
Endothelial Barrier Function and Leukocyte Transmigration in Atherosclerosis

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
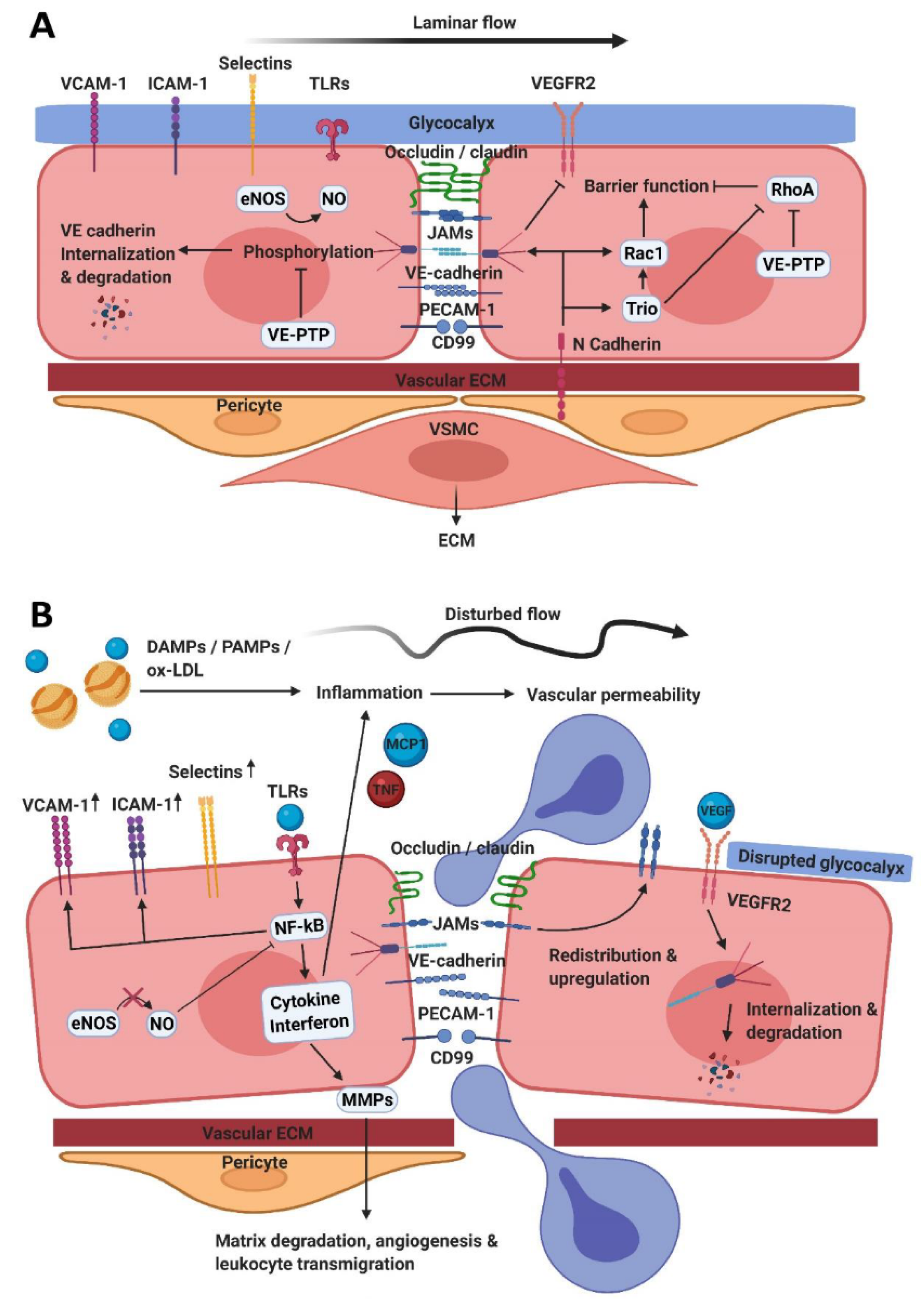
2. The Endothelial Barrier Function
2.1. Tight Junctions
2.2. Adherens Junctions
2.3. Tissue-Specific Endothelium
2.4. Vasomotor Function
3. Endothelial Activation and Leukocyte Transendothelial Migration
3.1. Finding Sites of Extravasation
3.2. Opening Endothelial Cell Junctions
3.3. Leukocyte Migration into the Vessel Wall
3.4. Vascular Bed Specific TEM
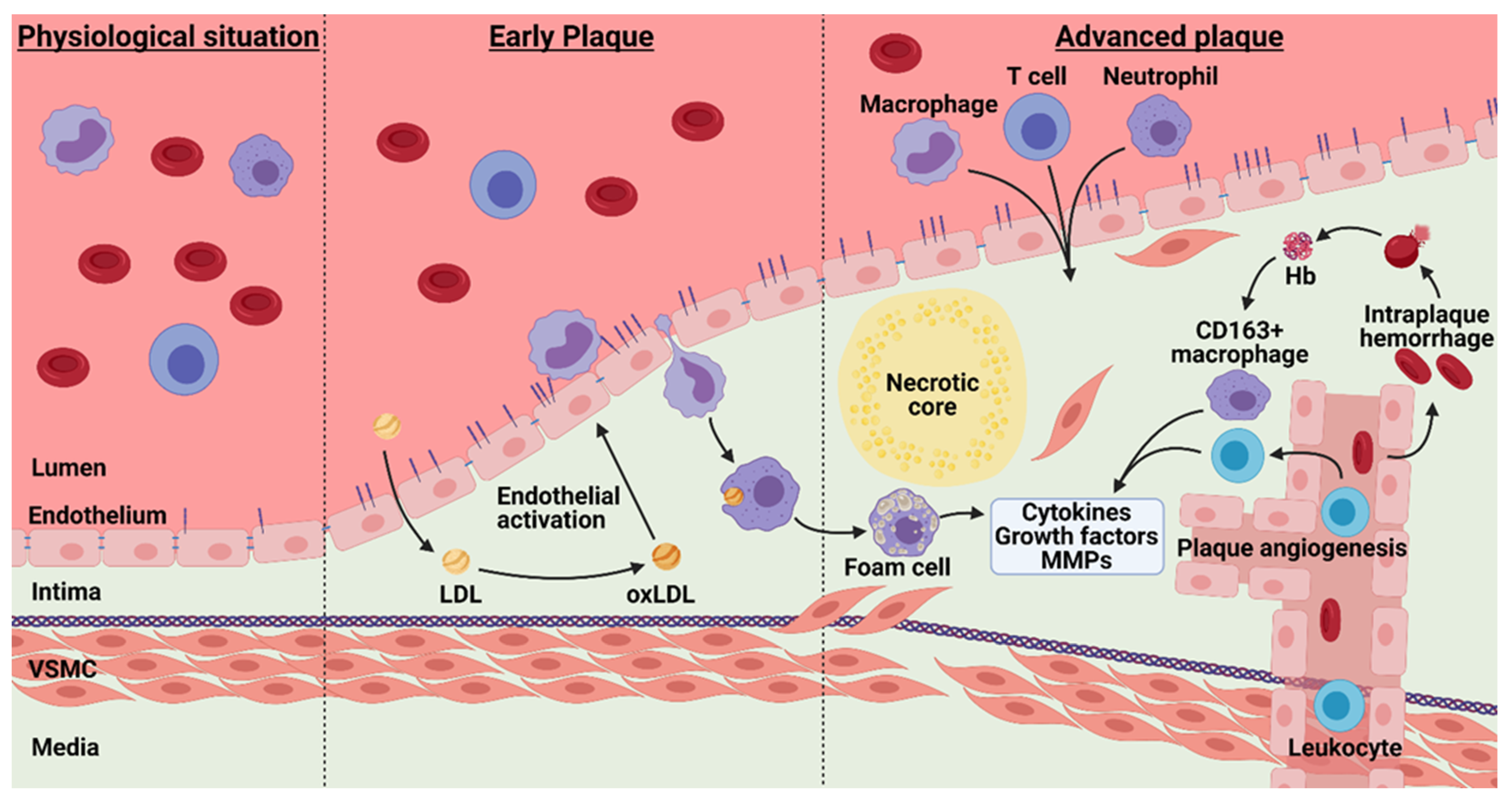
4. Atherosclerosis and Barrier Function
4.1. Plaque Hypoxia
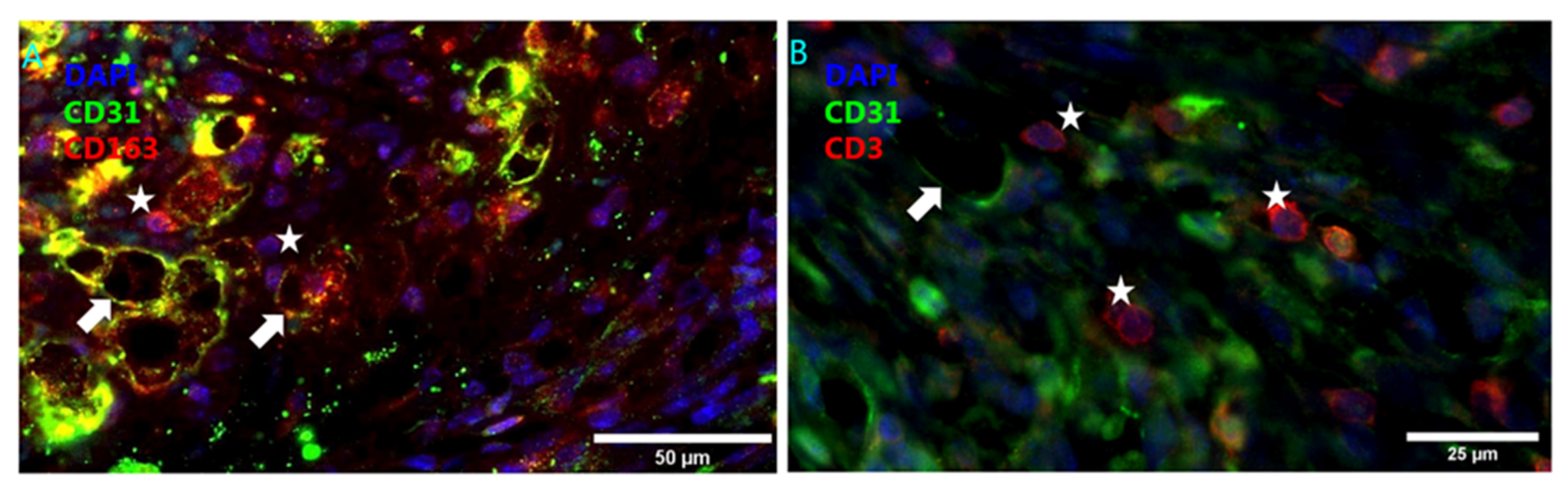
4.2. Intraplaque Angiogenesis and Intraplaque Hemorrhage
4.3. Angiogenesis Associated Macrophages
4.4. Statins
4.5. Atherosclerosis Heterogeneity
5. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Ethical Approval
References
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 156, 92–704. [Google Scholar] [CrossRef]
- Owen-Woods, C.; Joulia, R.; Barkaway, A.; Rolas, L.; Ma, B.; Nottebaum, A.F.; Arkill, K.P.; Stein, M.; Girbl, T.; Golding, M.; et al. Local microvascular leakage promotes trafficking of activated neutrophils to remote organs. J. Clin. Investig. 2020, 1302, 301–318. [Google Scholar] [CrossRef]
- De Vries, M.R.; Quax, P.H.A. Inflammation in Vein Graft Disease. Front. Cardiovasc. Med. 2018, 5, 3. [Google Scholar] [CrossRef]
- Heemskerk, N.; Schimmel, L.; Oort, C.; van Rijssel, J.; Yin, T.; Ma, B.; van Unen, J.; Pitter, B.; Huveneers, S.; Goedhart, J.; et al. F-actin-rich contractile endothelial pores prevent vascular leakage during leukocyte diapedesis through local RhoA signalling. Nat. Commun. 2016, 71, 0493. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, E.E. Intravital microscopy on atherosclerosis in apolipoprotein e-deficient mice establishes microvessels as major entry pathways for leukocytes to advanced lesions. Circulation 2011, 1242, 129–138. [Google Scholar] [CrossRef] [Green Version]
- De Vries, M.R.; Niessen, H.W.; Löwik, C.W.; Hamming, J.F.; Jukema, J.W.; Quax, P.H. Plaque rupture complications in murine atherosclerotic vein grafts can be prevented by TIMP-1 overexpression. PLoS ONE 2012, 7, e47134. [Google Scholar] [CrossRef] [Green Version]
- Sluimer, J.C.; Kolodgie, F.D.; Bijnens, A.P.; Maxfield, K.; Pacheco, E.; Kutys, B.; Duimel, H.; Frederik, P.M.; van Hinsbergh, V.M.; Virmani, R.; et al. Thin-walled microvessels in human coronary atherosclerotic plaques show incomplete endothelial junctions relevance of compromised structural integrity for intraplaque microvascular leakage. J. Am. Coll. Cardiol. 2009, 531, 517–527. [Google Scholar]
- Radeva, M.Y.; Waschke, J. Mind the gap: Mechanisms regulating the endothelial barrier. Acta Physiol. Oxf. 2018, 222, e12860. [Google Scholar] [CrossRef]
- Komarova, Y.A.; Kruse, K.; Mehta, D.; Malik, A.B. Protein Interactions at Endothelial Junctions and Signaling Mechanisms Regulating Endothelial Permeability. Circ. Res. 2017, 1201, 79–206. [Google Scholar] [CrossRef] [Green Version]
- Qu, Y.; Dahl, G. Accessibility of cx46 hemichannels for uncharged molecules and its modulation by voltage. Biophys. J. 2004, 861, 502–509. [Google Scholar] [CrossRef] [Green Version]
- Augustin, H.G.; Koh, G.Y. Organotypic vasculature: From descriptive heterogeneity to functional pathophysiology. Science 2017, 357. [Google Scholar] [CrossRef] [Green Version]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ. Res. 2007, 1001, 74–90. [Google Scholar] [CrossRef] [Green Version]
- Shakib, M.; Cunha-Vaz, J.G. Studies on the permeability of the blood-retinal barrier. IV. Junctional complexes of the retinal vessels and their role in the permeability of the blood-retinal barrier. Exp. Eye Res. 1966, 52, 29–34. [Google Scholar]
- Abdullahi, W.; Tripathi, D.; Ronaldson, P.T. Blood-brain barrier dysfunction in ischemic stroke: Targeting tight junctions and transporters for vascular protection. Am. J. Physiol. Cell Physiol. 2018, 315, C343–C356. [Google Scholar] [CrossRef]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol. 2003, 1616, 53–60. [Google Scholar] [CrossRef]
- Ebnet, K. Junctional Adhesion Molecules (JAMs): Cell Adhesion Receptors With Pleiotropic Functions in Cell Physiology and Development. Physiol. Rev. 2017, 971, 529–554. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Furuse, M.; Morita, K.; Kubota, K.; Saitou, M.; Tsukita, S. Direct binding of three tight junction-associated MAGUKs.; ZO-1.; ZO-2.; and ZO-3.; with the COOH termini of claudins. J. Cell Biol. 1999, 1471, 351–363. [Google Scholar] [CrossRef] [Green Version]
- Tornavaca, O.; Chia, M.; Dufton, N.; Almagro, L.O.; Conway, D.E.; Randi, A.M.; Schwartz, M.A.; Matter, K.; Balda, M.S. ZO-1 controls endothelial adherens junctions.; cell-cell tension.; angiogenesis.; and barrier formation. J. Cell Biol. 2015, 2088, 21–38. [Google Scholar] [CrossRef] [Green Version]
- Shigetomi, K.; Ono, Y.; Inai, T.; Ikenouchi, J. Adherens junctions influence tight junction formation via changes in membrane lipid composition. J. Cell Biol. 2018, 2172, 373–381. [Google Scholar] [CrossRef] [Green Version]
- Taddei, A.; Giampietro, C.; Conti, A.; Orsenigo, F.; Breviario, F.; Pirazzoli, V.; Potente, M.; Daly, C.; Dimmeler, S.; Dejana, E. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat. Cell Biol. 2008, 109, 23–34. [Google Scholar] [CrossRef]
- Dejana, E.; Orsenigo, F.; Molendini, C.; Baluk, P.; McDonald, D.M. Organization and signaling of endothelial cell-to-cell junctions in various regions of the blood and lymphatic vascular trees. Cell Tissue Res. 2009, 3351, 7–25. [Google Scholar] [CrossRef] [Green Version]
- Lertkiatmongkol, P.; Liao, D.; Mei, H.; Hu, Y.; Newman, P.J. Endothelial functions of platelet/endothelial cell adhesion molecule-1 (CD31). Curr. Opin. Hematol. 2016, 232, 53–59. [Google Scholar] [CrossRef] [Green Version]
- Yap, A.S.; Niessen, C.M.; Gumbiner, B.M. The juxtamembrane region of the cadherin cytoplasmic tail supports lateral clustering.; adhesive strengthening.; and interaction with p120ctn. J. Cell Biol. 1998, 1417, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Angulo-Urarte, A.; van der Wal, T.; Huveneers, S. Cell-cell junctions as sensors and transducers of mechanical forces. Biochim. Biophys. Acta Biomembr. 2020, 18621, 83316. [Google Scholar]
