
Membrane Carriers and Transporters in Kidney Physiology and Disease




Abstract
:1. Introduction
2. Function of Drug Transporters and Carriers
3. Effects on Endogenous Substrates
4. Effects on Drug Pharmacokinetics and Drug–Drug Interactions (DDIs)
5. Transporters as Therapeutic Drug Targets
6. Effects of Kidney Failure on Renal Drug Transporters
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette transporter |
| ADMA | asymmetric dimethylarginine |
| BCRP | breast cancer resistance protein |
| CrCl | creatinine clearance |
| CKD | chronic kidney disease |
| CMPF | 3-carboxy-4-methyl-5-propyl-2-furanopropanoic acid |
| CRF | chronic renal failure |
| DDI | drug–drug interaction |
| ENT1/SLC29A1 | equilibrative nucleoside transporter 1 |
| ENT2/SLC29A2 | equilibrative nucleoside transporter 2 |
| FDA | US Food and Drug Administration |
| GFR | glomerular filtration rate |
| GLUT9/SLC2A9 | facilitative glucose transporter 9 |
| GSA | guanidine succinate |
| GSH | glutathione |
| HNF1α/HNF1A | hepatocyte nuclear factor 1α |
| HNF4α/HNF4A | hepatocyte nuclear factor 4α |
| MATE1/SLC47A1 | multidrug and toxin extrusion protein 1 |
| MATE2/SLC47A2 | multidrug and toxin extrusion protein 2 |
| MATE2-K/SLC47A2 | multidrug and toxin extrusion protein 2 kidney-specific |
| MRP1/ABCC1 | multidrug resistance-associated protein 2 |
| MRP2/ABCC2 | multidrug resistance-associated protein 2 |
| MRP3/ABCC3 | multidrug resistance-associated protein 3 |
| MRP4/ABCC4 | multidrug resistance-associated protein 4 |
| MRP5/ABCC5 | multidrug resistance-associated protein 5 |
| MRP6/ABCC6 | multidrug resistance-associated protein 6 |
| MTX | methotrexate |
| NSAIDs | non-steroidal anti-inflammatory drugs |
| OAT1/SLC22A6 | organic anion transporter 1 |
| OAT2/SLC22A7 | organic anion transporter 2 |
| OAT3/SLC22A8 | organic anion transporter 3 |
| OAT4/SLC22A11 | organic anion transporter 4 |
| OATP1A2/SLCO1A2 | kidney organic anion transporting polypeptide 1A2 |
| OATP2B1/SLCO2B1 | kidney organic anion transporting polypeptide 2B1 |
| OATP4C1/SLCO4C1 | kidney organic anion transporting polypeptide 4C1 |
| OCT1/SLC22A1 | organic cation transporter 1 |
| OCT2/SLC22A2 | organic cation transporter 2 |
| OCT3/SLC22A3 | organic cation transporter 3 |
| OCTN1/SLC22A4 | organic cation/carnitine transporter 1 |
| OCTN2/SLC22A5 | organic cation/carnitine transporter 2 |
| OSTα/SLC51A | organic solute transporter α |
| OSTβ/SLC51B | organic solute transporter β |
| PAH | p-aminohippurate |
| PAN | puromycin aminonucleoside |
| PEPT1/SLC15A1 | peptide transporter 1 |
| PEPT2/SLC15A2 | peptide transporter 2 |
| P-gp. MDR1/ABCB1 | glycoprotein, multidrug resistance protein 1 |
| PMAT/SLC29A4 | plasma membrane amino-acid transporter |
| SGLT2/SLC5A2 | sodium-glucose co-transporter 2 |
| SLC | solute carrier |
| SLCO4A1/OATP4A1 | kidney organic anion transporting polypeptide 4A1 |
| URAT1/SLC22A12 | urate transporter 1 |
| XO | xanthine oxidase |
References
- Prasad, B.; Johnson, K.; Billington, S.; Lee, C.; Chung, G.W.; Brown, C.D.; Kelly, E.J.; Himmelfarb, J.; Unadkat, J.D. Abundance of drug transporters in the human kidney cortex as quantified by quantitative targeted proteomics. Drug Metab. Dispos. 2016, 44, 1920–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oswald, S.; Müller, J.; Neugebauer, U.; Schröter, R.; Herrmann, E.; Pavenstädt, H.; Ciarimboli, G. Protein abundance of clinically relevant drug transporters in the human kidneys. Int. J. Mol. Sci. 2019, 24, 5303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russel, F.G.; Masereeuw, R.; van Aubel, R.A. Molecular aspects of renal anionic drug transport. Annu. Rev. Physiol. 2002, 64, 563–594. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, K.M.; Stocker, S.L.; Wittwer, M.B.; Xu, L.; Giacomini, K.M. Renal transporters in drug development. Annu. Rev. Pharmacol. Toxicol. 2012, 53, 503–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacomini, K.M.; Galetin, A.; Huang, S.-M. The international transporter consortium: Summarizing advances in the role of transporters in drug development. Clin. Pharmacol. Ther. 2018, 104, 766–771. [Google Scholar] [CrossRef] [PubMed]
- FDA. In Vitro Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research. Available online: https://www.fda.gov/drugs/guidance-compliance-regulatory-information/guidances-drugs (accessed on 10 March 2020).
