
Novel Ex Vivo Model to Examine the Mechanism and Relationship of Esophageal Microbiota and Disease

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Esophageal Microbiome
2.1.1. Study Participants
2.1.2. Clinical Characteristics
2.1.3. Endoscopy
2.1.4. DNA Extraction
2.1.5. Next Generation Sequencing
2.1.6. Bioinformatics for Ion Torrent
2.1.7. qPCR Evaluation by EMB
2.1.8. Statistical Analysis
2.2. Transcriptome Profile
2.2.1. Study Participants and Sample Collection
2.2.2. RNA Isolation
2.2.3. Transcriptome Analysis
2.3. Ex Vivo Model
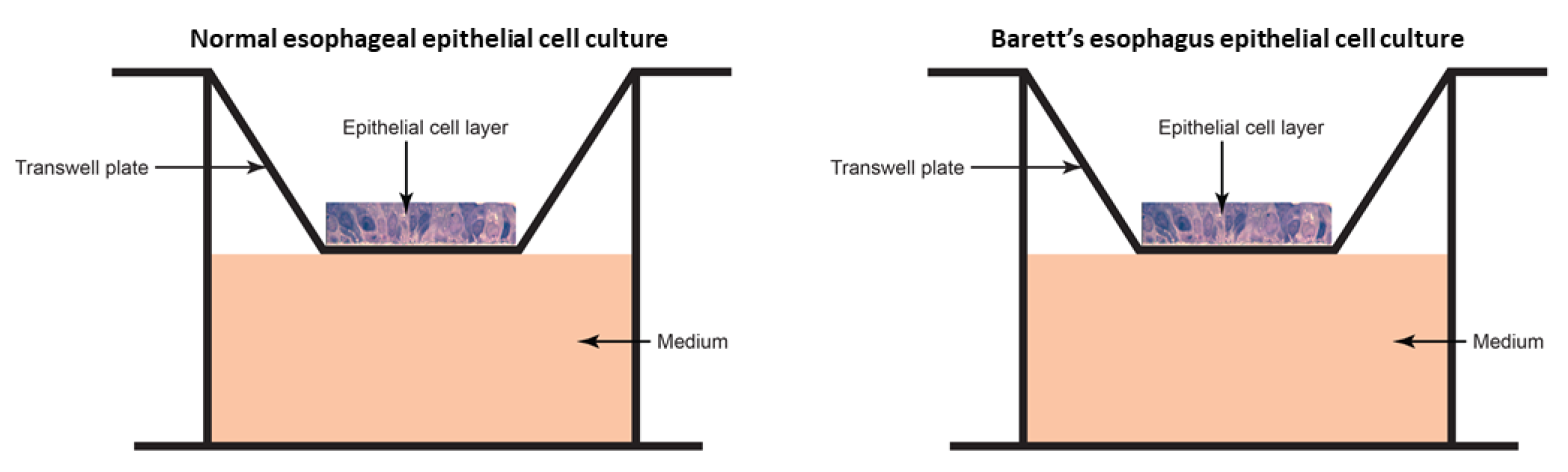
2.3.1. Epithelial Cell Culture
2.3.2. Microbiome Transplant
3. Results
3.1. Esophageal Microbiome
3.1.1. Demographics
3.1.2. Microbiota Detection Patterns in Patients with and without Barrett’s Esophagus
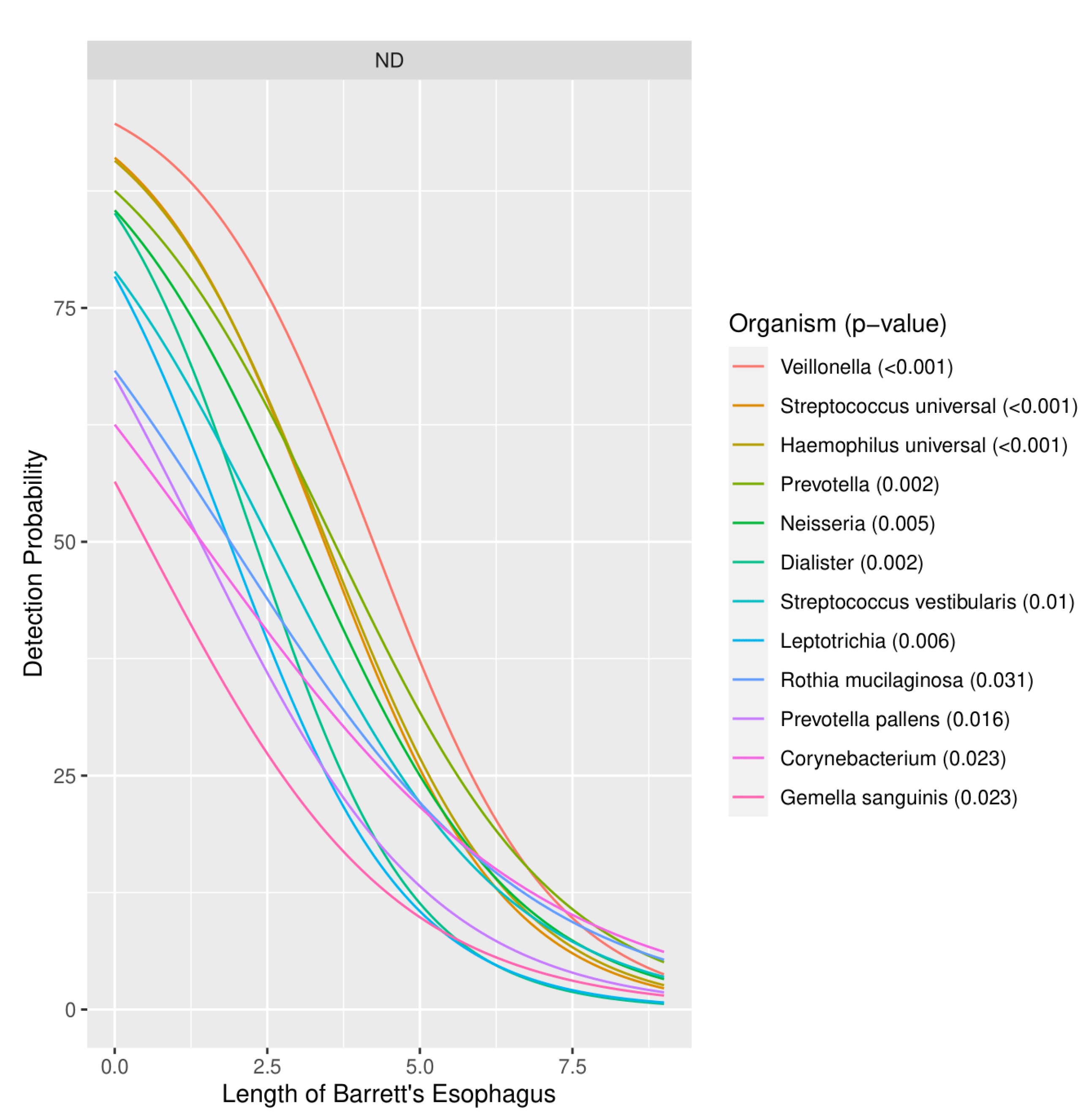
3.1.3. Severity of Barrett’s Esophagus versus Microbiome Pattern
3.2. Transcriptome Profile
3.2.1. Demographics
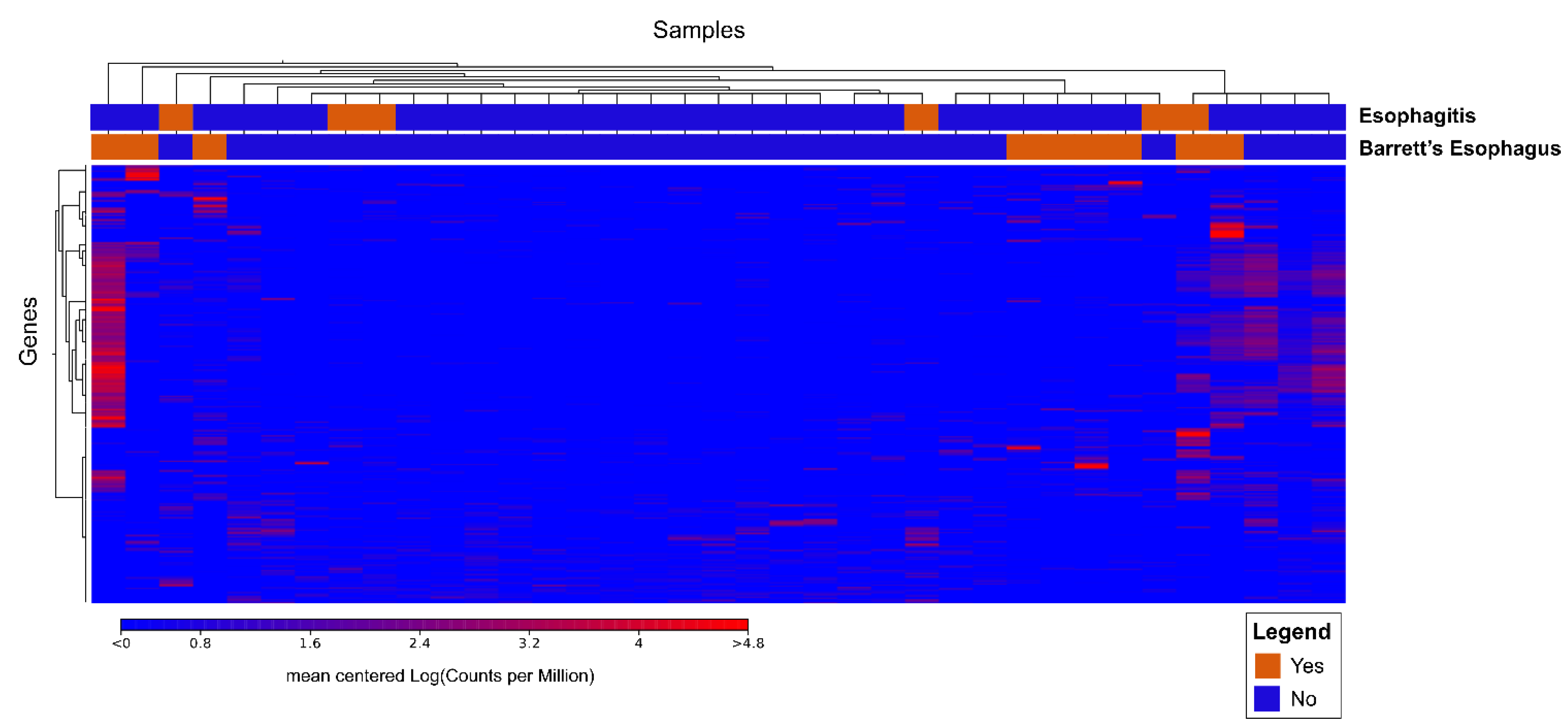
3.2.2. Cluster Analysis
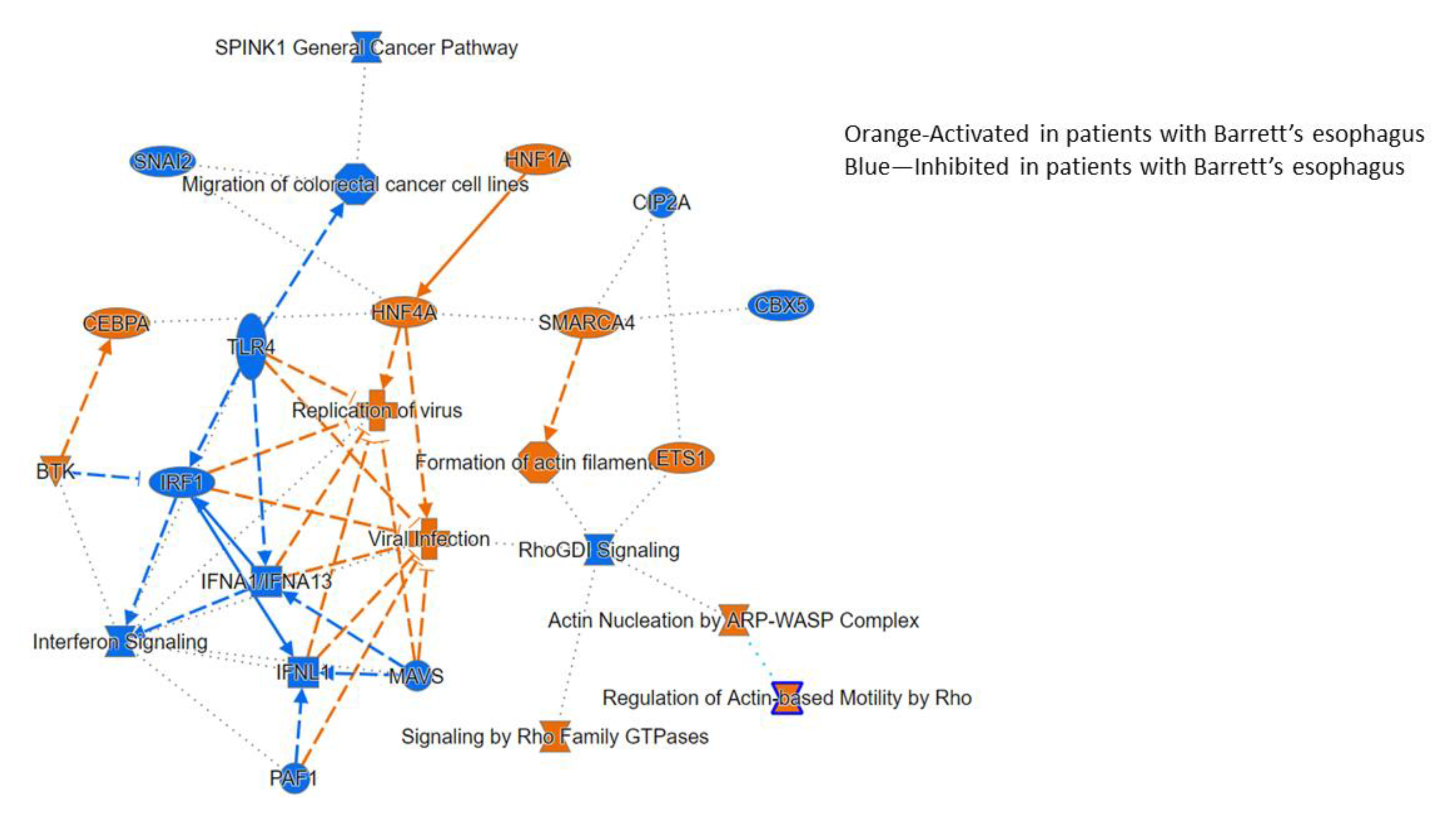
3.2.3. Ingenuity Pathway Analysis
3.3. Ex Vivo Model
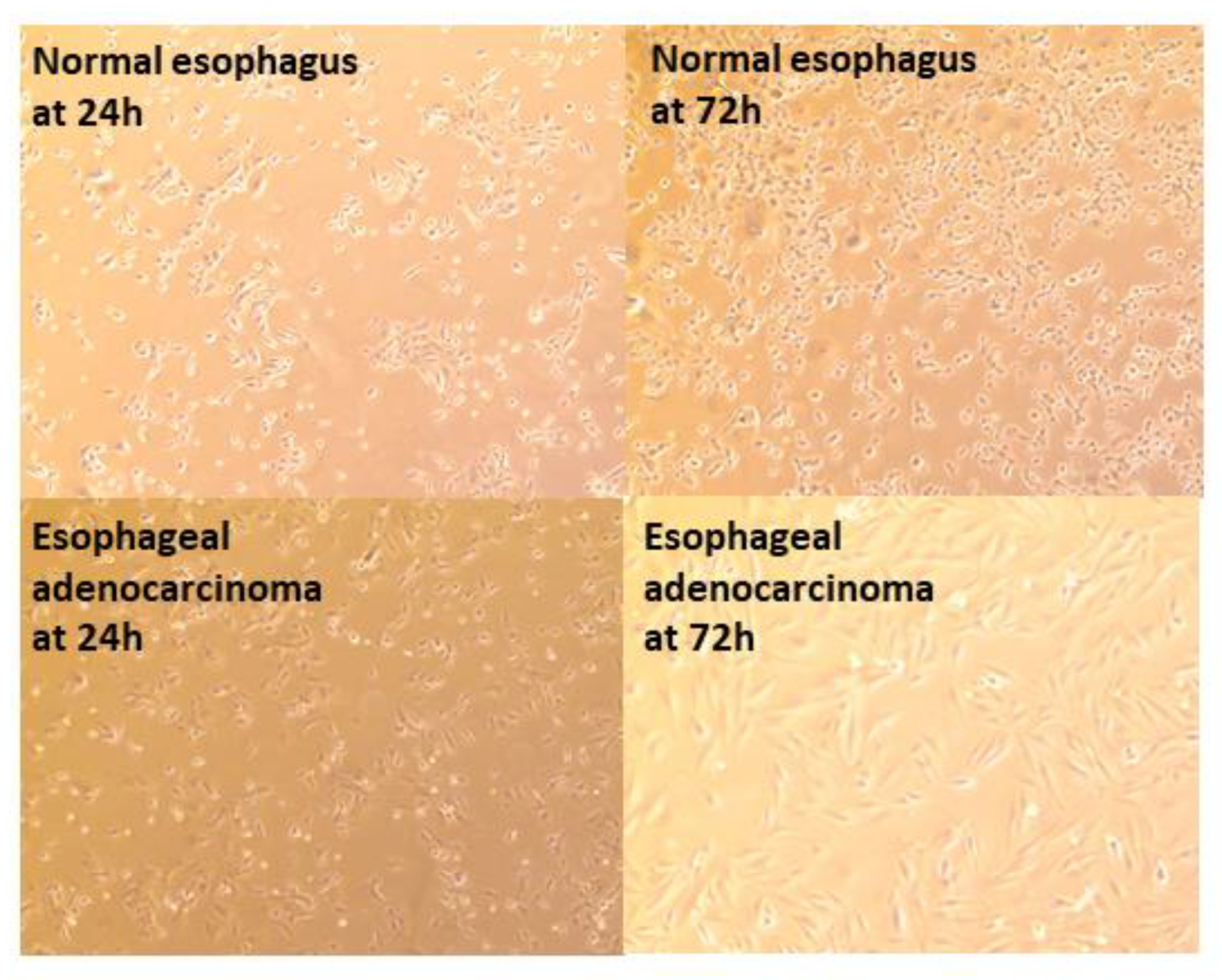
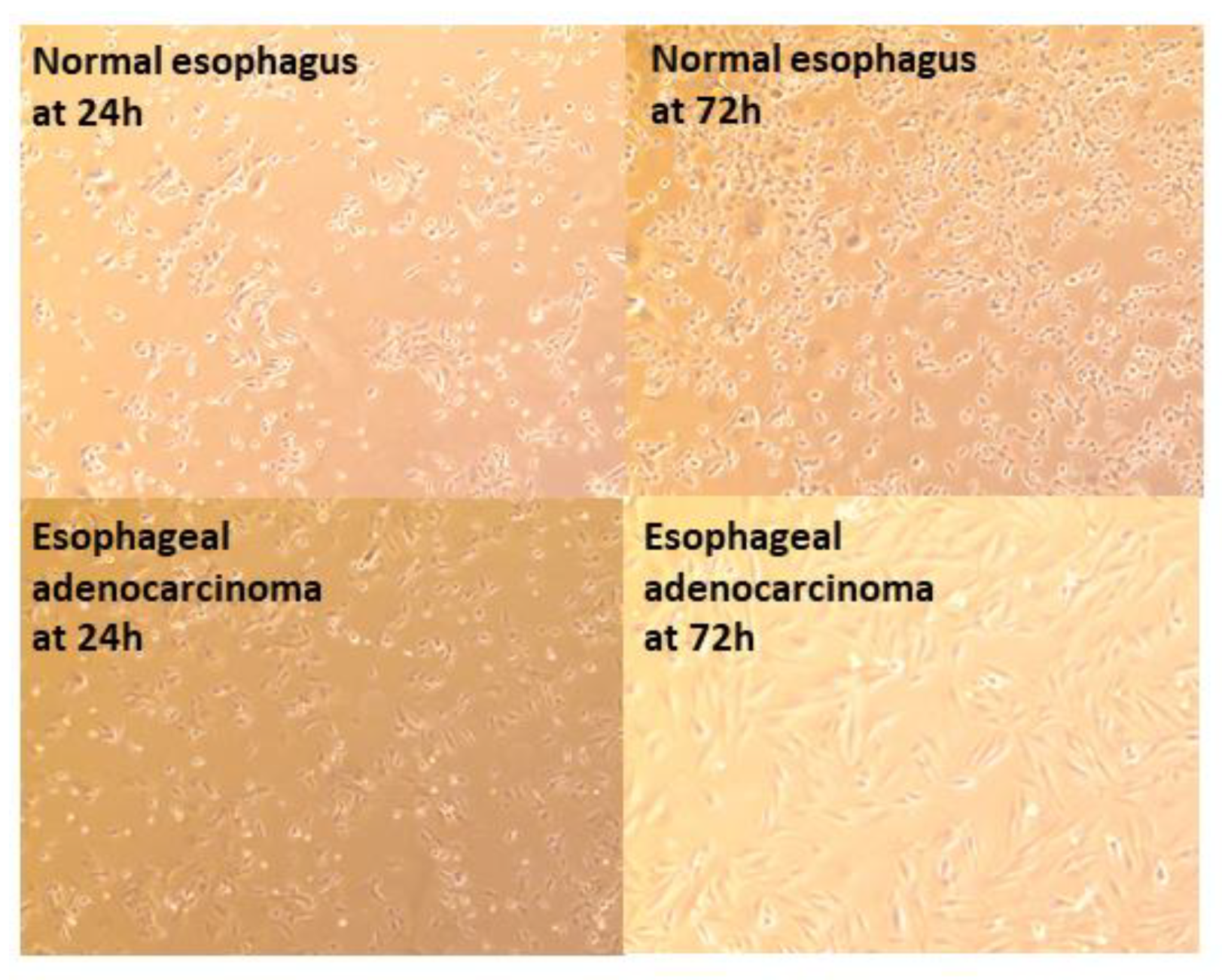
3.3.1. Epithelial Cell Layer
3.3.2. Transcriptome Analysis after Transplant
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rustgi, A.; El-Serag, H. Esophageal Carcinoma. N. Engl. J. Med. 2014, 71, 2499–2509. [Google Scholar] [CrossRef] [PubMed]
- Hur, C.; Miller, M.; Kong, C.Y.; Dowling, E.; Nattinger, K.; Dunn, M.; Feuer, E. Trends in Esophageal Adenocarcinoma Incidence and Mortality. Cancer 2013, 119, 1149–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, H.; Xie, S.H.; Lagergren, J. The Epidemiology of Esophageal Adenocarcinoma. Gastroenterology 2018, 154, 390–405. [Google Scholar] [PubMed]