- Schulte, D.; Küppers, V.; Dartsch, N.; Broermann, A.; Li, H.; Zarbock, A.; Kamenyeva, O.; Kiefer, F.; Khandoga, A.; Massberg, S.; et al. Stabilizing the VE-cadherin-catenin complex blocks leukocyte extravasation and vascular permeability. EMBO J. 2011, 304, 157–170. [Google Scholar] [CrossRef] [Green Version]
- Orlova, V.V.; Economopoulou, M.; Lupu, F.; Santoso, S.; Chavakis, T. Junctional adhesion molecule-C regulates vascular endothelial permeability by modulating VE-cadherin-mediated cell-cell contacts. J. Exp. Med. 2006, 2032, 703–714. [Google Scholar]
- Potter, M.D.; Barbero, S.; Cheresh, D.A. Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and beta-catenin and maintains the cellular mesenchymal state. J. Biol. Chem. 2005, 2803, 1906–1912. [Google Scholar]
- Baganha, F.; Jong, R.C.M.D.; Peters, E.A.; Voorham, W.; Jukema, J.W.; Delibegovic, M.; de Vries, M.R.; Quax, P.H.A. Atorvastatin pleiotropically decreases intraplaque angiogenesis and intraplaque haemorrhage by inhibiting ANGPT2 release and VE-Cadherin internalization. Angiogenesis 2021. [Google Scholar] [CrossRef]
- Lampugnani, M.G.; Corada, M.; Andriopoulou, P.; Esser, S.; Risau, W.; Dejana, E. Cell confluence regulates tyrosine phosphorylation of adherens junction components in endothelial cells. J. Cell Sci. 1997, 110 Pt 17, 2065–2077. [Google Scholar]
- Wessel, F.; Winderlich, M.; Holm, M.; Frye, M.; Rivera-Galdos, R.; Vockel, M.; Linnepe, R.; Ipe, U.; Stadtmann, A.; Zarbock, A.; et al. Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat. Immunol. 2014, 152, 23–30. [Google Scholar] [CrossRef]
- Nawroth, R.; Poell, G.; Ranft, A.; Kloep, S.; Samulowitz, U.; Fachinger, G.; Golding, M.; Shima, D.T.; Deutsch, U.; Vestweber, D. VE-PTP and VE-cadherin ectodomains interact to facilitate regulation of phosphorylation and cell contacts. EMBO J. 2002, 214, 885–895. [Google Scholar] [CrossRef]
- Juettner, V.V.; Kruse, K.; Dan, A.; Vu, V.H.; Khan, Y.; Le, J.; Le, J.; Leckband, D.; Komarova, Y.; Malik, A.B. VE-PTP stabilizes VE-cadherin junctions and the endothelial barrier via a phosphatase-independent mechanism. J. Cell Biol. 2019, 2181, 725–742. [Google Scholar] [CrossRef] [Green Version]
- Van Buul, J.D.; Geerts, D.; Huveneers, S. Rho GAPs and GEFs: Controling switches in endothelial cell adhesion. Cell Adh. Migr. 2014, 81, 08–24. [Google Scholar] [CrossRef] [Green Version]
- Timmerman, I.; Heemskerk, N.; Kroon, J.; Schaefer, A.; van Rijssel, J.; Hoogenboezem, M.; van Unen, J.; Goedhart, J.; Gadella, T.W., Jr.; Yin, T.; et al. A local VE-cadherin and Trio-based signaling complex stabilizes endothelial junctions through Rac1. J. Cell Sci. 2015, 1283, 041–054. [Google Scholar]
- Privratsky, J.R.; Paddock, C.M.; Florey, O.; Newman, D.K.; Muller, W.A.; Newman, P.J. Relative contribution of PECAM-1 adhesion and signaling to the maintenance of vascular integrity. J. Cell Sci. 2011, 124 Pt 9, 1477–1485. [Google Scholar] [CrossRef] [Green Version]
- Machida, K.; Thompson, C.M.; Dierck, K.; Jablonowski, K.; Kärkkäinen, S.; Liu, B.; Zhang, H.; Nash, P.D.; Newman, D.K.; Nollau, P.; et al. High-throughput phosphotyrosine profiling using SH2 domains. Mol. Cell 2007, 268, 99–915. [Google Scholar] [CrossRef]
- Caligiuri, G. Mechanotransduction, immunoregulation, and metabolic functions of CD31 in cardiovascular pathophysiology. Cardiovasc. Res. 2019, 1151, 425–434. [Google Scholar] [CrossRef] [Green Version]
- Biswas, P.; Canosa, S.; Schoenfeld, D.; Schoenfeld, J.; Li, P.; Cheas, L.C.; Zhang, J.; Cordova, A.; Sumpio, B.; Madri, J.A. PECAM-1 affects GSK-3beta-mediated beta-catenin phosphorylation and degradation. Am. J. Pathol. 2006, 1693, 14–24. [Google Scholar]
- Tzima, E.; Irani-Tehrani, M.; Kiosses, W.B.; Dejana, E.; Schultz, D.A.; Engelhardt, B.; Cao, G.; DeLisser, H.; Schwartz, M.A. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 2005, 4374, 26–31. [Google Scholar] [CrossRef]
- Garnacho, C.; Shuvaev, V.; Thomas, A.; McKenna, L.; Sun, J.; Koval, M.; Albelda, S.; Muzykantov, V.; Muro, S. RhoA activation and actin reorganization involved in endothelial CAM-mediated endocytosis of anti-PECAM carriers: Critical role for tyrosine 686 in the cytoplasmic tail of PECAM-1. Blood 2008, 1113, 024–033. [Google Scholar] [CrossRef] [Green Version]
- Claesson-Welsh, L.; Dejana, E.; McDonald, D.M. Permeability of the Endothelial Barrier: Identifying and Reconciling Controversies. Trends Mol. Med. 2020. [Google Scholar] [CrossRef]
- Baganha, F.; Ritsma, L.; Quax, P.H.A.; de Vries, M.R. Assessment of Microvessel Permeability in Murine Atherosclerotic Vein Grafts Using Two-Photon Intravital Microscopy. Int. J. Mol. Sci. 2020, 21, 9244. [Google Scholar] [CrossRef] [PubMed]
- Mitra, R.; O’Neil, G.L.; Harding, I.C.; Cheng, M.J.; Mensah, S.A.; Ebong, E.E. Glycocalyx in Atherosclerosis-Relevant Endothelium Function and as a Therapeutic Target. Curr. Atheroscler. Rep. 2017, 196, 3. [Google Scholar] [CrossRef] [Green Version]
- Morris, A.W.; Sharp, M.M.; Albargothy, N.J.; Fernandes, R.; Hawkes, C.A.; Verma, A.; Weller, R.O.; Carare, R.O. Vascular basement membranes as pathways for the passage of fluid into and out of the brain. Acta Neuropathol. 2016, 1317, 25–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengillo, J.D.; Winkler, E.A.; Walker, C.T.; Sullivan, J.S.; Johnson, M.; Zlokovic, B.V. Deficiency in Mural Vascular Cells Coincides with Blood–Brain Barrier Disruption in Alzheimer’s Disease. Brain Pathol. 2013, 233, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrot, C.Y.; Herrera, J.L.; Fournier-Goss, A.E.; Komatsu, M. Prostaglandin E2 breaks down pericyte-endothelial cell interaction via EP1 and EP4-dependent downregulation of pericyte N-cadherin, connexin-43, and R-Ras. Sci. Rep. 2020, 101, 1186. [Google Scholar] [CrossRef]
- Kruse, K.; Lee, Q.S.; Sun, Y.; Klomp, J.; Yang, X.; Huang, F.; Sun, M.Y.; Zhao, S.; Hong, Z.; Vogel, S.M.; et al. N-cadherin signaling via Trio assembles adherens junctions to restrict endothelial permeability. J. Cell Biol. 2019, 2182, 99–316. [Google Scholar] [CrossRef]
- Kivelä, R.; Hemanthakumar, K.A.; Vaparanta, K.; Robciuc, M.; Izumiya, Y.; Kidoya, H.; Takakura, N.; Peng, X.; Sawyer, D.B.; Elenius, K.; et al. Endothelial Cells Regulate Physiological Cardiomyocyte Growth via VEGFR2-Mediated Paracrine Signaling. Circulation 2019, 1392, 570–584. [Google Scholar] [CrossRef]
- Davis, G.E.; Senger, D.R. Endothelial Extracellular Matrix. Circ. Res. 2005, 971, 093–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Torres, M.P.; Pérez-Rivero, G.; Rodríguez-Puyol, M.; Rodríguez-Puyol, D.; Díez-Marqués, M.L. The leukocyte-endothelial cell interactions are modulated by extracellular matrix proteins. Cell Physiol. Biochem. 2006, 17, 221–232. [Google Scholar] [CrossRef]