- Kremer, J.M.; Hamilton, R.A. The effects of nonsteroidal anti-inflammatory drugs on methotrexate (MTX) pharmacokinetics: Impairment of renal clearance of MTX at weekly maintenance doses but not at 7.5 mg. J. Rheumatol. 1995, 22, 2072–2077. [Google Scholar] [PubMed]
- Bourre-Tessier, J.; Haraoui, B. Methotrexate drug interactions in the treatment of rheumatoid arthritis: A systematic review. J. Rheumatol. 2010, 37, 1416–1421. [Google Scholar] [CrossRef]
- Belz, G.G.; Doering, W.; Munkes, R.; Matthews, J. Interaction between digoxin and calcium antagonists and antiarrhythmic drugs. Clin. Pharm. Ther. 1983, 33, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Song, I.H.; Zong, J.; Borland, J.; Jerva, F.; Wynne, B.; Zamek-Gliszczynski, M.J.; Humphreys, J.E.; Bowers, G.D.; Choukour, M. The effect of dolutegravir on the pharmacokinetics of metformin in healthy subjects. J. Acquir. Immune Defic. Syndr. 2016, 72, 400–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Guo, D.; Dong, Z.; Zhang, W.; Zhang, L.; Huang, S.-M.; Polli, J.E.; Shua, Y. Ondansetron can enhance cisplatin-induced nephrotoxicity via inhibition of multiple toxin and extrusion proteins (MATEs). Toxicol. Appl. Pharmacol. 2013, 273, 100–109. [Google Scholar] [CrossRef] [Green Version]
- Roth, M.; Obaidat, A.; Hagenbuch, B. OATPs, OATs and OCTs: The organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br. J. Pharmacol. 2012, 165, 1260–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huls, M.; Brown, C.D.A.; Windass, A.S.; Heemskerk, S.; Russel, F.G.M.; Masereeuw, R. The breast cancer resistance protein transporter ABCG2 is expressed in the human kidney proximal tubule apical membrane. Kidney Int. 2008, 73, P220–P225. [Google Scholar] [CrossRef] [Green Version]
- Yabuuchi, H.; Tamai, I.; Nezu, J.; Sakamoto, K.; Oku, A.; Shimane, M.; Sai, Y.; Tsuji, A. Novel membrane transporter OCTN1 mediates multispecific, bidirectional, and pH-dependent transport of organic cations. J. Pharmacol. Exp. Ther. 1999, 289, 768–773. [Google Scholar] [PubMed]
- Tamai, I.; Ohashi, R.; Nezu, J.; Yabuuchi, H.; Oku, A.; Shimane, M.; Sai, Y.Z.; Tsuji, A. Molecular and functional identification of sodium ion-dependent, high affinity human carnitine transporter OCTN2. J. Biol. Chem. 1998, 273, 20378–20382. [Google Scholar] [CrossRef] [Green Version]
- Nigam, S.K.; Wu, W.; Bush, K.T.; Hoenig, M.P.; Blantz, R.C.; Bhatnagar, V. Handling of drugs, metabolites, and uremic toxins by kidney proximal tubule drug transporters. Clin. J. Am. Soc. Nephrol. 2015, 10, 2039–2049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enomoto, A.; Kimura, H.; Chairoungdua, A.; Shigeta, Y.; Jutabha, P.; Cha, S.H.; Hosoyamada, M.; Takeda, M.; Sekine, T.; Igarashi, T.; et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002, 417, 447–452. [Google Scholar] [CrossRef]
- Hagos, Y.; Stein, D.; Ugele, B.; Burckhardt, G.; Bahn, A. Human renal organic anion transporter 4 operates as an asymmetric urate transporter. J. Am. Soc. Nephrol. 2007, 18, 430–439. [Google Scholar] [CrossRef]
- Hagenbuch, B.; Stieger, B. The SLCO (former SLC21) superfamily of transporters. Mol. Asp. Med. 2013, 34, 396–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballatori, N.; Christian, W.V.; Lee, J.Y.; Dawson, P.A.; Soroka, C.J.; Boyer, J.L.; Madejczyk, M.S.; Li, N. OSTalpha-OSTbeta: A major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology 2005, 42, 1270–1279. [Google Scholar] [CrossRef]
- Scheffer, G.L.; Kool, M.; de Haas, M.; de Vree, J.M.; Pijnenborg, A.C.; Bosman, D.K.; Elferink, R.; van der Valk, P.; Borst, P.; Scheper, R.J. Tissue distribution and induction of human multidrug resistant protein. Lab. Investig. 2002, 82, 193–201. [Google Scholar] [CrossRef]
