein Retention as the Initiating Process in Atherosclerosis. Update and Therapeutic Implications. Circulation 2007, 116, 1832–1844. [Google Scholar] [CrossRef]
- Østerud, B.; Bjørklid, E. Role of Monocytes in Atherogenesis. Physiol. Rev. 2003, 83, 1069–1112. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.J.; Tabas, I. Macrophages in the Pathogenesis of Atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [Green Version]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodorou, K.; Boon, R.A. Endothelial Cell Metabolism in Atherosclerosis. Front. Cell Dev. Biol. 2018, 6, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredman, G.; Hellmann, J.; Proto, J.D.; Kuriakose, G.; Colas, R.A.; Dorweiler, B.; Connolly, E.S.; Solomon, R.; Jones, D.M.; Heyer, E.J.; et al. An Imbalance Between Specialized Pro-Resolving Lipid Mediators and Pro-Inflammatory Leukotrienes Promotes Instability of Atherosclerotic Plaques. Nat. Commun. 2017, 120, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Newby, A.C. Macrophage Heterogeneity in Atherosclerotic Plaques. Curr. Opin. Lipidol. 2009, 20, 370–378. [Google Scholar] [CrossRef] [PubMed]
- de Gaetano, M.; Crean, D.; Barry, M.; Belton, O. M1- and M2-Type Macrophage Responses Are Predictive of Adverse Outcomes in Human Atherosclerosis. Front. Immunol. 2016, 7, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, T.J. Macrophages in Atherosclerosis Regression. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.W.; Zaitsev, K.; Kim, K.W.; Ivanov, S.; Saunders, B.T.; Schrank, P.R.; Kim, K.; Elvington, A.; Kim, S.H.; Tucker, C.G.; et al. Limited Proliferation Capacity of Aortic Intima Resident Macrophages Requires Monocyte Recruitment for Atherosclerotic Plaque Progression. Nat. Immunol. 2020, 21, 1194–1204. [Google Scholar] [CrossRef]
- Tabas, I.; Bornfeldt, K.E. Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circ. Res. 2016, 118, 653–667. [Google Scholar] [CrossRef] [Green Version]
- Poznyak, A.V.; Nikiforov, N.G.; Starodubova, A.V.; Popkova, T.V.; Orekhov, A.N. Macrophages and Foam Cells: Brief Overview of Their Role, Linkage, and Targeting Potential in Atherosclerosis. Biomedicines 2021, 9, 1221. [Google Scholar] [CrossRef]
- Wu, H.; Gower, R.M.; Wang, H.; Perrard, X.Y.; Ma, R.; Bullard, D.C.; Burns, A.R.; Paul, A.; Smith, C.W.; Simon, S.I.; et al. Functional Role of CD11c+ Monocytes in Atherogenesis Associated with Hypercholesterolemia. Circulation 2009, 119, 2708–2717. [Google Scholar] [CrossRef] [Green Version]
- Foster, G.A.; Xu, L.; Chidambaram, A.A.; Soderberg, S.R.; Armstrong, E.J.; Wu, H.; Simon, S.I. CD11c/CD18 Signals very Late Antigen-4 Activation to Initiate Foamy Monocyte Recruitment during the Onset of Hypercholesterolemia. J. Immunol. 2015, 195, 5380–5392. [Google Scholar] [CrossRef] [Green Version]
- Robbins, C.S.; Hilgendorf, I.; Weber, G.F.; Theurl, I.; Iwamoto, Y.; Figueiredo, J.L.; Gorbatov, R.; Sukhova, G.K.; Gerhardt, L.M.; Smyth, D.; et al. Local Proliferation Dominates Lesional Macrophage Accumulation in Atherosclerosis. Nat. Med. 2013, 19, 1166–1172. [Google Scholar] [CrossRef]
- Xu, L.; Perrard, X.D.; Perrard, J.L.; Yang, D.; Xiao, X.; Teng, B.B.; Simon, S.I.; Ballantyne, C.M.; Wu, H. Foamy Monocytes form Early and Contribute to Nascent Atherosclerosis in Mice with Hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1787–1797. [Google Scholar] [CrossRef] [Green Version]