- MacLeod, D.C.; Strauss, B.H.; de Jong, M.; Escaned, J.; Umans, V.A.; van Suylen, R.J.; Verkerk, A.; de Feyter, P.J.; Serruys, P.W. Proliferation and extracellular matrix synthesis of smooth muscle cells cultured from human coronary atherosclerotic and restenotic lesions. J. Am. Coll. Cardiol. 1994, 235, 9–65. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.; Shu, B.; Zhang, Y.; Wang, M. Endothelial Response to Pathophysiological Stress. Arterioscler. Thromb. Vasc. Biol. 2019, 39, e233–e243. [Google Scholar] [CrossRef]
- Zhang, C.; Zhou, T.; Chen, Z.; Yan, M.; Li, B.; Lv, H.; Wang, C.; Xiang, S.; Shi, L.; Zhu, Y.; et al. Coupling of Integrin α5 to Annexin A2 by Flow Drives Endothelial Activation. Circ. Res. 2020, 1271, 074–090. [Google Scholar] [CrossRef] [PubMed]
- Cardillo, C.; Kilcoyne, C.M.; Cannon, R.O., 3rd; Panza, J.A. Interactions between nitric oxide and endothelin in the regulation of vascular tone of human resistance vessels in vivo. Hypertension 2000, 351, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Fleming, I. Molecular mechanisms underlying the activation of eNOS. Pflugers Arch. 2010, 4597, 93–806. [Google Scholar] [CrossRef]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 1186, 20–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, P.; d’Uscio, L.V.; Shaw, S.; Takase, H.; Barton, M.; Lüscher, T.F. Angiotensin II Increases Tissue Endothelin and Induces Vascular Hypertrophy. Circulation 1997, 961, 593–597. [Google Scholar] [CrossRef]
- Vidal, F.; Colomé, C.; Martínez-González, J.; Badimon, L. Atherogenic concentrations of native low-density lipoproteins down-regulate nitric-oxide-synthase mRNA and protein levels in endothelial cells. Eur. J. Biochem. 1998, 2523, 78–84. [Google Scholar] [CrossRef]
- Salvador, B.; Arranz, A.; Francisco, S.; Córdoba, L.; Punzón, C.; Llamas, M.; Fresno, M. Modulation of endothelial function by Toll like receptors. Pharmacol. Res. 2016, 1084, 6–56. [Google Scholar] [CrossRef]
- Karper, J.C.; Vries, M.R.D.; Brand, B.T.V.D.; Hoefer, I.E.; Fischer, J.W.; Jukema, J.W.; Niessen, H.W.M.; Quax, P.H.A. Toll-Like Receptor 4 Is Involved in Human and Mouse Vein Graft Remodeling.; and Local Gene Silencing Reduces Vein Graft Disease in Hypercholesterolemic APOE*3Leiden Mice. Arterioscler. Thromb. Vasc. Biol. 2011, 311, 033–040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, K.H.; Peters, H.A.B.; Jukema, J.W.; de Vries, M.R.; Quax, P.H.A. A protective role of IRF3 and IRF7 signalling downstream TLRs in the development of vein graft disease via type I interferons. J. Intern. Med. 2017, 2825, 22–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajan, S.; Ye, J.; Bai, S.; Huang, F.; Guo, Y.L. NF-kappaB, but not p38 MAP kinase.; is required for TNF-alpha-induced expression of cell adhesion molecules in endothelial cells. J. Cell Biochem. 2008, 1054, 77–86. [Google Scholar]
- Jersmann, H.P.A.; Hii, C.S.T.; Ferrante, J.V.; Ferrante, A. Bacterial Lipopolysaccharide and Tumor Necrosis Factor Alpha Synergistically Increase Expression of Human Endothelial Adhesion Molecules through Activation of NF-κB and p38 Mitogen-Activated Protein Kinase Signaling Pathways. Infect. Immun. 2001, 691, 273–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denk, A.; Goebeler, M.; Schmid, S.; Berberich, I.; Ritz, O.; Lindemann, D.; Ludwig, S.; Wirth, T. Activation of NF-kappa B via the Ikappa B kinase complex is both essential and sufficient for proinflammatory gene expression in primary endothelial cells. J. Biol. Chem. 2001, 2762, 8451–8458. [Google Scholar]
- Sehnert, B.; Burkhardt, H.; Wessels, J.T.; Schröder, A.; May, M.J.; Vestweber, D.; Zwerina, J.; Warnatz, K.; Nimmerjahn, F.; Schett, G.; et al. NF-κB inhibitor targeted to activated endothelium demonstrates a critical role of endothelial NF-κB in immune-mediated diseases. Proc. Natl. Acad. Sci. USA 2013, 1101, 6556–6561. [Google Scholar]
- Lockyer, J.M.; Colladay, J.S.; Alperin-Lea, W.L.; Hammond, T.; Buda, A.J. Inhibition of nuclear factor-kappaB-mediated adhesion molecule expression in human endothelial cells. Circ. Res. 1998, 823, 14–20. [Google Scholar]
- Ranta, V.; Orpana, A.; Carpén, O.; Turpeinen, U.; Ylikorkala, O.; Viinikka, L. Human vascular endothelial cells produce tumor necrosis factor-alpha in response to proinflammatory cytokine stimulation. Crit. Care Med. 1999, 272, 184–187. [Google Scholar]
- Schmitt, M.M.; Megens, R.T.; Zernecke, A.; Bidzhekov, K.; van den Akker, N.M.; Rademakers, T.; van Zandvoort, M.A.; Hackeng, T.M.; Koenen, R.R.; Weber, C. Endothelial junctional adhesion molecule-a guides monocytes into flow-dependent predilection sites of atherosclerosis. Circulation 2014, 1296, 6–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, M.M.; Fraemohs, L.; Hackeng, T.M.; Weber, C.; Koenen, R.R. Atherogenic mononuclear cell recruitment is facilitated by oxidized lipoprotein-induced endothelial junctional adhesion molecule-A redistribution. Atherosclerosis 2014, 2342, 54–64. [Google Scholar] [CrossRef]
- Reglero-Real, N.; Colom, B.; Bodkin, J.V.; Nourshargh, S. Endothelial Cell Junctional Adhesion Molecules: Role and Regulation of Expression in Inflammation. Arterioscler. Thromb. Vasc. Biol. 2016, 362, 048–057. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.H.; Harith, H.H.; Israf, D.A.; Tham, C.L. Differential Regulation of LPS-Mediated VE-Cadherin Disruption in Human Endothelial Cells and the Underlying Signaling Pathways: A Mini Review. Front. Cell Dev. Biol. 2020, 7, 280. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Kataoka, N.; Nakamura, E.; Tsujioka, K.; Kajiya, F. Oxidized LDL specifically promotes the initiation of monocyte invasion during transendothelial migration with upregulated PECAM-1 and downregulated VE-cadherin on endothelial junctions. Atherosclerosis 2007, 194, e9–e17. [Google Scholar] [CrossRef] [PubMed]
- Hordijk, P.L. Recent insights into endothelial control of leukocyte extravasation. Cell Mol. Life Sci. 2016, 731, 591–608. [Google Scholar] [CrossRef]
- Schnoor, M.; Alcaide, P.; Voisin, M.B.; van Buul, J.D. Crossing the Vascular Wall: Common and Unique Mechanisms Exploited by Different Leukocyte Subsets during Extravasation. Mediat. Inflamm. 2015, 20159, 46509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinamon, G.; Shinder, V.; Alon, R. Shear forces promote lymphocyte migration across vascular endothelium bearing apical chemokines. Nat. Immunol. 2001, 25, 15–22. [Google Scholar] [CrossRef]