- Miyamoto, M.; Yoshida, Y.; Taguchi, I.; Nagasaka, Y.; Tasaki, M.; Zhang, Y.; Xu, B.; Nameta, M.; Sezaki, H.; Cuellar, L.; et al. In-depth proteomic profiling of the normal human kidney glomerulus using two-dimensional protein prefractionation in combination with liquid chromatography-tandem mass spectrometry. J. Proteome Res. 2007, 6, 3680–3690. [Google Scholar] [CrossRef] [PubMed]
- Zennaro, C.; Rastaldi, M.P.; Pascolo, L.; Stebel, M.; Trevisan, E.; Artero, M.; Tiribelli, C.; Di Maso, V.; Carraro, M. Podocyte expression of membrane transporters involved in puromycin aminonucleoside-mediated injury. PLoS ONE 2013, 8, e66159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zennaro, C.; Artero, M.; Di Maso, V.; Carraro, M. Small molecule membrane transporters in the mammalian podocyte: A pathogenic and therapeutic target. Int. J. Mol. Sci. 2014, 15, 21366–21380. [Google Scholar] [CrossRef] [Green Version]
- Xia, L.; Zhou, M.; Kalhorn, T.F.; Ho, H.T.; Wang, J. Podocyte-specific expression of organic cation transporter PMAT: Implication in puromycin aminonucleoside nephrotoxicity. Am. J. Physiol. Renal Physiol. 2009, 296, F1307–F1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasik, A.A.; Lehtonen, S. Glucose transporters in diabetic kidney disease—Friends or foes? Front. Endocrinol. 2018, 9, 155. [Google Scholar] [CrossRef] [Green Version]
- Motohashi, H.; Inui, K. Organic cation transporter OCTs (SLC22) and MATEs (SLC47) in the human kidney. AAPS J. 2013, 15, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Seegmiller, J.C.; Burns, B.E.; Schinstock, C.A.; Lieske, J.C.; Larson, T.S. Discordance between iothalamate and iohexol urinary clearances. Am. J. Kidney Dis. 2016, 67, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Boger, C.A.; Gorski, M.; Li, M.; Hoffmann, M.M.; Huang, C.; Yang, Q.; Teumer, A.; Krane, V.; O’Seaghdha, C.; Kutalik, Z.; et al. Association of eGFR-related loci identified by GWAS with incident CKD and ESRD. PLoS Genet. 2011, 7, e1002292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepist, E.I.; Zhang, X.; Hao, J.; Huang, J.; Kosaka, A.; Birkus, G.; Murray, B.P.; Bannister, R.; Cihlar, T.; Huang, Y.; et al. Contribution of the organic anion transporter OAT2 to the renal active tubular secretion of creatinine and mechanism for serum creatinine elevations caused by cobicistat. Kidney Int. 2014, 86, 350–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochs, H.; Gugler, R.; Guthoff, T.; Greenblatt, D. Effect of cimetidine on digoxin kinetics and creatinine clearance. Am. Heart, J. 1984, 107, 170–172. [Google Scholar] [CrossRef]
- Berglund, F.; Killander, J.; Pompeius, R. Effect of trimethoprim-sulfamethoxazole on the renal excretion of creatinine in man. J. Urol. 1975, 114, 802–808. [Google Scholar] [CrossRef]
- Opravil, M.; Keusch, G.; Luthy, R. Pyrimethamine inhibits renal secretion of creatinine. Antimicrob. Agents Chemother. 1993, 37, 1056–1060. [Google Scholar] [CrossRef] [Green Version]
- Scotcher, D.; Arya, V.; Yang, X.; Zhao, P.; Zhang, L.; Huang, S.-M.; Rostami-Hodjegan, A.; Galetin, A. Mechanistic models as framework for understanding biomarker disposition: Prediction of creatinine-drug interactions. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Shi, Y.; Zhuang, S.; Liu, N. Recent advances on uric acid transporters. Oncotarget 2017, 8, 100852–100862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, A.K.; Mount, D.B. The molecular physiology of uric acid homeostasis. Annu. R. Physiol. 2015, 77, 323–345. [Google Scholar] [CrossRef]