- Guijas, C.; Meana, C.; Astudillo, A.M.; Balboa, M.A.; Balsinde, J. Foamy Monocytes Are Enriched in Cis-7-Hexadecenoic Fatty Acid (16:1n-9), a Possible Biomarker for Early Detection of Cardiovascular Disease. Cell Chem. Biol. 2016, 23, 689–699. [Google Scholar] [CrossRef] [Green Version]
- Tertov, V.V.; Kalenich, O.S.; Orekhov, A.N. Lipid-Laden White Blood Cells in the Circulation of Patients with Coronary Heart Disease. Exp. Mol. Pathol. 1992, 57, 22–28. [Google Scholar] [CrossRef]
- Dresel, H.A.; Via, D.P.; Stöhr, M.; Elchner, U.; Gnasso, A.; Postiglione, A.; Blin, N.; Augustin, J.; Schettler, G. Observations on Leukocytes from Patients with Severe Familial Hypercholesterolemia. Arteriosclerosis 1986, 6, 259–264. [Google Scholar] [CrossRef] [Green Version]
- Varela, L.M.; Ortega, A.; Bermúdez, B.; López, S.; Pacheco, Y.M.; Villar, J.; Abia, R.; Muriana, F.J. A High-Fat Meal Promotes Lipid-Load and Apolipoprotein B-48 Receptor Transcriptional Activity in Circulating Monocytes. Am. J. Clin. Nutr. 2011, 93, 918–925. [Google Scholar] [CrossRef] [PubMed]
- den Hartigh, L.J.; Connolly-Rohrbach, J.E.; Fore, S.; Huser, T.R.; Rutledge, J.C. Fatty Acids from very Low-Density Lipoprotein Lipolysis Products Induce Lipid Droplet Accumulation in Human Monocytes. J. Immunol. 2010, 184, 3927–3936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Hartigh, L.J.; Altman, R.; Norman, J.E.; Rutledge, J.C. Postprandial VLDL Lipolysis Products Increase Monocyte Adhesion and Lipid Droplet Formation Via Activation of ERK2 and NFκB. Am. J. Physiol. 2014, 306, H109–H120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, S.; Jaramillo, S.; Varela, L.M.; Ortega, A.; Bermúdez, B.; Abia, R.; Muriana, F.J.G. MAPK Protects Human Monocytes from Postprandial Triglyceride-Rich Lipoprotein-Induced Toxicity. J. Nutr. 2013, 143, 620–626. [Google Scholar] [CrossRef] [Green Version]
- Alipour, A.; van Oostrom, A.J.; Izraeljan, A.; Verseyden, C.; Collins, J.M.; Frayn, K.N.; Plokker, T.W.; Elte, J.W.; Castro Cabezas, M. Leukocyte Activation by Triglyceride-Rich Lipoproteins. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Lian, Z.; Perrard, X.D.; Peng, X.; Raya, J.L.; Hernandez, A.A.; Johnson, C.G.; Lagor, W.R.; Pownall, H.J.; Hoogeveen, R.C.; Simon, S.I.; et al. Replacing Saturated Fat with Unsaturated Fat in Western Diet Reduces Foamy Monocytes and Atherosclerosis in Male Ldlr Mice. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Gower, R.M.; Wu, H.; Foster, G.A.; Devaraj, S.; Jialal, I.; Ballantyne, C.M.; Knowlton, A.A.; Simon, S.I. CD11c/CD18 Expression Is Upregulated on Blood Monocytes during Hypertriglyceridemia and Enhances Adhesion to Vascular Cell Adhesion Molecule-1. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 160–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, A.A.; Foster, G.A.; Soderberg, S.R.; Fernandez, A.; Reynolds, M.B.; Orser, M.K.; Bailey, K.A.; Rogers, J.H.; Singh, G.D.; Wu, H.; et al. An Allosteric Shift in CD11c Affinity Activates a Proatherogenic State in Arrested Intermediate Monocytes. J. Immunol. 2020, 205, 2806–2820. [Google Scholar] [CrossRef]
- Sprecher, H. Metabolism of Highly Unsaturated N-3 and N-6 Fatty Acids. Biochim. Biophys. Acta 2000, 1486, 219–231. [Google Scholar] [CrossRef]
- Pérez-Chacón, G.; Astudillo, A.M.; Balgoma, D.; Balboa, M.A.; Balsinde, J. Control of Free Arachidonic Acid Levels by Phospholipases A2 and Lysophospholipid Acyltransferases. Biochim. Biophys. Acta 2009, 1791, 1103–1113. [Google Scholar] [CrossRef] [Green Version]