- Miao, H.; Hu, Y.L.; Shiu, Y.T.; Yuan, S.; Zhao, Y.; Kaunas, R.; Wang, Y.; Jin, G.; Usami, S.; Chien, S. Effects of flow patterns on the localization and expression of VE-cadherin at vascular endothelial cell junctions: In vivo and in vitro investigations. J. Vasc. Res. 2005, 427, 7–89. [Google Scholar] [CrossRef]
- Finsterbusch, M.; Voisin, M.B.; Beyrau, M.; Williams, T.J.; Nourshargh, S. Neutrophils recruited by chemoattractants in vivo induce microvascular plasma protein leakage through secretion of TNF. J. Exp. Med. 2014, 2111, 307–314. [Google Scholar] [CrossRef]
- Zarbock, A.; Ley, K.; McEver, R.P.; Hidalgo, A. Leukocyte ligands for endothelial selectins: Specialized glycoconjugates that mediate rolling and signaling under flow. Blood 2011, 1186, 743–751. [Google Scholar] [CrossRef] [Green Version]
- Alon, R.; Shulman, Z. Chemokine triggered integrin activation and actin remodeling events guiding lymphocyte migration across vascular barriers. Exp. Cell Res. 2011, 3176, 32–41. [Google Scholar] [CrossRef]
- Carman, C.V.; Martinelli, R.T. Lymphocyte–Endothelial Interactions: Emerging Understanding of Trafficking and Antigen-Specific Immunity. Front. Immunol. 2015, 6, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillipson, M.; Heit, B.; Colarusso, P.; Liu, L.; Ballantyne, C.M.; Kubes, P. Intraluminal crawling of neutrophils to emigration sites: A molecularly distinct process from adhesion in the recruitment cascade. J. Exp. Med. 2006, 2032, 569–575. [Google Scholar]
- Auffray, C.; Fogg, D.; Garfa, M.; Elain, G.; Join-Lambert, O.; Kayal, S.; Sarnacki, S.; Cumano, A.; Lauvau, G.; Geissmann, F. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 2007, 3176, 66–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumagin, R.; Prizant, H.; Lomakina, E.; Waugh, R.E.; Sarelius, I.H. LFA-1 and Mac-1 define characteristically different intralumenal crawling and emigration patterns for monocytes and neutrophils in situ. J. Immunol. 2010, 1857, 057–066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carman, C.V.; Springer, T.A. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J. Cell Biol. 2004, 1673, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Carman, C.V.; Jun, C.D.; Salas, A.; Springer, T.A. Endothelial cells proactively form microvilli-like membrane projections upon intercellular adhesion molecule 1 engagement of leukocyte LFA-1. J. Immunol. 2003, 1716, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Barreiro, O.; Yanez-Mo, M.; Serrador, J.M.; Montoya, M.C.; Vicente-Manzanares, M.; Tejedor, R.; Furthmayr, H.; Sanchez-Madrid, F. Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J. Cell Biol. 2002, 1571, 233–245. [Google Scholar] [CrossRef] [Green Version]
- Van Buul, J.D.; Allingham, M.J.; Samson, T.; Meller, J.; Boulter, E.; García-Mata, R.; Burridge, K. RhoG regulates endothelial apical cup assembly downstream from ICAM1 engagement and is involved in leukocyte trans-endothelial migration. J. Cell Biol. 2007, 1781, 279–293. [Google Scholar] [CrossRef] [Green Version]
- Van Rijssel, J.; Kroon, J.; Hoogenboezem, M.; van Alphen, F.P.; de Jong, R.J.; Kostadinova, E.; Geerts, D.; Hordijk, P.L.; van Buul, J.D. The Rho-guanine nucleotide exchange factor Trio controls leukocyte transendothelial migration by promoting docking structure formation. Mol. Biol. Cell 2012, 232, 831–844. [Google Scholar] [CrossRef]
- Woodfin, A.; Voisin, M.B.; Beyrau, M.; Colom, B.; Caille, D.; Diapouli, F.M.; Nash, G.B.; Chavakis, T.; Albelda, S.M.; Rainger, G.E.; et al. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat. Immunol. 2011, 127, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Ostermann, G.; Weber, K.S.; Zernecke, A.; Schröder, A.; Weber, C. JAM-1 is a ligand of the beta integrin LFA-1 involved in transendothelial migration of leukocytes. Nat. Immunol. 2002, 31, 51–58. [Google Scholar]
- Babinska, A.; Azari, B.M.; Salifu, M.O.; Liu, R.; Jiang, X.C.; Sobocka, M.B.; Boo, D.; Al Khoury, G.; Deitch, J.S.; Marmur, J.D.; et al. The F11 receptor (F11R/JAM-A) in atherothrombosis: Overexpression of F11R in atherosclerotic plaques. Thromb. Haemost. 2007, 972, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Chavakis, T.; Keiper, T.; Matz-Westphal, R.; Hersemeyer, K.; Sachs, U.J.; Nawroth, P.P.; Preissner, K.T.; Santoso, S. The junctional adhesion molecule-C promotes neutrophil transendothelial migration in vitro and in vivo. J. Biol. Chem. 2004, 2795, 5602–5608. [Google Scholar]
- Cera, M.R.; Fabbri, M.; Molendini, C.; Corada, M.; Orsenigo, F.; Rehberg, M.; Reichel, C.A.; Krombach, F.; Pardi, R.; Dejana, E. JAM-A promotes neutrophil chemotaxis by controlling integrin internalization and recycling. J. Cell Sci. 2009, 122 Pt 2, 268–277. [Google Scholar] [CrossRef] [Green Version]
- Ferrero, E.; Ferrero, M.E.; Pardi, R.; Zocchi, M.R. The platelet endothelial cell adhesion molecule-1 (PECAM1) contributes to endothelial barrier function. FEBS Lett. 1995, 3743, 23–26. [Google Scholar]
- Marelli-Berg, F.M.; Clement, M.; Mauro, C.; Caligiuri, G. An immunologist’s guide to CD31 function in T-cells. J. Cell Sci. 2013, 126 Pt 11, 2343–2352. [Google Scholar] [CrossRef] [Green Version]
- Tada, Y.; Koarada, S.; Morito, F.; Ushiyama, O.; Haruta, Y.; Kanegae, F.; Ohta, A.; Ho, A.; Mak, T.W.; Nagasawa, K. Acceleration of the onset of collagen-induced arthritis by a deficiency of platelet endothelial cell adhesion molecule 1. Arthritis Rheum. 2003, 483, 280–290. [Google Scholar] [CrossRef]
- Wong, M.X.; Hayball, J.D.; Hogarth, P.M.; Jackson, D.E. The inhibitory co-receptor.; PECAM-1 provides a protective effect in suppression of collagen-induced arthritis. J. Clin. Immunol. 2005, 251, 9–28. [Google Scholar] [CrossRef]
- Graesser, D.; Solowiej, A.; Bruckner, M.; Osterweil, E.; Juedes, A.; Davis, S.; Rudlle, N.H.; Engelhardt, B.; Madri, J.A. Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1-deficient mice. J. Clin. Investig. 2002, 1093, 83–92. [Google Scholar] [CrossRef]
- Lou, O.; Alcaide, P.; Luscinskas, F.W.; Muller, W.A. CD99 is a key mediator of the transendothelial migration of neutrophils. J. Immunol. 2007, 1781, 136–143. [Google Scholar] [CrossRef]
- Schenkel, A.R.; Mamdouh, Z.; Chen, X.; Liebman, R.M.; Muller, W.A. CD99 plays a major role in the migration of monocytes through endothelial junctions. Nat. Immunol. 2002, 31, 43–50. [Google Scholar] [CrossRef]