- Hvid-Jensen, F.; Pedersen, L.; Drewes, A.M.; Sorensen, H.T.; Funch-Jensen, P. Incidence of adenocarcinoma among patients with Barrett’s esophagus. N. Engl. J. Med. 2011, 365, 1375–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okereke, I.C.; Hamilton, C.; Reep, G.; Krill, T.; Booth, A.; Ghouri, Y.; Jala, V.; Andersen, C.; Pyles, R. Microflora composition in the gastrointestinal tract in patients with Barrett’s esophagus. J. Thorac. Dis. 2019, 11, S1581–S1587. [Google Scholar] [CrossRef]
- Snider, E.J.; Freedberg, D.E.; Abrams, J.A. Potential role of the microbiome in Barrett’s esophagus and esophageal adenocarcinoma. Dig. Dis. Sci. 2016, 61, 2217–2225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baba, Y.; Iwatsuki, M.; Yoshida, N.; Watanabe, M.; Baba, H. Review of the gut microbiome and esophageal cancer: Pathogenesis and potential clinical implications. Ann. Gastroenterol. Surg. 2017, 1, 99–104. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Francois, F.; Pei, Z. Molecular pathways: Pathogenesis and clinical implications of microbiome alteration in esophagitis and Barrett esophagus. Clin. Cancer Res. 2012, 18, 2138–2144. [Google Scholar]
- Koike, Y.; Li, B.; Lee, C.; Cheng, S.; Miyake, H.; Welsh, C.; Hock, A.; Belik, J.; Zani, A.; Perro, A. Gastric emptying is reduced in experimental NEC and correlates with the severity of intestinal damage. J. Pediatr. Surg. 2017, 52, 744–748. [Google Scholar] [CrossRef]
- Takakura, K.; Hasegawa, K.; Goto, Y.; Muramatsu, I. Nitric oxide produced by inducible nitric oxide synthase delays gastric emptying in lipopolysaccharide-treated rats. Anesthesiology 1997, 87, 652–657. [Google Scholar] [CrossRef]