- Astudillo, A.M.; Balgoma, D.; Balboa, M.A.; Balsinde, J. Dynamics of Arachidonic Acid Mobilization by Inflammatory Cells. Biochim. Biophys. Acta 2012, 1821, 249–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guijas, C.; Astudillo, A.M.; Gil-de-Gómez, L.; Rubio, J.M.; Balboa, M.A.; Balsinde, J. Phospholipid Sources for Adrenic Acid Mobilization in RAW 264.7 Macrophages: Comparison with Arachidonic Acid. Biochim. Biophys. Acta 2012, 1821, 1386–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monge, P.; Garrido, A.; Rubio, J.M.; Magrioti, V.; Kokotos, G.; Balboa, M.A.; Balsinde, J. The Contribution of Cytosolic Group IVA and Calcium-Independent Group VIA Phospholipase A2s to Adrenic Acid Mobilization in Murine Macrophages. Biomolecules 2020, 10, 542. [Google Scholar] [CrossRef] [Green Version]
- Brouwers, H.; Jónasdóttir, H.S.; Kuipers, M.E.; Kwekkeboom, J.C.; Auger, J.L.; González-Torres, M.; López-Vicario, C.; Clària, J.; Freysdottir, J.; Hardardottir, I.; et al. Anti-Inflammatory and Proresolving Effects of the Omega-6 Polyunsaturated Fatty Acid Adrenic Acid. J. Immunol. 2020, 205, 2840–2849. [Google Scholar] [CrossRef]
- Harkewicz, R.; Fahy, E.; Andreyev, A.; Dennis, E.A. Arachidonate-Derived Dihomoprostaglandin Production Observed in Endotoxin-Stimulated Macrophage-like Cells. J. Biol. Chem. 2007, 282, 2899–2910. [Google Scholar] [CrossRef] [Green Version]
- Sprecher, H.; Van Rollins, M.; Sun, F.; Wyche, A.; Needleman, P. Dihomo-Prostaglandins and -Thromboxane. A Prostaglandin Family from Adrenic Acid That May Be Preferentially Synthesized in the Kidney. J. Biol. Chem. 1982, 257, 3912–3918. [Google Scholar] [CrossRef]
- Kopf, P.G.; Zhang, D.X.; Gauthier, K.M.; Nithipatikom, K.; Yi, X.Y.; Falck, J.R.; Campbell, W.B. Adrenic Acid Metabolites as Endogenous Endothelium-Derived and Zona Glomerulosa-Derived Hyperpolarizing Factors. Hypertension 2010, 55, 547–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, R. Atherosclerosis—an Inflammatory Disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Wong, J.T.; Tran, K.; Pierce, G.N.; Chan, A.C.; O., K.; Choy, P.C. Lysophosphatidylcholine Stimulates the Release of Arachidonic Acid in Human Endothelial Cells. J. Biol. Chem. 1998, 273, 6830–6836. [Google Scholar] [CrossRef] [Green Version]
- Bogatcheva, N.V.; Sergeeva, M.G.; Dudek, S.M.; Verin, A.D. Arachidonic Acid Cascade in Endothelial Pathobiology. Microvasc. Res. 2005, 69, 107–127. [Google Scholar] [CrossRef]
- Badimon, L.; Vilahur, G.; Rocca, B.; Patrono, C. The Key Contribution of Platelet and Vascular Arachidonic Acid Metabolism to the Pathophysiology of Atherothrombosis. Cardiovasc. Res. 2021, 117, 2001–2015. [Google Scholar] [CrossRef]
- Hanasaki, K.; Yamada, K.; Yamamoto, S.; Ishimoto, Y.; Saiga, A.; Ono, T.; Ikeda, M.; Notoya, M.; Kamitani, S.; Arita, H. Potent Modification of Low Density Lipoprotein by Group X Secretory Phospholipase A2 Is Linked to Macrophage Foam Cell Formation. J. Biol. Chem. 2002, 277, 29116–29124. [Google Scholar] [CrossRef] [Green Version]
- Balsinde, J.; Balboa, M.A.; Yedgar, S.; Dennis, E.A. Group V Phospholipase A2-Mediated Oleic Acid Mobilization in Lipopolysaccharide-Stimulated P388D1 Macrophages. J. Biol. Chem. 2000, 275, 4783–4786. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Kato, R.; Isogai, Y.; Saka, G.; Ohtsuki, M.; Taketomi, Y.; Yamamoto, K.; Tsutsumi, K.; Yamada, J.; Masuda, S.; et al. Analyses of Group III Secreted Phospholipase A2 Transgenic Mice Reveal Potential Participation of This Enzyme in Plasma Lipoprotein Modification, Macrophage Foam Cell Formation, and Atherosclerosis. J. Biol. Chem. 2008, 283, 23483–33497. [Google Scholar] [CrossRef] [Green Version]