- Woodfin, A.; Voisin, M.B.; Imhof, B.A.; Dejana, E.; Engelhardt, B.; Nourshargh, S. Endothelial cell activation leads to neutrophil transmigration as supported by the sequential roles of ICAM-2.; JAM-A.; and PECAM-1. Blood 2009, 1136, 246–257. [Google Scholar] [CrossRef]
- Broermann, A.; Winderlich, M.; Block, H.; Frye, M.; Rossaint, J.; Zarbock, A.; Cagna, G.; Linnepe, R.; Schulte, D.; Nottebaum, A.F.; et al. Dissociation of VE-PTP from VE-cadherin is required for leukocyte extravasation and for VEGF-induced vascular permeability in vivo. J. Exp. Med. 2011, 2082, 393–401. [Google Scholar]
- Nottebaum, A.F.; Cagna, G.; Winderlich, M.; Gamp, A.C.; Linnepe, R.; Polaschegg, C.; Filippova, K.; Lyck, R.; Engelhardt, B.; Kamenyeva, O.; et al. VE-PTP maintains the endothelial barrier via plakoglobin and becomes dissociated from VE-cadherin by leukocytes and by VEGF. J. Exp. Med. 2008, 2052, 929–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, R.D.; Noble, K.E.; Larbi, K.Y.; Dewar, A.; Duncan, G.S.; Mak, T.W.; Nourshargh, S. Platelet-endothelial cell adhesion molecule-1 (PECAM-1)-deficient mice demonstrate a transient and cytokine-specific role for PECAM-1 in leukocyte migration through the perivascular basement membrane. Blood 2001, 971, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, O.; Ushiki, T. Neutrophil extravasation in rat mesenteric venules induced by the chemotactic peptide N-formyl-methionyl-luecylphenylalanine (fMLP).; with special attention to a barrier function of the vascular basal lamina for neutrophil migration. Arch. Histol. Cytol. 2004, 671, 7–14. [Google Scholar]
- Wu, C.; Ivars, F.; Anderson, P.; Hallmann, R.; Vestweber, D.; Nilsson, P.; Robenek, H.; Tryggvason, K.; Song, J.; Korpos, E.; et al. Endothelial basement membrane laminin α5 selectively inhibits T lymphocyte extravasation into the brain. Nat. Med. 2009, 155, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Sixt, M.; Engelhardt, B.; Pausch, F.; Hallmann, R.; Wendler, O.; Sorokin, L.M. Endothelial Cell Laminin Isoforms.; Laminins 8 and 10.; Play Decisive Roles in T Cell Recruitment across the Blood–Brain Barrier in Experimental Autoimmune Encephalomyelitis. J. Cell Biol. 2001, 1539, 33–46. [Google Scholar] [CrossRef]
- Wang, S.; Voisin, M.B.; Larbi, K.Y.; Dangerfield, J.; Scheiermann, C.; Tran, M.; Maxwell, P.H.; Sorokin, L.; Nourshargh, S. Venular basement membranes contain specific matrix protein low expression regions that act as exit points for emigrating neutrophils. J. Exp. Med. 2006, 2031, 519–532. [Google Scholar]
- Song, J.; Zhang, X.; Buscher, K.; Wang, Y.; Wang, H.; Di Russo, J.; Li, L.; Lutke-Enking, S.; Zarbock, A.; Stadtmann, A.; et al. Endothelial Basement Membrane Laminin 511 Contributes to Endothelial Junctional Tightness and Thereby Inhibits Leukocyte Transmigration. Cell Rep. 2017, 181, 256–269. [Google Scholar] [CrossRef] [Green Version]
- Pruessmeyer, J.; Hess, F.M.; Alert, H.; Groth, E.; Pasqualon, T.; Schwarz, N.; Nyamoya, S.; Kollert, J.; van der Vorst, E.; Donners, M. Leukocytes require ADAM10 but not ADAM17 for their migration and inflammatory recruitment into the alveolar space. Blood 2014, 1234, 077–088. [Google Scholar] [CrossRef] [Green Version]
- Morsing, S.K.H.; Rademakers, T.; Brouns, S.L.N.; Stalborch, A.D.V.; Donners, M.; van Buul, J.D. ADAM10-Mediated Cleavage of ICAM-1 Is Involved in Neutrophil Transendothelial Migration. Cells 2021, 10, 232. [Google Scholar] [CrossRef]
- Kalucka, J.; de Rooij, L.; Goveia, J.; Rohlenova, K.; Dumas, S.J.; Meta, E.; Conchinha, N.V.; Taverna, F.; Teuwen, L.A.; Veys, K.; et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 2020, 1807, 64–79.e20. [Google Scholar] [CrossRef]
- Maas, S.L.; Soehnlein, O.; Viola, J.R. Organ-Specific Mechanisms of Transendothelial Neutrophil Migration in the Lung, Liver, Kidney, and Aorta. Front. Immunol. 2018, 9, 2739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virmani, R.; Kolodgie, F.D.; Burke, A.P.; Farb, A.; Schwartz, S.M. Lessons from Sudden Coronary Death. Arterioscler. Thromb. Vasc. Biol. 2000, 201, 262–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parthasarathy, S.; Wieland, E.; Steinberg, D. A role for endothelial cell lipoxygenase in the oxidative modification of low density lipoprotein. Proc. Natl. Acad. Sci. USA 1989, 861, 046–050. [Google Scholar] [CrossRef] [Green Version]
- Hartley, A.; Haskard, D.; Khamis, R. Oxidized LDL and anti-oxidized LDL antibodies in atherosclerosis—Novel insights and future directions in diagnosis and therapy. Trends Cardiovasc. Med. 2019, 292, 2–6. [Google Scholar] [CrossRef]
- Maher, B.M.; Dhonnchu, T.N.; Burke, J.P.; Soo, A.; Wood, A.E.; Watson, R.W.G. Statins alter neutrophil migration by modulating cellular Rho activity—a potential mechanism for statins-mediated pleotropic effects? J. Leukoc. Biol. 2009, 851, 86–93. [Google Scholar] [CrossRef]
- Park, J.G.; Ryu, S.Y.; Jung, I.H.; Lee, Y.H.; Kang, K.J.; Lee, M.R.; Lee, M.N.; Sohn, S.K.; Lee, J.H.; Lee, H.; et al. Evaluation of VCAM-1 antibodies as therapeutic agent for atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis 2013, 2263, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Cybulsky, M.I.; Iiyama, K.; Li, H.; Zhu, S.; Chen, M.; Iiyama, M.; Davis, V.; Gutierrez-Ramos, J.C.; Connelly, P.W.; Milstone, D.S. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J. Clin. Investig. 2001, 1071, 255–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitagawa, K.; Matsumoto, M.; Sasaki, T.; Hashimoto, H.; Kuwabara, K.; Ohtsuki, T.; Hori, M. Involvement of ICAM-1 in the progression of atherosclerosis in APOE-knockout mice. Atherosclerosis 2002, 1603, 05–10. [Google Scholar] [CrossRef]
- Samson, T.; van Buul, J.D.; Kroon, J.; Welch, C.; Bakker, E.N.; Matlung, H.L.; van den Berg, T.K.; Sharek, L.; Doerschuk, C.; Hahn, K.; et al. The guanine-nucleotide exchange factor SGEF plays a crucial role in the formation of atherosclerosis. PLoS ONE 2013, 8, e55202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Z.M.; Chapman, S.M.; Brown, A.A.; Frenette, P.S.; Hynes, R.O.; Wagner, D.D. The combined role of P- and E-selectins in atherosclerosis. J. Clin. Investig. 1998, 1021, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Gareus, R.; Kotsaki, E.; Xanthoulea, S.; van der Made, I.; Gijbels, M.J.; Kardakaris, R.; Polykratis, A.; Kollias, G.; de Winther, M.P.; Pasparakis, M.; et al. Endothelial cell-specific NF-kappaB inhibition protects mice from atherosclerosis. Cell Metab. 2008, 83, 72–83. [Google Scholar]