- Caulfield, M.J.; Munroe, P.B.; O’Neill, D.; Witkowska, K.; Charchar, F.J.; Doblado, M.; Evans, S.; Eyheramendy, S.; Onipinla, A.; Howard, P.; et al. SLC2A9 is a high-capacity urate transporter in humans. PLoS Med. 2008, 5, e197. [Google Scholar] [CrossRef] [PubMed]
- Burckhardt, G. Drug transport by organic anion transporters (OATs). Pharmacol. Ther. 2012, 136, 106–130. [Google Scholar] [CrossRef]
- Shen, Z.; Rowlings, C.; Kerr, B.; Hingorani, V.; Manhard, K.; Quart, B.; Yeh, L.T.; Storgard, C. Pharmacokinetics, pharmacodynamics, and safety of lesinurad, a selective uric acid reabsorption inhibitor, in healthy adult males. Drug Des. Dev. Ther. 2015, 9, 3423–3434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derosa, G.; Maffioli, P.; Sahebkar, A. Plasma uric acid concentrations are reduced by fenofibrate: A systematic review and meta-analysis of randomized placebo-controlled trials. Pharm. Res. 2015, 102, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Wolff, M.L.; Cruz, J.L.; Vanderman, A.J.; Brown, J.N. The effect of angiotensin II receptor blockers on hyperuricemia. Ther. Adv. Chronic Dis. 2015, 6, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Mihaila, S.M.; Faria, J.; Stefens, M.F.J.; Stamatialis, D.; Verhaar, M.C.; Gerritsen, K.G.F.; Masereeuw, R. Drugs commonly applied to kidney patients may compromise renal tubular uremic toxins excretion. Toxins 2020, 12, 391. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.P.; Sweet, D.H.; Peng, Y.-H.; Hsieh, Y.-W.; Chao, P.-D.; Hou, Y.-C.; Lin, S.-P. Effects of nonsteroidal anti-inflammatory drugs on the renal excretion of indoxyl sulfate, a nephro-cardiovascular toxin, in rats. Eur. J. Pharm. Sci. 2017, 101, 66–70. [Google Scholar] [CrossRef]
- Yaxley, J. Common analgesic agents and their roles in analgesic nephropathy: A commentary on the evidence. Korean J. Fam. Med. 2016, 37, 310–331. [Google Scholar] [CrossRef] [PubMed]
- Toyohara, T.; Suzuki, T.; Morimoto, R.; Akiyama, Y.; Souma, T.; Shiwaku, H.O.; Takeuchi, Y.; Mishima, E.; Abe, M.; Tanemoto, M.; et al. SLCO4C1 transporter eliminates uremic toxins and attenuates hypertension and renal inflammation. J. Am. Soc. Nephrol. 2009, 20, 2546–2555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koepsell, H. Organic cation transporters in health and disease. Pharmacol. Rev. 2020, 72, 253–319. [Google Scholar] [CrossRef]
- Liang, X.; Giacomini, K.M. Transporters involved in metformin pharmacokinetics and treatment. J. Pharmacol. Sci. 2017, 106, 2245–2250. [Google Scholar] [CrossRef] [Green Version]
- Bourdet, D.L.; Pritchard, J.B.; Thakker, D.R. Differential substrate and inhibitory activities of ranitidine and famotidine toward human organic cation transporter 1 (hOCT1; SLC22A1), hOCT2 (SLC22A2), and hOCT3 (SLC22A3). J. Pharmacol. Exp. Ther. 2005, 315, 1288–1297. [Google Scholar] [CrossRef] [Green Version]
- Jung, N.; Lehmann, C.; Rubbert, A.; Knispel, M.; Hartmann, P.; van Lunzen, J.; Stellbrink, H.-J.; Faetkenheuer, G.; Taubert, D. Relevance of the organic cation transporters 1 and 2 for antiretroviral drug therapy in human immunodeficiency virus infection. Drug Metab. Dispos. 2008, 36, 1616–1623. [Google Scholar] [CrossRef]
- Dudley, A.J.; Bleasby, K.; Brown, C.D. The organic cation transporter OCT2 mediates the uptake of ß-adrenoceptor antagonists across the apical membrane of renal LLC-PK1 cell monolayers. Br. J. Pharmacol. 2000, 131, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Yokoo, S.; Yonezawa, A.; Masuda, S.; Fukatsu, A.; Katsura, T.; Inui, K. Differential contribution of organic cation transporters, OCT2 and MATE1, in platinum agent-induced nephrotoxicity. Biochem. Pharmacol. 2007, 74, 477–487. [Google Scholar] [CrossRef]