- Karabina, S.A.; Brochériou, I.; Le Naour, G.; Agrapart, M.; Durand, H.; Gelb, M.; Lambeau, G.; Ninio, E. Atherogenic Properties of LDL Particles Modified by Human Group X Secreted Phospholipase A2 on Human Endothelial Cell Function. FASEB J. 2006, 20, 2547–2549. [Google Scholar] [CrossRef]
- Tallima, H.; El Ridi, R. Arachidonic Acid: Physiological Roles and Potential Health Benefits–A Review. J. Adv. Res. 2018, 11, 33–41. [Google Scholar] [CrossRef]
- Guijas, C.; Bermúdez, M.A.; Meana, C.; Astudillo, A.M.; Pereira, L.; Fernández-Caballero, L.; Balboa, M.A.; Balsinde, J. Neutral Lipids Are Not a Source of Arachidonic Acid for Lipid Mediator Signaling in Human Foamy Monocytes. Cells 2019, 8, 941. [Google Scholar] [CrossRef] [Green Version]
- Astudillo, A.M.; Meana, C.; Guijas, C.; Pereira, L.; Lebrero, R.; Balboa, M.A.; Balsinde, J. Occurrence and Biological Activity of Palmitoleic Acid Isomers in Phagocytic Cells. J. Lipid Res. 2018, 59, 237–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astudillo, A.M.; Meana, C.; Bermúdez, M.A.; Pérez-Encabo, A.; Balboa, M.A.; Balsinde, J. Release of Anti-Inflammatory Palmitoleic Acid and Its Positional Isomers by Mouse Peritoneal Macrophages. Biomedicines 2020, 8, 480. [Google Scholar] [CrossRef] [PubMed]
- Scanferlato, R.; Bortolotti, M.; Sansone, A.; Chatgilialoglu, C.; Polito, L.; De Spirito, M.; Maulucci, G.; Bolognesi, A.; Ferreri, C. Hexadecenoic Fatty Acid Positional Isomers and De Novo PUFA Synthesis in Colon Cancer Cells. Int. J. Mol. Sci. 2019, 20, 832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aryal, P.; Syed, I.; Lee, J.; Patel, R.; Nelson, A.T.; Siegel, D.; Saghatelian, A.; Kahn, B.A. Distinct Biological Activities of Isomers from Several Families of Branched Fatty Acid Esters of Hydroxy Fatty Acids (FAHFAs). J. Lipid Res. 2021, 62, 100108. [Google Scholar] [CrossRef]
- Young, R.S.E.; Bowman, A.P.; Williams, E.D.; Tousignant, K.D.; Bidgood, C.L.; Narreddula, V.R.; Gupta, R.; Marshall, D.L.; Poad, B.L.J.; Nelson, C.C.; et al. Apocryphal FADS2 Activity Promotes Fatty Acid Diversification in Cancer. Cell Rep. 2021, 34, 108738. [Google Scholar] [CrossRef]
- Nomura, D.K.; Morrison, B.E.; Blankman, J.L.; Long, J.Z.; Kinsey, S.G.; Marcondes, M.C.; Ward, A.M.; Hahn, Y.K.; Lichtman, A.H.; Conti, B.; et al. Endocannabinoid Hydrolysis Generates Brain Prostaglandins That Promote Neuroinflammation. Science 2011, 334, 809–813. [Google Scholar] [CrossRef] [Green Version]
- Grabner, G.F.; Eichmann, T.O.; Wagner, B.; Gao, Y.; Farzi, A.; Taschler, U.; Radner, F.P.; Schweiger, M.; Lass, A.; Holzer, P.; et al. Deletion of Monoglyceride Lipase in Astrocytes Attenuates Lipopolysaccharide-Induced Neuroinflammation. J. Biol. Chem. 2016, 291, 913–923. [Google Scholar] [CrossRef] [Green Version]
- Schlager, S.; Vujic, N.; Korbelius, M.; Duta-Mare, M.; Dorow, J.; Leopold, C.; Rainer, S.; Wegscheider, M.; Reicher, H.; Ceglarek, U.; et al. Lysosomal Lipid Hydrolysis Provides Substrates for Lipid Mediator Synthesis in Murine Macrophages. Oncotarget 2017, 8, 40037–40051. [Google Scholar] [CrossRef] [Green Version]
- Dichlberger, A.; Schlager, S.; Maaninka, K.; Schneider, W.J.; Kovanen, P.T. Adipose Triglyceride Lipase Regulates Eicosanoid Production in Activated Human Mast Cells. J. Lipid Res. 2014, 55, 2471–2478. [Google Scholar] [CrossRef] [Green Version]