- Beldman, T.J.; Malinova, T.S.; Desclos, E.; Grootemaat, A.E.; Misiak, A.L.S.; van der Velden, S.; van Roomen, C.; Beckers, L.; van Veen, H.A.; Krawczyk, P.M.; et al. Nanoparticle-Aided Characterization of Arterial Endothelial Architecture during Atherosclerosis Progression and Metabolic Therapy. ACS Nano 2019, 131, 3759–3774. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, Q.; Long, L.; Zhang, N.; Liu, Y. Asn563Ser polymorphism of CD31/PECAM-1 is associated with atherosclerotic cerebral infarction in a southern Han population. Neuropsychiatr. Dis. Treat. 2015, 111, 5–20. [Google Scholar]
- Elrayess, M.A.; Webb, K.E.; Bellingan, G.J.; Whittall, R.A.; Kabir, J.; Hawe, E.; Syvanne, M.; Taskinen, M.R.; Frick, M.H.; Nieminen, M.S.; et al. R643G polymorphism in PECAM-1 influences transendothelial migration of monocytes and is associated with progression of CHD and CHD events. Atherosclerosis 2004, 1771, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Harrison, M.; Smith, E.; Ross, E.; Krams, R.; Segers, D.; Buckley, C.D.; Nash, G.B.; Rainger, G.E. The Role of Platelet-Endothelial Cell Adhesion Molecule-1 in Atheroma Formation Varies Depending on the Site-Specific Hemodynamic Environment. Arterioscler. Thromb. Vasc. Biol. 2013, 336, 94–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harry, B.L.; Sanders, J.M.; Feaver, R.E.; Lansey, M.; Deem, T.L.; Zarbock, A.; Bruce, A.C.; Pryor, A.W.; Gelfand, B.D.; Balckman, B.R.; et al. Endothelial cell PECAM-1 promotes atherosclerotic lesions in areas of disturbed flow in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2008, 282, 003–008. [Google Scholar] [CrossRef]
- Goel, R.; Schrank, B.R.; Arora, S.; Boylan, B.; Fleming, B.; Miura, H.; Newman, P.J.; Molthen, R.C.; Newman, D.K. Site-Specific Effects of PECAM-1 on Atherosclerosis in LDL Receptor–Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2008, 281, 996–2002. [Google Scholar] [CrossRef] [Green Version]
- Fornasa, G.; Groyer, E.; Clement, M.; Dimitrov, J.; Compain, C.; Gaston, A.T.; Varthaman, A.; Khallou-Laschet, J.; Newman, D.K.; Graff-Dubois, S.; et al. TCR stimulation drives cleavage and shedding of the ITIM receptor CD31. J. Immunol. 2010, 1845, 485–492. [Google Scholar] [CrossRef] [Green Version]
- Caligiuri, G.; Rossignol, P.; Julia, P.; Groyer, E.; Mouradian, D.; Urbain, D.; Misra, N.; Ollivier, V.; Sapoval, M.; Boutouyrie, P.; et al. Reduced immunoregulatory CD31+ T cells in patients with atherosclerotic abdominal aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 2006, 266, 18–23. [Google Scholar] [CrossRef] [Green Version]
- Groyer, E.; Nicoletti, A.; Ait-Oufella, H.; Khallou-Laschet, J.; Varthaman, A.; Gaston, A.T.; Thaunat, O.; Kaveri, S.V.; Blatny, R.; Stockinger, H.; et al. Atheroprotective effect of CD31 receptor globulin through enrichment of circulating regulatory T-cells. J. Am. Coll. Cardiol. 2007, 503, 44–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ait-Oufella, H.; Salomon, B.L.; Potteaux, S.; Robertson, A.K.; Gourdy, P.; Zoll, J.; Merval, R.; Esposito, B.; Cohen, J.L.; Fisson, S.; et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat. Med. 2006, 121, 78–80. [Google Scholar] [CrossRef]
- De Jong, A.; de Jong, R.C.M.; Peters, E.A.; Arens, R.; Jukema, J.W.; de Vries, M.R.; Quax, P.H.A. P300/CBP Associated Factor (PCAF) Deficiency Enhances Diet-Induced Atherosclerosis in ApoE3*Leiden Mice via Systemic Inhibition of Regulatory T Cells. Front. Cardiovasc. Med. 2021, 7, 604821. [Google Scholar] [CrossRef]
- Klingenberg, R.; Gerdes, N.; Badeau, R.M.; Gisterå, A.; Strodthoff, D.; Ketelhuth, D.F.; Lundberg, A.M.; Rudling, M.; Nilsson, S.K.; Olivecrona, G.; et al. Depletion of FOXP3+ regulatory T cells promotes hypercholesterolemia and atherosclerosis. J. Clin. Investig. 2013, 1231, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Saigusa, R.; Winkels, H.; Ley, K. T cell subsets and functions in atherosclerosis. Nat. Rev. Cardiol. 2020, 173, 87–401. [Google Scholar] [CrossRef]
- Simons, K.H.; de Jong, A.; Jukema, J.W.; de Vries, M.R.; Arens, R.; Quax, P.H.A. T cell co-stimulation and co-inhibition in cardiovascular disease: A double-edged sword. Nat. Rev. Cardiol. 2019, 163, 25–43. [Google Scholar] [CrossRef]
- Cochain, C.; Chaudhari, S.M.; Koch, M.; Wiendl, H.; Eckstein, H.H.; Zernecke, A. Programmed cell death-1 deficiency exacerbates T cell activation and atherogenesis despite expansion of regulatory T cells in atherosclerosis-prone mice. PLoS ONE 2014, 9, e93280. [Google Scholar] [CrossRef] [Green Version]
- Van Gils, J.M.; Zwaginga, J.J.; Hordijk, P.L. Molecular and functional interactions among monocytes.; platelets.; and endothelial cells and their relevance for cardiovascular diseases. J. Leukoc. Biol. 2009, 851, 95–204. [Google Scholar] [CrossRef] [PubMed]
- Karshovska, E.; Zhao, Z.; Blanchet, X.; Schmitt, M.M.; Bidzhekov, K.; Soehnlein, O.; von Hundelshausen, P.; Mattheij, N.J.; Cosemans, J.M.; Megens, R.T.; et al. Hyperreactivity of junctional adhesion molecule A-deficient platelets accelerates atherosclerosis in hyperlipidemic mice. Circ. Res. 2015, 1165, 87–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillgruber, C.; Pöppelmann, B.; Weishaupt, C.; Steingräber, A.K.; Wessel, F.; Berdel, W.E.; Gessner, J.E.; Ho-Tin-Noe, B.; Vestweber, D.; Goerge, T. Blocking neutrophil diapedesis prevents hemorrhage during thrombocytopenia. J. Exp. Med. 2015, 2121, 255–266. [Google Scholar]
- Collot-Teixeira, S.; Martin, J.; McDermott-Roe, C.; Poston, R.; McGregor, J.L. CD36 and macrophages in atherosclerosis. Cardiovasc. Res. 2007, 754, 68–77. [Google Scholar]