- di Pietro, M.; Chan, D.; Fitzgerald, R.C.; Wang, K.K. Screening for Barrett’s esophagus. Gastroenterology 2015, 148, 912–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Blankenburg, D.; Gordon, A.; Von Kuster, G.; Coraor, N.; Taylor, J.; Nekrutenko, A.; Galaxy Team. Manipulation of FASTQ data with Galaxy. Bioinformatics 2010, 26, 1783–1785. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glockner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Chaudhary, N.; Baghdadi, J.; Pei, Z. Microbiome in reflux disorders and esophageal adenocarcinoma. Cancer J. 2014, 20, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Kaakoush, N.; Morris, M. The oesophageal microbiome: An unexplored link in obesity-associated oesophageal adenocarcinoma. FEMS Microbiol. Ecol. 2016, 92. [Google Scholar] [CrossRef] [Green Version]
- Paull, G.; Yardley, J. Gastric and esophageal Campylobacter pylori in patients with Barrett’s esophagus. Gastroenterology 1988, 95, 216–218. [Google Scholar] [CrossRef]
- Deshpande, N.; Riordan, S.; Castaño-Rodríguez, N.; Wilkins, M.R.; Kaakoush, N.O. Signatures within the esophageal microbiome are associated with host genetics, age, and disease. Microbiome 2018, 6, 227. [Google Scholar] [CrossRef] [Green Version]
- Blackett, K.L.; Siddhi, S.S.; Cleary, S.; Steed, H.; Miller, M.H.; Macfarlane, S.; Macfarlane, G.T.; Dillon, J.F. Oesophageal bacterial biofilm changes in gastro-oesophageal reflux disease, Barrett’s and oesophageal carcinoma: Association or causality? Aliment. Pharm. Ther. 2013, 37, 1084–1092. [Google Scholar] [CrossRef]