- Tanihara, Y.; Masuda, S.; Sato, T.; Katsura, T.; Ogawa, O.; Inui, K.I. Substrate specificity of MATE1 and MATE2-K, human multidrug and toxin extrusions/H+-organic cation antiporters. Biochem. Pharmacol. 2007, 74, 359–371. [Google Scholar] [CrossRef] [Green Version]
- Harrach, S.; Ciarimboli, G. Role of transporters in the distribution of platinum-based drugs. Front. Pharmacol. 2015, 6, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohta, K.Y.; Imamura, Y.; Okudaira, N.; Atsumi, R.; Inoue, K.; Yuasa, H. Functional characterization of multidrug and toxin extrusion protein 1 as a facilitative transporter for fluoroquinolones. J. Pharmacol. Exp. Ther. 2009, 328, 628–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taub, M.E.; Podila, L.; Ely, D.; Almeida, I. Functional assessment of multiple P-glycoprotein (P-gp) probe substrates: Influence of cell line and modulator concentration on P-gp activity. Drug Metab. Dispos. 2005, 33, 1679–1687. [Google Scholar] [CrossRef]
- Wang, R.; Sun, X.; Deng, Y.-S.; Qiu, X.-W. Effects of MDR1 1236C > T-2677G > T-3435C > T polymorphisms on the intracellular accumulation of tacrolimus, cyclosporine A, sirolimus and everolimus. Xenobiotica 2019, 49, 1373–1378. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Gyémánt, N.; Molnár, J.; Hilgeroth, A. Comparative effects on intestinal absorption in situ by P-glycoprotein-modifying HIV protease inhibitors. Pharm. Res. 2004, 21, 1862–1866. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Kwan, P.; Zuo, Z.; Baum, L. In vitro concentration dependent transport of phenytoin and phenobarbital, but not ethosuximide, by human P-glycoprotein. Life Sci. 2010, 86, 899–905. [Google Scholar] [CrossRef]
- Bogman, K.; Peyer, A.K.; Török, M.; Küsters, E.; Drewe, J. HMG-CoA reductase inhibitors and P-glycoprotein modulation. Br. J. Pharmacol. 2001, 132, 1183–1192. [Google Scholar] [CrossRef] [Green Version]
- Takeda, M.; Khamdang, S.; Narikawa, S.; Kimura, H.; Hosoyamada, M.; Cha, S.H.; Sekine, T.; Endou, H. Characterization of methotrexate transport and its drug interactions with human organic anion transporters. J. Pharmacol. Exp. Ther. 2002, 302, 666–671. [Google Scholar] [CrossRef] [Green Version]
- El-Sheikh, A.A.K.; van den Heuvel, J.J.M.W.; Koenderink, J.B.; Russel, F.G.M. Interaction of nonsteroidal anti-inflammatory drugs with multidrug resistance protein (MRP) 2/ABCC2- and MRP4/ABCC4-mediated methotrexate transport. J. Pharmacol. Exp. Ther. 2007, 320, 229–235. [Google Scholar] [CrossRef] [Green Version]
- Uwai, Y.; Saito, H.; Hashimoto, Y.; Inui, K.I. Interaction and transport of thiazide diuretics, loop diuretics, and acetazolamide via rat renal organic anion transporter rOAT1. J. Pharmacol. Exp. Ther. 2000, 295, 261–265. [Google Scholar]
- Yin, J.; Wagner, D.J.; Prasad, B.; Isoherranen, N.; Thummel, K.E.; Wang, J. Renal secretion of hydrochlorothiazide involves organic anion transporter 1/3, organic cation transporter 2, and multidrug and toxin extrusion protein 2-K. Am. J. Physiol. Renal Physiol. 2019, 317, F805–F814. [Google Scholar] [CrossRef]
- Khamdang, S.; Takeda, M.; Noshiro, R.; Narikawa, S.; Enomoto, A.; Anzai, N.; Piyachaturawat, P.; Endou, H. Interactions of human organic anion transporters and human organic cation transporters with nonsteroidal anti-inflammatory drugs. J. Pharmacol. Exp. Ther. 2002, 303, 534–539. [Google Scholar] [CrossRef]
- Sato, M.; Iwanaga, T.; Mamada, H.; Ogihara, T.; Yabuuchi, H.; Maeda, T.; Tamai, I. Involvement of uric acid transporters in alteration of serum uric acid level by angiotensin II receptor blockers. Pharm. Res. 2008, 25, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Babu, E.; Narikawa, S.; Endou, H. Interaction of human organic anion transporters with various cephalosporin antibiotics. Eur. J. Pharmacol. 2002, 438, 137–142. [Google Scholar] [CrossRef]