- Schlager, S.; Goeritzer, M.; Jandl, K.; Frei, R.; Vujic, N.; Kolb, D.; Strohmaier, H.; Dorow, J.; Eichmann, T.O.; Rosenberger, A.; et al. Adipose Triglyceride Lipase Acts on Neutrophil Lipid Droplets to Regulate Substrate Availability for Lipid Mediator Synthesis. J. Leukoc. Biol. 2015, 98, 837–850. [Google Scholar] [CrossRef] [Green Version]
- Dennis, E.A.; Cao, J.; Hsu, Y.H.; Magrioti, V.; Kokotos, G. Phospholipase A2 Enzymes: Physical Structure, Biological Function, Disease Implication, Chemical Inhibition, and Therapeutic Intervention. Chem. Rev. 2011, 111, 6130–6185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, M. Novel Functions of Phospholipase A2s: Overview. Biochim. Biophys. Acta 2019, 1864, 763–765. [Google Scholar]
- Mouchlis, V.D.; Dennis, E.A. Phospholipase A2 Catalysis and Lipid Mediator Lipidomics. Biochim. Biophys. Acta 2019, 1864, 766–771. [Google Scholar] [CrossRef] [PubMed]
- Kita, Y.; Shindou, H.; Shimizu, T. Cytosolic Phospholipase A2 and Lysophospholipid Acyltransferases. Biochim. Biophys. Acta 2019, 1864, 838–845. [Google Scholar] [CrossRef]
- Turk, J.; White, T.D.; Nelson, A.J.; Lei, X.; Ramanadham, S. iPLA2β and Its Role in Male Fertility, Neurological Disorders, Metabolic Disorders, and Inflammation. Biochim. Biophys. Acta 2019, 1864, 846–860. [Google Scholar]
- Murakami, M.; Sato, H.; Miki, Y.; Yamamoto, K.; Taketomi, Y. A New Era of Secreted Phospholipase A2. J. Lipid Res. 2015, 56, 1248–1261. [Google Scholar] [CrossRef] [Green Version]
- Murakami, M.; Sato, H.; Taketomi, Y. Updating Phospholipase A2 Biology. Biomolecules 2020, 10, 1457. [Google Scholar] [CrossRef] [PubMed]
- White, T.D.; Almutairi, A.; Tusing, Y.G.; Lei, X.; Ramanadham, S. The Impact of the Ca2+-Independent Phospholipase A2β on Immune Cells. Biomolecules 2021, 11, 577. [Google Scholar] [CrossRef] [PubMed]
- Dabral, D.; van den Bogaart, G. The Roles of Phospholipase A2 in Phagocytes. Front. Cell Dev. Biol. 2021, 9, 673502. [Google Scholar] [CrossRef]
- Peng, Z.; Chang, Y.; Fan, J.; Ji, W.; Su, C. Phospholipase A2 Superfamily in Cancer. Cancer Lett. 2021, 497, 165–177. [Google Scholar] [CrossRef]
- Sun, G.Y.; Geng, X.; Teng, T.; Yang, B.; Appenteng, M.K.; Greenlief, C.M. Dynamic Role of Phospholipases A2 in Health and Diseases in the Central Nervous System. Cells 2021, 10, 2963. [Google Scholar] [CrossRef]
- Pindado, J.; Balsinde, J.; Balboa, M.A. TLR3-Dependent Induction of Nitric Oxide Synthase in RAW 264.7 Macrophage-like Cells Via a Cytosolic Phospholipase A2/Cyclooxygenase-2 Pathway. J. Immunol. 2007, 179, 4821–4828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astudillo, A.M.; Rodríguez, J.P.; Guijas, C.; Rubio, J.M.; Balboa, M.A.; Balsinde, J. Choline Glycerophospholipid-Derived Prostaglandins Attenuate TNFα Gene Expression in Macrophages Via a cPLA2α/COX-1 Pathway. Cells 2021, 10, 447. [Google Scholar] [CrossRef] [PubMed]
- Mouchlis, V.D.; Chen, Y.; McCammon, J.A.; Dennis, E.A. Membrane Allostery and Unique Hydrophobic Sites Promote Enzyme Substrate Specificity. J. Am. Chem. Soc. 2018, 140, 3285–3291. [Google Scholar] [CrossRef] [Green Version]