- Pei, Z.; Bini, E.J.; Yang, L.; Zhou, M.; Francois, F.; Blaser, M.J. Bacterial biota in the human distal esophagus. Proc. Natl. Acad. Sci. USA 2004, 101, 4250–4255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okereke, I.C.; Miller, A.; Hamilton, C.; Booth, A.L.; Reep, G.L.; Andersen, C.L.; Reynolds, S.T.; Pyles, R.B. Microbiota of the oropharynx and endoscope compared to the esophagus. Sci. Rep. 2019, 9, 10201. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.H.; Yu, X.; Liao, W. The expensive-tissue hypothesis in vertebrates: Gut microbiota effect, a review. Int. J. Mol. Sci. 2018, 19, 1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, J.W.; Zhu, C.; Jolivet, P.; Zhu, S.X.; Lu, P.; Meaney, M.J.; West, R.B. Gene expression profiling of single cells from archival tissue with laser-capture microdissection and Smart-3SEQ. Genome Res. 2019, 29, 1816–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.; Wang, J.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Charles, D.D.; Fisher, J.; Hoskinson, S.M.; Medina-Colorado, A.A.; Shen, Y.C.; Chaaban, M.R.; Widen, S.G.; Eaves-Pyles, T.D.; Maxwell, C.A.; Miller, A.L.; et al. Development of a novel ex vivo nasal epithelial cell model supporting colonization with human nasal microbiota. Front. Cell. Infect. Microbiol. 2019, 21, 165. [Google Scholar] [CrossRef] [Green Version]
- Amerson-Brown, M.H.; Miller, A.L.; Maxwell, C.A.; White, M.M.; Vincent, K.L.; Bourne, N.; Pyles, R.B. Cultivated human vaginal microbiome communities impact Zika and herpes simplex virus replication in ex vivo vaginal mucosal cultures. Front. Microbiol. 2019, 9, 3340. [Google Scholar] [CrossRef] [Green Version]
- Gentry, M.; Taormina, J.; Pyles, R.B.; Yeager, L.; Kirtley, M.; Popov, V.L.; Klimpel, G.; Eaves-Pyles, T. Role of primary human alveolar epithelial cells in host defense against Francisella tularensis infection. Infect. Immun. 2007, 75, 3969–3978. [Google Scholar] [CrossRef] [Green Version]