- Newby, A.C. Metalloproteinase Expression in Monocytes and Macrophages and its Relationship to Atherosclerotic Plaque Instability. Arterioscler. Thromb. Vasc. Biol. 2008, 282, 108–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parathath, S.; Mick, S.L.; Feig, J.E.; Joaquin, V.; Grauer, L.; Habiel, D.M.; Gassmann, M.; Gardner, L.B.; Fisher, E.A. Hypoxia is present in murine atherosclerotic plaques and has multiple adverse effects on macrophage lipid metabolism. Circ. Res. 2011, 1091, 141–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aarup, A.; Pedersen, T.X.; Junker, N.; Christoffersen, C.; Bartels, E.D.; Madsen, M.; Nielsen, C.H.; Nielsen, L.B. Hypoxia-Inducible Factor-1α Expression in Macrophages Promotes Development of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 361, 782–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björnheden, T.; Levin, M.; Evaldsson, M.; Wiklund, O. Evidence of Hypoxic Areas within the Arterial Wall In Vivo. Arterioscler. Thromb. Vasc. Biol. 1999, 198, 70–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sluimer, J.C.; Gasc, J.M.; van Wanroij, J.L.; Kisters, N.; Groeneweg, M.; Sollewijn Gelpke, M.D.; Cleutjes, J.P.; van den Akker, L.H.; Corvol, P.; Wouters, B.G.; et al. Hypoxia, hypoxia-inducible transcription factor.; and macrophages in human atherosclerotic plaques are correlated with intraplaque angiogenesis. J. Am. Coll. Cardiol. 2008, 511, 258–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parma, L.; Peters, H.A.B.; Baganha, F.; Sluimer, J.C.; de Vries, M.R.; Quax, P.H.A. Prolonged Hyperoxygenation Treatment Improves Vein Graft Patency and Decreases Macrophage Content in Atherosclerotic Lesions in ApoE3*Leiden Mice. Cells 2020, 9, 336. [Google Scholar] [CrossRef] [Green Version]
- Hutter, R.; Speidl, W.S.; Valdiviezo, C.; Sauter, B.; Corti, R.; Fuster, V.; Badimon, J.J. Macrophages transmit potent proangiogenic effects of oxLDL in vitro and in vivo involving HIF-1α activation: A novel aspect of angiogenesis in atherosclerosis. J. Cardiovasc. Transl. Res. 2013, 65, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Chaudhari, S.M.; Sluimer, J.C.; Koch, M.; Theelen, T.L.; Manthey, H.D.; Busch, M.; Caballero-Franco, C.; Vogel, F.; Cochain, C.; Pelisek, J.; et al. Deficiency of HIF1α in Antigen-Presenting Cells Aggravates Atherosclerosis and Type 1 T-Helper Cell Responses in Mice. Arterioscler. Thromb. Vasc. Biol. 2015, 352, 316–325. [Google Scholar] [CrossRef] [Green Version]
- Herbert, S.P.; Stainier, D.Y. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat. Rev. Mol. Cell Biol. 2011, 125, 51–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavard, J.; Gutkind, J.S. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat. Cell Biol. 2006, 81, 223–234. [Google Scholar]
- Lampugnani, M.G.; Orsenigo, F.; Gagliani, M.C.; Tacchetti, C.; Dejana, E. Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J. Cell Biol. 2006, 1745, 93–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donners, M.M.; Wolfs, I.M.; Olieslagers, S.; Mohammadi-Motahhari, Z.; Tchaikovski, V.; Heeneman, S.; van Buul, J.D.; Caolo, V.; Molin, D.G.; Post, M.J.; et al. A disintegrin and metalloprotease 10 is a novel mediator of vascular endothelial growth factor-induced endothelial cell function in angiogenesis and is associated with atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2010, 302, 188–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellings, W.E.; Peeters, W.; Moll, F.L.; Piers, S.R.; van Setten, J.; Van der Spek, P.J.; de Vries, J.P.; Seldenrijk, K.A.; de Bruin, P.C.; Vink, A.; et al. Composition of carotid atherosclerotic plaque is associated with cardiovascular outcome: A prognostic study. Circulation 2010, 1211, 941–950. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.P.; Tournaire, R.; Pouyssegur, J. The angiopoietin-2 gene of endothelial cells is up-regulated in hypoxia by a HIF binding site located in its first intron and by the central factors GATA-2 and Ets-1. J. Cell Physiol. 2008, 2178, 09–18. [Google Scholar] [CrossRef]
- Gamble, J.R.; Drew, J.; Trezise, L.; Underwood, A.; Parsons, M.; Kasminkas, L.; Rudge, J.; Yancopoulos, G.; Vadas, M.A. Angiopoietin-1 is an antipermeability and anti-inflammatory agent in vitro and targets cell junctions. Circ. Res. 2000, 876, 03–07. [Google Scholar] [CrossRef] [Green Version]
- Parma, L.; Baganha, F.; Quax, P.H.A.; de Vries, M.R. Plaque angiogenesis and intraplaque hemorrhage in atherosclerosis. Eur. J. Pharmacol. 2017, 8161, 07–15. [Google Scholar] [CrossRef]
- Melder, R.J.; Yuan, J.; Munn, L.L.; Jain, R.K. Erythrocytes enhance lymphocyte rolling and arrest in vivo. Microvasc. Res. 2000, 593, 16–22. [Google Scholar] [CrossRef]
- Nagy, E.; Eaton, J.W.; Jeney, V.; Soares, M.P.; Varga, Z.; Galajda, Z.; Szentmiklosi, J.; Mehes, G.; Csonka, T.; Smith, A.; et al. Red cells, hemoglobin, heme, iron, and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2010, 301, 347–353. [Google Scholar] [CrossRef]
- Jeney, V.; Balla, G.; Balla, J. Red blood cell, hemoglobin and heme in the progression of atherosclerosis. Front. Physiol. 2014, 5, 379. [Google Scholar] [CrossRef] [Green Version]
- Boyle, J.J.; Harrington, H.A.; Piper, E.; Elderfield, K.; Stark, J.; Landis, R.C.; Haskard, D.O. Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype. Am. J. Pathol. 2009, 1741, 097–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Akahori, H.; Harari, E.; Smith, S.L.; Polavarapu, R.; Karmali, V.; Otsuka, F.; Gannon, R.L.; Braumann, R.E.; Dickinson, M.H.; et al. CD163+ macrophages promote angiogenesis and vascular permeability accompanied by inflammation in atherosclerosis. J. Clin. Investig. 2018, 1281, 106–124. [Google Scholar] [CrossRef] [PubMed]
- De Vries, M.R.; Parma, L.; Peters, H.A.B.; Schepers, A.; Hamming, J.F.; Jukema, J.W.; Goumans, M.J.T.H.; Guo, L.; Finn, A.V.; Virmani, R.; et al. Blockade of vascular endothelial growth factor receptor 2 inhibits intraplaque haemorrhage by normalization of plaque neovessels. J. Intern. Med. 2019, 2855, 9–74. [Google Scholar] [CrossRef]
- Taqueti, V.R.; Di Carli, M.F.; Jerosch-Herold, M.; Sukhova, G.K.; Murthy, V.L.; Folco, E.J.; Kwong, R.Y.; Ozaki, C.K.; Belkin, M.; Nahrendorf, M.; et al. Increased microvascularization and vessel permeability associate with active inflammation in human atheromata. Circ. Cardiovasc. Imag. 2014, 79, 20–29. [Google Scholar] [CrossRef] [Green Version]
- Oksala, N.; Levula, M.; Airla, N.; Pelto-Huikko, M.; Ortiz, R.M.; Järvinen, O.; Salenius, J.P.; Ozsait, B.; Komurcu-Bayrak, E.; Erginel-Unaltuna, N.; et al. ADAM-9, ADAM-15, and ADAM-17 are upregulated in macrophages in advanced human atherosclerotic plaques in aorta and carotid and femoral arteries--Tampere vascular study. Ann. Med. 2009, 412, 79–90. [Google Scholar] [CrossRef]
- Satoh, M.; Ishikawa, Y.; Itoh, T.; Minami, Y.; Takahashi, Y.; Nakamura, M. The expression of TNF-alpha converting enzyme at the site of ruptured plaques in patients with acute myocardial infarction. Eur. J. Clin. Investig. 2008, 389, 7–105. [Google Scholar]
- De Vries, M.R.; Quax, P.H. Plaque angiogenesis and its relation to inflammation and atherosclerotic plaque destabilization. Curr. Opin. Lipidol. 2016, 274, 99–506. [Google Scholar] [CrossRef]