- Gil-de-Gómez, L.; Astudillo, A.M.; Guijas, C.; Magrioti, V.; Kokotos, G.; Balboa, M.A.; Balsinde, J. Cytosolic Group IVA and Calcium-Independent Group VIA Phospholipase A2s Act on Distinct Phospholipid Pools in Zymosan-Stimulated Mouse Peritoneal Macrophages. J. Immunol. 2014, 192, 752–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebrero, P.; Astudillo, A.M.; Rubio, J.M.; Fernández-Caballero, J.; Kokotos, G.; Balboa, M.A.; Balsinde, J. Cellular Plasmalogen Content Does Not Influence Arachidonic Acid Levels or Distribution in Macrophages: A Role for Cytosolic Phospholipase A2γ in Phospholipid Remodeling. Cells 2019, 8, 799. [Google Scholar] [CrossRef] [Green Version]
- Gil-de-Gómez, L.; Monge, P.; Rodríguez, J.P.; Astudillo, A.M.; Balboa, M.A.; Balsinde, J. Phospholipid Arachidonic acid Remodeling during Phagocytosis in Mouse Peritoneal Macrophages. Biomedicines 2020, 8, 274. [Google Scholar] [CrossRef]
- Murakami, M.; Lambeau, G. Emerging Roles of Secreted Phospholipase A2 Enzymes: An Update. Biochimie 2013, 95, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Samuchiwal, S.K.; Balestrieri, B. Harmful and Protective Roles of Group V Phospholipase A2: Current Perspectives and Future Directions. Biochim. Biophys. Acta 2019, 1864, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Balboa, M.A.; Shirai, Y.; Gaietta, G.; Ellisman, M.H.; Balsinde, J.; Dennis, E.A. Localization of Group V Phospholipase A2 in Caveolin-Enriched Granules in Activated P388D1 Macrophage-like Cells. J. Biol. Chem. 2003, 278, 48059–48065. [Google Scholar] [CrossRef] [Green Version]
- Bingham, C.O.; Fijneman, R.J.; Friend, D.S.; Goddeau, R.P.; Rogers, R.A.; Austen, K.F.; Arm, J.P. Low Molecular Weight Group IIA and Group V Phospholipase A2 Enzymes Have Different Intracellular Locations in Mouse Bone Marrow-Derived Mast Cells. J. Biol. Chem. 1999, 274, 31476–31484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruipérez, V.; Astudillo, M.A.; Balboa, M.A.; Balsinde, J. Coordinate Regulation of TLR-Mediated Arachidonic Acid Mobilization in Macrophages by Group IVA and Group V Phospholipase A2s. J. Immunol. 2009, 182, 3877–3883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balboa, M.A.; Pérez, R.; Balsinde, J. Amplification Mechanisms of Inflammation: Paracrine Stimulation of Arachidonic Acid Mobilization by Secreted Phospholipase A2 Is Regulated by Cytosolic Phospholipase A2-Derived Hydroperoxyeicosatetraenoic Acid. J. Immunol. 2003, 171, 989–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikawada, E.; Bonventre, J.V.; Arm, J.P. Group V Secretory PLA2 Regulates TLR2-Dependent Eicosanoid Generation in Mouse Mast Cells through Amplification of ERK and cPLA2α Activation. Blood 2007, 110, 561–567. [Google Scholar] [CrossRef]
- Murakami, M.; Miki, Y.; Sato, H.; Murase, R.; Taketomi, Y.; Yamamoto, K. Group IID, IIE, IIF and III Secreted Phospholipase A2s. Biochim. Biophys. Acta 2019, 1864, 803–818. [Google Scholar] [CrossRef]
- Miki, Y.; Yamamoto, K.; Taketomi, Y.; Sato, H.; Shimo, K.; Kobayashi, T.; Ishikawa, Y.; Ishii, T.; Nakanishi, H.; Ikeda, K.; et al. Lymphoid Tissue Phospholipase A2 Group IID Resolves Contact Hypersensitivity by Driving Antiinflammatory Lipid Mediators. J. Exp. Med. 2013, 210, 1217–1234. [Google Scholar] [CrossRef] [Green Version]
- Ait-Oufella, H.; Herbin, O.; Lahoute, C.; Coatrieux, C.; Loyer, X.; Joffre, J.; Laurans, L.; Ramkhelawon, B.; Blanc-Brude, O.; Karabina, S.; et al. Group X Secreted Phospholipase A2 Limits the Development of Atherosclerosis in LDL Receptor-Null Mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 466–473. [Google Scholar] [CrossRef] [Green Version]