- Lynch, S.V.; Pedersen, O. The human intestinal microbiome in health and disease. N. Engl. J. Med. 2016, 15, 2369–2379. [Google Scholar] [CrossRef] [Green Version]
- Young, V.B. The role of the microbiome in human health and disease: An introduction for clinicians. BMJ 2017, 356, j831. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Kim, C.M.; Ramakrishnan, V. Microbiome and disease in the upper airway. Curr. Opin. Allergy Clin. Immunol. 2019, 19, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Xavier, J.B.; Young, V.B.; Skufca, J.; Ginty, F.; Testerman, T.; Pearson, A.T.; Macklin, P.; Mitchell, A.; Shmulevich, I.; Xie, L.; et al. The Cancer Microbiome: Distinguishing Direct and Indirect Effects Requires a Systemic View. Trends Cancer 2020, 6, 192–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaser, M.J.; Atherton, J.C. Helicobacter pylori persistence: Biology and disease. J. Clin. Investig. 2004, 113, 321–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Vuai, M.S.; Zhong, M. The role of bacteria in cancer therapy-enemies in the past, but allies at present. Infect. Agent. Cancer 2018, 13, 9. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Chang, W.; Jin, G.; Wang, T.C. Gastrin and upper GI cancers. Curr. Opin. Pharm. 2016, 31, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Elias, P.S.; Castell, D.O. The role of acid suppression in Barrett’s esophagus. Am. J. Med. 2017, 130, 525–529. [Google Scholar] [CrossRef]