- Uwai, Y.; Ida, H.; Tsuji, Y.; Katsura, T.; Inui, K.I. Renal transport of adefovir, cidofovir, and tenofovir by SLC22A family members (hOAT1, hOAT3, and hOCT2). Pharm. Res. 2007, 24, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Srimaroeng, C.; Perry, J.L.; Pritchard, J.B. Physiology, structure, and regulation of the cloned organic anion transporters. Xenobiotica 2008, 38, 889–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanyuk, A.; Livio, F.; Biollaz, J.; Buclin, T. Renal Drug Transporters and Drug Interactions. Clin. Pharmacokinet. 2017, 56, 825–892. [Google Scholar] [CrossRef]
- Lepist, I.; Ray, A.S. Renal transporter-mediated drug-drug interactions: Are they clinically relevant? J. Clin. Pharmacol. 2016, 56, S73–S81. [Google Scholar] [CrossRef] [Green Version]
- Zelniker, T.A.; Wiviott, S.D.; Raz, I.; Im, K.; Goodrich, E.L.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Furtado, R.H.M.; et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet 2019, 393, 31–39. [Google Scholar] [CrossRef]
- Tsuda, M.; Terada, T.; Ueba, M.; Sato, T.; Masuda, S.; Katsura, T.; Inui, K.-I. Involvement of human multidrug and toxin extrusion 1 in the drug interaction between cimetidine and metformin in renal epithelial cells. J. Pharmacol. Exp. Ther. 2009, 329, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Somogyi, A.; Stockley, C.; Keal, J.; Rolan, P.; Bochner, F. Reduction of metformin renal tubular secretion by cimetidine in man. Br. J. Clin. Pharmacol. 1987, 23, 545–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusuhara, H.; Ito, S.; Kumagai, Y.; Jiang, M.; Shiroshita, T.; Moriyama, Y.; Inoue, K.; Yuasa, H.; Sugiyama, Y. Effects of aMATE protein inhibitor, pyrimethamine, on the renal elimination of metformin at oral microdose and at therapeutic dose in healthy subjects. Clin. Pharmacol. Ther. 2011, 89, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Grun, B.; Kiessling, M.K.; Burhenne, J.; Riedel, K.-D.; Weiss, J.; Rauch, G.; Haefeli, W.E.; Czock, D. Trimethoprimmetformin interaction and its genetic modulation by OCT2 and MATE1 transporters. Br. J. Clin. Pharmacol. 2013, 76, 787–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Jia, Y.; Song, Y.; Lu, C.; Li, Y.; Chen, M.; Wang, M.; Wen, A. The effect of lansoprazole, an OCT inhibitor, on metformin pharmacokinetics in healthy subjects. Eur. J. Clin. Pharmacol. 2014, 70, 141–146. [Google Scholar] [CrossRef]
- Sanchez-Niño, M.D.; Zheng-Lin, B.; Valiño-Rivas, L.; Sanz, A.B.; Ramos, A.M.; Luño, J.; Goicoechea, M.; Ortiz, A. Lesinurad: What the nephrologist should know. Clin. Kidney J. 2017, 10, 679–687. [Google Scholar] [CrossRef]
- Shitara, Y.; Sato, H.; Sugiyama, Y. Evaluation of drug-drug interaction in the hepatobiliary and renal transport of drugs. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 689–723. [Google Scholar] [CrossRef]
- Hamada, T.; Ichida, K.; Hosoyamada, M.; Mizuta, E.; Yanagihara, K.; Sonoyama, K.; Sugihara, S.; Igawa, O.; Hosoya, T.; Ohtahara, A.; et al. Uricosuric action of losartan via the inhibition of urate transporter 1 (URAT 1) in hypertensive patients. Am. J. Hypertens. 2008, 21, 1157–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uetake, D.; Ohno, I.; Ichida, K.; Yamaguchi, Y.; Saikawa, H.; Endou, H.; Hosoya, T. Effect of fenofibrate on uric acid metabolism and urate transporter 1. Intern. Med. 2010, 49, 9–94. [Google Scholar] [CrossRef] [Green Version]
- FDA. U.S. Food and Drug Administration. Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. Available online: https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers (accessed on 10 March 2021).