- Murase, R.; Sato, H.; Yamamoto, K.; Ushida, A.; Nishito, Y.; Ikeda, K.; Kobayashi, T.; Yamamoto, T.; Taketomi, Y.; Murakami, M. Group X Secreted Phospholipase A2 Releases ω3 Polyunsaturated Fatty Acids, Suppresses Colitis, and Promotes Sperm Fertility. J. Biol. Chem. 2016, 291, 6895–6911. [Google Scholar] [CrossRef] [Green Version]
- Murphy, R.C.; Folco, G. Lysophospholipid Acyltransferases and Leukotriene Biosynthesis: Intersection of the Lands Cycle and the Arachidonate PI Cycle. J. Lipid Res. 2019, 60, 219–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patton-Vogt, J.; de Kroon, A.I.P.M. Phospholipid Turnover and Acyl Chain Remodeling in the Yeast ER. Biochim. Biophys. Acta 2020, 1865, 158462. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Hayashi, Y.; Nemoto-Sasaki, Y.; Ito, M.; Oka, S.; Tanikawa, T.; Waku, K.; Sugiura, T. Acyltransferases and Transacylases That Determine the Fatty Acid Composition of Glycerolipids and the Metabolism of Bioactive Lipid Mediators in Mammalian Cells and Model Organisms. Prog. Lipid Res. 2014, 53, 18–81. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Hayashi, Y.; Matsumoto, N.; Nemoto-Sasaki, Y.; Koizumi, T.; Inagaki, Y.; Oka, S.; Tanikawa, T.; Sugiura, T. Coenzyme-A-Independent Transacylation System; Possible Involvement of Phospholipase A2 in Transacylation. Biology 2017, 6, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, M.; Tucker, D.E.; Burchett, S.A.; Leslie, C.C. Properties of the Group IV Phospholipase A2 Family. Prog. Lipid Res. 2006, 45, 487–510. [Google Scholar] [CrossRef]
- Gil-de-Gómez, L.; Astudillo, A.M.; Lebrero, P.; Balboa, M.A.; Balsinde, J. Essential Role for Ethanolamine Plasmalogen Hydrolysis in Bacterial Lipopolysaccharide Priming of Macrophages for Enhanced Arachidonic Acid Release. Front. Immunol. 2017, 8, 1251. [Google Scholar] [CrossRef]
- Rubio, J.M.; Astudillo, A.M.; Casas, J.; Balboa, M.A.; Balsinde, J. Regulation of Phagocytosis in Macrophages by Membrane Ethanolamine Plasmalogens. Front. Immunol. 2018, 9, 1723. [Google Scholar] [CrossRef]
- Pereira-Dutra, F.S.; Teixeira, J.; de Souza Costa, M.F.; Bozza, P.T. Fat, Fight, and Beyond: The Multiple Roles of Lipid Droplets in Infections and Inflammation. J. Leukoc. Biol. 2019, 106, 563–580. [Google Scholar] [CrossRef]
- Pereira-Dutra, F.S.; Bozza, P.T. Lipid Droplets Diversity and Functions in Inflammation and Immune Response. Expert. Rev. Proteom. 2021, 18, 809–825. [Google Scholar] [CrossRef] [PubMed]
- Boucher, D.M.; Vijithakumar, V.; Ouimet, M. Lipid Droplets as Regulators of Metabolism and Immunity. Immunometabolism 2021, 3, e210021. [Google Scholar]
- Castoldi, A.; Monteiro, L.B.; Bakker, N.T.; Sanin, D.E.; Rana, N.; Corrado, M.; Cameron, A.M.; Hässler, F.; Matsushita, M.; Caputa, G.; et al. Triacylglycerol Synthesis Enhances Macrophage Inflammatory Function. Nat. Commun. 2020, 11, 4107. [Google Scholar] [CrossRef]
- Dichlberger, A.; Schlager, S.; Kovanen, P.T.; Schneider, W.J. Lipid Droplets in Activated Mast Cells—A Significant Source of Triglyceride-Derived Arachidonic Acid for Eicosanoid Production. Eur. J. Pharmacol. 2016, 785, 59–69. [Google Scholar] [CrossRef]
- Hayashi, D.; Mouchlis, V.D.; Dennis, E.A. Omega-3 Versus Omega-6 Fatty Acid Availability Is Controlled by Hydrophobic Site Geometries of Phospholipase A2s. J. Lipid Res. 2021, 62, 100113. [Google Scholar] [CrossRef] [PubMed]
- Zizza, P.; Iurisci, C.; Bonazzi, M.; Cossart, P.; Leslie, C.C.; Corda, D.; Mariggio, S. Phospholipase A2 IVa Regulates Phagocytosis Independent of Its Enzymatic Activity. J. Biol. Chem. 2012, 287, 16849–16859. [Google Scholar] [CrossRef] [Green Version]