- Souza, R.F. From reflux esophagitis to esophageal adenocarcinoma. Dig. Dis. 2016, 34, 483–490. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Neves, A.A.; Di Pietro, M.; O’Donovan, M.; Waterhouse, D.J.; Bohndiek, S.E.; Brindle, K.M.; Fitzgerald, R.C. Detection of early neoplasia in Barrett’s esophagus using lectin-based near-infrared imaging: An ex vivo study on human tissue. Endoscopy 2018, 50, 618–625. [Google Scholar] [CrossRef] [Green Version]
- Herbst-Kralovetz, M.M.; Pyles, R.B.; Ratner, A.J.; Sycuro, L.K.; Mitchell, C. New systems for studying intercellular interactions in bacterial vaginosis. J. Infect. Dis. 2016, 214, S6–S13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasnim, S.; Miller, A.; Jupiter, D.; Hamilton, C.; Reep, G.; Krill, T.; Pyles, R.; Okereke, I.C. Effects of Proton Pump Inhibitor Use on the Esophageal Microbial Community. BMC Gastroenterol. 2020, 20, 312. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Barrett’s Esophagus | GERD without Barrett’s | p-Value | |
|---|---|---|---|
| N | 34 | 40 | |
| Male | 62% (18/29) | 50% (20/40) | 0.32 |
| Age, years (mean) | 61.7 ± 10.7 | 59.0 ± 8.9 | 0.26 |
| BMI (mean) | 31.5 ± 8.7 | 31.1 ± 5.5 | 0.83 |
| Hiatal hernia | 52% (15/29) | 35% (14/40) | 0.16 |
| Current smoker | 24% (7/29) | 20% (8/40) | 0.61 |
| Current PPI use | 97% (28/29) | 88% (35/40) | 0.23 |
| Mean PPI dose (milligrams) | 46.2 | 37.0 | 0.11 |
| Actinomyces | Lactobacillus | Rothia mucilaginosa | Campylobacter | Lautropia | sal 337 |
|---|---|---|---|---|---|
| Campylobacter consius | Leptotrichia unspecified | Streptococcus anginosis | Campylobacter showae | Leptotrichia wadei | Streptococcus mutans |
| Capnocytophaga | Mycoplasma faucium | Streptococcus oralis | Corynebacterium | Neisseria | Streptococcus pneumoniae |