- Cunningham, R.F.; Israili, Z.H.; Dayton, P.G. Clinical pharmacokinetics of probenecid. Clin. Pharmacokinet. 1981, 6, 135–151. [Google Scholar] [CrossRef]
- Robbins, N.; Koch, S.E.; Tranter, M.; Rubinstein, J. The history and future of probenecid. Cardiovasc. Toxicol. 2012, 12, 1–9. [Google Scholar] [CrossRef]
- Cundy, K.C.; Petty, B.G.; Flaherty, J.; Fisher, P.E.; Polis, M.A.; Wachsman, M. Clinical pharmacokinetics of cidofovir in human immunodeficiency virus–infected patients. Antimicrob. Agents Chemother. 1995, 39, 1247–1252. [Google Scholar] [CrossRef] [Green Version]
- Hemmersbach, P. The Probenecid-story—A success in the fight against doping through out-of-competition testing. Drug Test. Anal. 2020, 12, 589–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.H. Sodium-sensitive, probenecid-insensitive p-aminohippuric acid uptake in cultured renal proximal tubule cells of the rabbit. Proc. Soc. Exp. Biol. Med. 1992, 199, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.; De Napoli, I.E.; Fedecostante, M.; Schophuizen, C.M.; Chevtchik, N.V.; Wilmer, M.J.; van Asbeck, A.H.; Croes, H.J.; PertijWetzels, J.C.; Hilbrands, L.B. Human proximal tubule epithelial cells cultured on hollow fibers: Living membranes that actively transport organic cations. Sci. Rep. 2015, 5, 16702. [Google Scholar] [CrossRef] [PubMed]
- Naud, J.; Michaud, J.; Beauchemin, S.; Hébert, M.-J.; Roger, M.; Lefrancois, S.; Leblond, F.; Pichette, V. Effects of chronic renal failure on kidney drug transporters and cytochrome P450 in rats. Drug Metab. Dispos. 2011, 39, 1363–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurai, Y.; Motohashi, H.; Ueo, H.; Masuda, S.; Saito, H.; Okuda, M.; Mori, N.; Matsuura, M.; Doi, T.; Fukatsu, A.; et al. Expression levels of renal organic anion transporters (OATs) and their correlation with anionic drug excretion in patients with renal diseases. Pharm. Res. 2004, 21, 61–67. [Google Scholar] [CrossRef]
- Zhong, K.; Li, X.; Xie, C.; Zhang, Y.; Zhong, D.; Chen, X. Effects of renal impairment on the pharmacokinetics of morinidazole: Uptake transporter-mediated renal clearance of the conjugated metabolites. Antimicrob. Agents Chemother. 2014, 58, 4153–4161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, O.; Hong, S.M.; Blouch, K. Alteration in renal organic anion transporter 1 after ischemia/reperfusion in cadaveric renal allografts. J. Histochem. Cytochem. 2007, 55, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Tin, A.; Marten, J.; Kuhns, V.; Li, Y.; Wuttke, M.; Kirsten, H.; Sieber, K.; Qiu, C.; Gorski, M.; Yu, Z. Target genes, variants, tissues and transcriptional pathways influencing human serum urate levels. Nat. Genet. 