- Virmani, R.; Kolodgie, F.D.; Burke, A.P.; Finn, A.V.; Gold, H.K.; Tulenko, T.N.; Wrenn, S.P.; Narula, J. Atherosclerotic Plaque Progression and Vulnerability to Rupture. Arterioscle. Thromb. Vasc. Biol. 2005, 252, 054–061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parma, L.; Peters, H.A.B.; Sluiter, T.J.; Simons, K.H.; Lazzari, P.; de Vries, M.R.; Quax, P.H.A. bFGF blockade reduces intraplaque angiogenesis and macrophage infiltration in atherosclerotic vein graft lesions in ApoE3*Leiden mice. Sci. Rep. 2020, 101, 5968. [Google Scholar] [CrossRef]
- Van der Vorst, E.P.; Zhao, Z.; Rami, M.; Holdt, L.M.; Teupser, D.; Steffens, S.; Weber, C. Contrasting effects of myeloid and endothelial ADAM17 on atherosclerosis development. Thromb. Haemost. 2017, 1176, 44–46. [Google Scholar]
- Members, T.F.; Montalescot, G.; Sechtem, U.; Achenbach, S.; Andreotti, F.; Arden, C.; Budaj, A.; Bugiardini, R.; Crea, F.; Cuisset, T.; et al. 2013 ESC guidelines on the management of stable coronary artery disease: The Task Force on the management of stable coronary artery disease of the European Society of Cardiology. Eur. Heart J. 2013, 342, 949–3003. [Google Scholar]
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S). Lancet 1994, 3441, 383–389.
- Nohria, A.; Grunert, M.E.; Rikitake, Y.; Noma, K.; Prsic, A.; Ganz, P.; Liao, J.K.; Creager, M.A. Rho kinase inhibition improves endothelial function in human subjects with coronary artery disease. Circ. Res. 2006, 991, 426–432. [Google Scholar] [CrossRef] [Green Version]
- Wagner, A.H.; Köhler, T.; Rückschloss, U.; Just, I.; Hecker, M. Improvement of nitric oxide-dependent vasodilatation by HMG-CoA reductase inhibitors through attenuation of endothelial superoxide anion formation. Arterioscler. Thromb. Vasc. Biol. 2000, 206, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weitz-Schmidt, G.; Welzenbach, K.; Brinkmann, V.; Kamata, T.; Kallen, J.; Bruns, C.; Cottens, S.; Takada, Y.; Hommel, U. Statins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin site. Nat. Med. 2001, 76, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.C.; Jiang, X.Z.; Bai, Q.K.; Deng, S.H.; Zhang, Y.; Zhang, Z.P.; Jiang, Q. Evaluating the Efficacy of Atorvastatin on Patients with Carotid Plaque by an Innovative Ultrasonography. J. Stroke Cerebrovasc. Dis. 2019, 288, 30–37. [Google Scholar] [CrossRef]
- Hochman, J.S.; Phillips, W.J.; Ruggieri, D.; Ryan, S.F. The distribution of atherosclerotic lesions in the coronary arterial tree: Relation to cardiac risk factors. Am. Heart J. 1988, 116 Pt 1, 1217–1222. [Google Scholar] [CrossRef]
- McGill, H.C.; McMahan, C.A.; Herderick, E.E.; Tracy, R.E.; Malcom, G.T.; Zieske, A.W.; Strong, J.P. Effects of Coronary Heart Disease Risk Factors on Atherosclerosis of Selected Regions of the Aorta and Right Coronary Artery. Arterioscler. Thromb. Vasc. Biol. 2000, 208, 36–45. [Google Scholar] [CrossRef] [Green Version]
- Chiu, J.J.; Chien, S. Effects of disturbed flow on vascular endothelium: Pathophysiological basis and clinical perspectives. Physiol. Rev. 2011, 913, 27–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moossy, J. Morphology, sites and epidemiology of cerebral atherosclerosis. Res. Publ. Assoc. Res. Nerv. Ment Dis. 1966, 411, 22. [Google Scholar]
- D’Armiento, F.P.; Bianchi, A.; de Nigris, F.; Capuzzi, D.M.; D’Armiento, M.R.; Crimi, G.; Abete, P.; Palinski, W.; Condorelli, M.; Napoli, C. Age-related effects on atherogenesis and scavenger enzymes of intracranial and extracranial arteries in men without classic risk factors for atherosclerosis. Stroke 2001, 322, 472–479. [Google Scholar] [CrossRef] [Green Version]
- Ritz, K.; Denswil, N.P.; Stam, O.C.G.; Lieshout, J.J.V.; Daemenm, M.J.A.P. Cause and Mechanisms of Intracranial Atherosclerosis. Circulation 2014, 1301, 407–414. [Google Scholar] [CrossRef] [Green Version]
- Napoli, C.; Paternò, R.; Faraci, F.M.; Taguchi, H.; Postiglione, A.; Heistad, D.D. Mildly Oxidized Low-Density Lipoprotein Impairs Responses of Carotid but Not Basilar Artery in Rabbits. Stroke 1997, 282, 266–272. [Google Scholar] [CrossRef]
- De Boer, O.J.; van der Wal, A.C.; Teeling, P.; Becker, A.E. Leucocyte recruitment in rupture prone regions of lipid-rich plaques: A prominent role for neovascularization? Cardiovasc. Res. 1999, 414, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Sluimer, J.C.; Daemen, M.J. Novel concepts in atherogenesis: Angiogenesis and hypoxia in atherosclerosis. J. Pathol. 2009, 2187, 29. [Google Scholar] [CrossRef]
- Steenman, M.; Espitia, O.; Maurel, B.; Guyomarch, B.; Heymann, M.F.; Pistorius, M.A.; Ory, B.; Heymann, D.; Houlgatte, R.; Goueffic, Y.; et al. Identification of genomic differences among peripheral arterial beds in atherosclerotic and healthy arteries. Sci. Rep. 2018, 83, 940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, D.; Matsumura, M.; Rundback, J.; Yoho, J.A.; Witzenbichler, B.; Stone, G.W.; Mintz, G.S.; Maehara, A. Comparison of plaque morphology between peripheral and coronary artery disease (from the CLARITY and ADAPT-DES IVUS substudies). Coron. Artery Dis. 2017, 283, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Takumi, T.; Mathew, V.; Chung, W.Y.; Barsness, G.W.; Rihal, C.S.; Gulati, R.; McCue, E.T.; Holmes, D.R.; Eeckhout, E.; et al. Plaque characteristics and arterial remodeling in coronary and peripheral arterial systems. Atherosclerosis 2012, 2233, 65–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildebrandt, H.A.; Gossl, M.; Mannheim, D.; Versari, D.; Herrmann, J.; Spendlove, D.; Bajanowski, T.; Malyar, N.M.; Erbel, R.; Lerman, L.O.; et al. Differential distribution of vasa vasorum in different vascular beds in humans. Atherosclerosis 2008, 1994, 7–54. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sluiter, T.J.; van Buul, J.D.; Huveneers, S.; Quax, P.H.A.; de Vries, M.R. Endothelial Barrier Function and Leukocyte Transmigration in Atherosclerosis. Biomedicines 2021, 9, 328. https://doi.org/10.3390/biomedicines9040328
Sluiter TJ, van Buul JD, Huveneers S, Quax PHA, de Vries MR. Endothelial Barrier Function and Leukocyte Transmigration in Atherosclerosis. Biomedicines. 2021; 9(4):328. https://doi.org/10.3390/biomedicines9040328
Chicago/Turabian StyleSluiter, Thijs J., Jaap D. van Buul, Stephan Huveneers, Paul H. A. Quax, and Margreet R. de Vries. 2021. "Endothelial Barrier Function and Leukocyte Transmigration in Atherosclerosis" Biomedicines 9, no. 4: 328. https://doi.org/10.3390/biomedicines9040328