| Dialister | Porphyromonas endodontalis | Streptococcus salivarius | Filifactor alocis | Porphyromonas gingivalis | Streptococcus sanguinis |
| Fusobacterium nucleatum | Prevotella denticola | Streptococcus thermophilus | Fusobacterium periodonticum | Prevotella intermedia | Streptococcus vestibularis |
| Gemella sanguinis | Prevotella melaninogenica | Streptococcus | Haemophilus haemolyticus | Prevotella nigrescens | Veillonella |
| Haemophilus influenza | Prevotella oris | Veillonella atypica | Haemophilus parahaemolyticus | Prevotella pallens | Veillonella parvula |
| Haemophilus | Prevotella | Haemophilus parainfluenzae | Prevotella timonensis | Total 16S | Human GAPDH |
| Barrett’s Esophagus Group | GERD without Barrett’s Group | p-Value | |
|---|---|---|---|
| N | 9 | 28 | N/A |
| Age (years) | 63.3 | 61.2 | 0.58 |
| BMI | 26.9 | 31.5 | 0.03 |
| Hiatal hernia | 56% (5/9) | 32% (9/28) | 0.21 |
| Current smoker | 22% (2/9) | 25% (7/28) | 0.99 |
| Current PPI use | 89% (8/9) | 86% (24/28) | 0.81 |
| Mean PPI dose (milligrams) | 37.8 | 36.4 | 0.87 |
| Transcription Factor | Function |
|---|---|
| FosB | DNA binding |
| Early growth response protein 1 (EGR1) | Tumor suppressor |
| Early growth response protein 3 (EGR3) | Tumor suppressor |
| Nuclear receptor subfamily 4 group A member 1 (NR4A1) | Energy homeostasis |
| Cyclic AMP-dependent factor ATF-3 (ATF3) | Transcription repressor |
| Hepatocyte nuclear factor 4-alpha (HNF4A) | Transcription repressor |
| Ankyrin repeat and SAM domain-containing protein 4B (ANKS4B) | Epithelial brush differentiation |
| Galectin-4 (LGALS4) | Cell assembly |
| Apoptosis facilitator Bcl-2-like protein 14 | BCL2L14 |
| Cyclin-dependent kinase inhibitor 2A (CDKN2A) | Cell proliferation |
| Matrix metalloproteinase 7 (MMP7) | Cell degradation |