- Casas, J.; Meana, C.; Esquinas, E.; Valdearcos, M.; Pindado, J.; Balsinde, J.; Balboa, M.A. Requirement of JNK-Mediated Phosphorylation for Translocation of Group IVA Phospholipase A2 to Phagosomes in Human Macrophages. J. Immunol. 2009, 183, 2767–2774. [Google Scholar] [CrossRef] [Green Version]
- San Pietro, E.; Capestrano, M.; Polishchuk, E.V.; DiPentima, A.; Trucco, A.; Zizza, P.; Mariggiò, S.; Pulvirenti, T.; Sallese, M.; Tete, S.; et al. Group IV Phospholipase A2α Controls the Formation of Inter-Cisternal Continuities Involved in Intra-Golgi Transport. PLoS Biol. 2009, 7, e1000194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balboa, M.A.; Balsinde, J.; Dillon, D.A.; Carman, G.M.; Dennis, E.A. Proinflammatory Macrophage-Activating Properties of the Novel Phospholipid Diacylglycerol Pyrophosphate. J. Biol. Chem. 1999, 274, 522–526. [Google Scholar] [CrossRef] [Green Version]
- Oörni, K.; Kovanen, P.T. Lipoprotein Modification by Secretory Phospholipase A2 Enzymes Contributes to the Initiation and Progression of Atherosclerosis. Curr. Opin. Lipidol. 2009, 20, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Triggiani, M.; Oriente, A.; Seeds, M.C.; Bass, D.A.; Marone, G.; Chilton, F.H. Migration of Human Inflammatory Cells into the Lung Results in the Remodeling of Arachidonic Acid into a Triglyceride Pool. J. Exp. Med. 1995, 182, 1181–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.M.; Vaughn, B.; Triggiani, M.; Swan, D.D.; Fonteh, A.N.; Chilton, F.H. Role of Arachidonyl Triglycerides within Lipid Bodies in Eicosanoid Formation by Human Polymorphonuclear Cells. Am. J. Respir. Cell Mol. Biol. 1999, 21, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Pompeia, C.; Lima, T.; Curi, R. Arachidonic Acid Cytotoxicity: Can Arachidonic Acid Be a Physiological Mediator of Cell Death? Cell Biochem. Funct. 2003, 21, 97–104. [Google Scholar] [CrossRef]
- Blank, M.L.; Smith, Z.L.; Snyder, F. Arachidonate-Containing Triacylglycerols: Biosynthesis and a Lipolytic Mechanism for the Release and Transfer of Arachidonate to Phospholipids in HL-60 Cells. Biochim. Biophys. Acta 1993, 1170, 275–282. [Google Scholar] [CrossRef]
- Schreiber, R.; Zechner, R. Lipolysis Meets Inflammation: Arachidonic Acid Mobilization from Fat. J. Lipid Res. 2014, 55, 2447–2449. [Google Scholar] [CrossRef] [Green Version]
- Sonnweber, T.; Pizzini, A.; Nairz, M.; Weiss, G.; Tancevski, I. Arachidonic Acid Metabolites in Cardiovascular and Metabolic Diseases. Int. J. Mol. Sci. 2018, 19, 3285. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bermúdez, M.A.; Balboa, M.A.; Balsinde, J. Lipid Droplets, Phospholipase A2, Arachidonic Acid, and Atherosclerosis. Biomedicines 2021, 9, 1891. https://doi.org/10.3390/biomedicines9121891
Bermúdez MA, Balboa MA, Balsinde J. Lipid Droplets, Phospholipase A2, Arachidonic Acid, and Atherosclerosis. Biomedicines. 2021; 9(12):1891. https://doi.org/10.3390/biomedicines9121891
Chicago/Turabian StyleBermúdez, Miguel A., María A. Balboa, and Jesús Balsinde. 2021. "Lipid Droplets, Phospholipase A2, Arachidonic Acid, and Atherosclerosis" Biomedicines 9, no. 12: 1891. https://doi.org/10.3390/biomedicines9121891
APA StyleBermúdez, M. A., Balboa, M. A., & Balsinde, J. (2021). Lipid Droplets, Phospholipase A2, Arachidonic Acid, and Atherosclerosis. Biomedicines, 9(12), 1891. https://doi.org/10.3390/biomedicines9121891