| E3 ubiquitin-protein ligase Mdm2 (MDM2) | Apoptosis |
| Regenerating islet-derived protein (REG) | Inflammation |
| Calpain 8 (CAPN8) | Cellular apoptosis |
| Defensin beta 103A (DEFB103A) | Regulation of cytokine production |
| Rho GTPase Activating Protein 26 (ARHGAP26) | Protein coding |
| Cell division cycle 25B (CDC25B) | Regulation of mitosis |
| G protein subunit beta-2 (GNB2) | Antiproliferative function |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cass, S.; Hamilton, C.; Miller, A.; Jupiter, D.; Khanipov, K.; Booth, A.; Pyles, R.; Krill, T.; Reep, G.; Okereke, I. Novel Ex Vivo Model to Examine the Mechanism and Relationship of Esophageal Microbiota and Disease. Biomedicines 2021, 9, 142. https://doi.org/10.3390/biomedicines9020142
Cass S, Hamilton C, Miller A, Jupiter D, Khanipov K, Booth A, Pyles R, Krill T, Reep G, Okereke I. Novel Ex Vivo Model to Examine the Mechanism and Relationship of Esophageal Microbiota and Disease. Biomedicines. 2021; 9(2):142. https://doi.org/10.3390/biomedicines9020142
Chicago/Turabian StyleCass, Samuel, Catherine Hamilton, Aaron Miller, Daniel Jupiter, Kamil Khanipov, Adam Booth, Richard Pyles, Timothy Krill, Gabriel Reep, and Ikenna Okereke. 2021. "Novel Ex Vivo Model to Examine the Mechanism and Relationship of Esophageal Microbiota and Disease" Biomedicines 9, no. 2: 142. https://doi.org/10.3390/biomedicines9020142
APA StyleCass, S., Hamilton, C., Miller, A., Jupiter, D., Khanipov, K., Booth, A., Pyles, R., Krill, T., Reep, G., & Okereke, I. (2021). Novel Ex Vivo Model to Examine the Mechanism and Relationship of Esophageal Microbiota and Disease. Biomedicines, 9(2), 142. https://doi.org/10.3390/biomedicines9020142