2019, 51, 1459–1474. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, T.; Watanabe, H.; Yoshitome, K.; Morisaki, T.; Hamada, A.; Nonoguchi, H.; Kohda, Y.; Tomita, K.; Inui, K.; Saito, H. Downregulation of organic anion transporters in rat kidney under ischemia/reperfusion-induced acute [corrected] renal failure. Kidney Int. 2007, 71, 539–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Meng, Q.; Wang, C.; Liu, Q.; Guo, X.; Sun, H.; Peng, J.; Ma, X.; Kaku, T.; Liu, K. Changes in expression of renal Oat1, Oat3 and Mrp2 in cisplatin-induced acute renal failure after treatment of JBP485 in rats. Toxicol. Appl. Pharmacol. 2012, 264, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.H.; Murakami, T.; Okochi, A.; Yumoto, R.; Nagai, J.; Takano, M. Expression and function of P-glycoprotein in rats with glycerol-induced acute renal failure. Eur. J. Pharmacol. 2000, 406, 453–460. [Google Scholar] [CrossRef]
- Hsueh, C.-H.; Yoshida, K.; Zhao, P.; Meyer, T.W.; Zhang, L.; Huang, S.-M.; Giacomini, K.M. Identification and quantitative assessment of uremic solutes as inhibitors of renal organic anion transporters, OAT1 and OAT3. Mol. Pharm. 2016, 13, 3130–3140. [Google Scholar] [CrossRef]
- Hsueh, C.-H.; Zhao, P.; Meyer, T.; Zhang, L.; Huang, S.-M.; Giacomini, K.M. Secretory clearance mediated by organic cation transporter 2 reduced in parallel with glomerular filtration rate in patients with chronic kidney disease despite the potential OCT2 inhibition effect by uremic solutes. In Proceedings of the AAPS Annual Meeting, Denver, CO, USA, 13–16 November 2016. [Google Scholar]


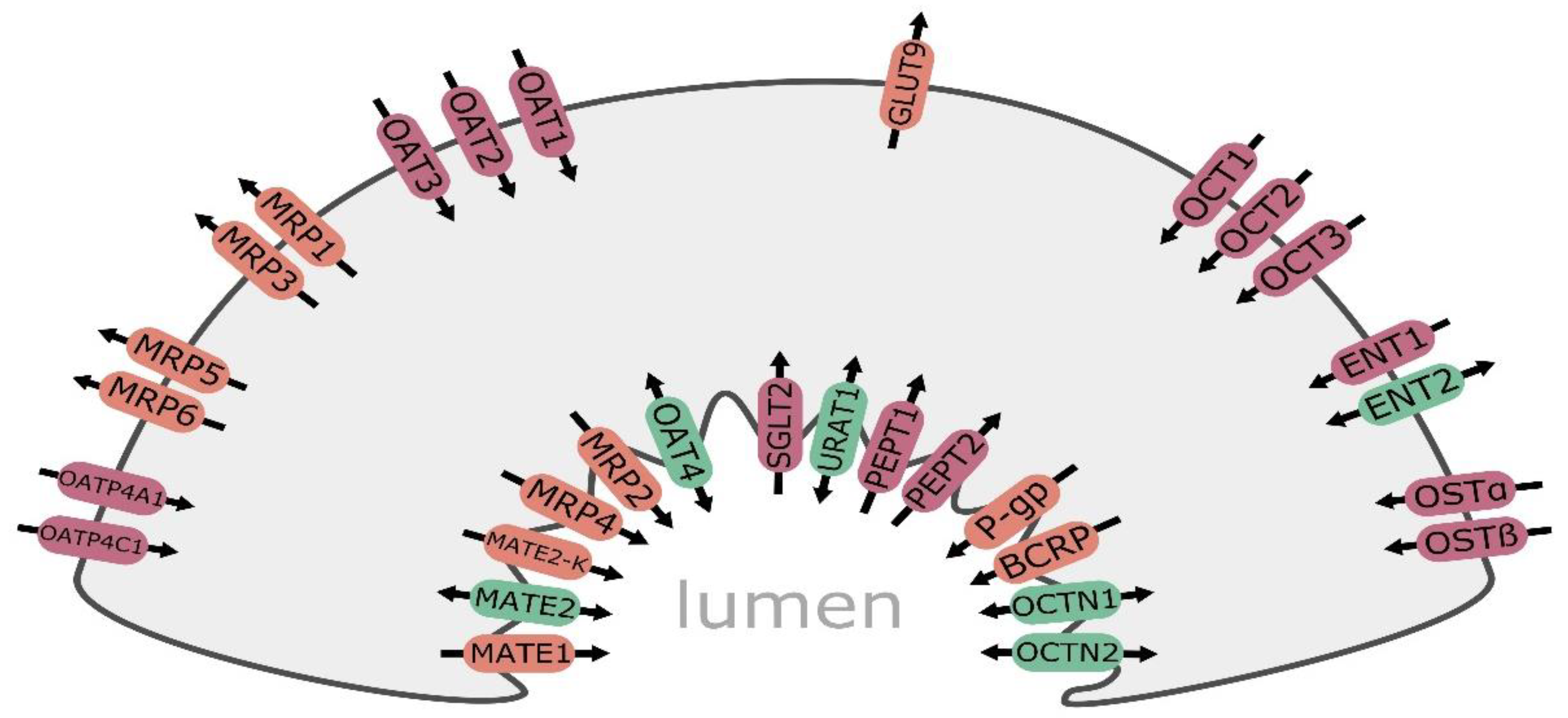{kind=link}
| Transporter | Substrate | |
|---|---|---|
| Endogenous | Drugs | |
| ABC transporters | ||
| ABCB1/MDR1 | Aldosterone, β-amyloid, corticosterone, cortisol | Actinomycin D, amitriptyline, amprenavir, atorvastatin, carbamazepine, celiprolol, chlorpromazine, clopidogrel, citalopram, colchicine, cyclosporin A, daunorubicin, dexamethasone, digoxin, diltiazem, doxycycline, doxorubicin, erythromycin, etoposide, fexofenadine, imatinib, indinavir, irinotecan, itraconazole, ketoconazole, lamotrigine, lansoprazole, levetiracetam, levofloxacin, loperamide, losartan, lovastatin, melphalan, methylprednisolone, mitomycin C, mitoxantrone, morphine, nelfinavir, omeprazole, ondansetron, paclitaxel, pantoprazole, pentazocine, phenobarbital, phenothiazine, phenytoin, propranolol, quinidine, ranitidine, rhodamine-123, rifampicin, ritonavir, saquinavir, simvastatin, sirolimus, sparfloxacin, tacrolimus, talinolol, 99mTc-MIBI, teniposide, terfenadine, tetracycline, topotecan, vecuronium, verapamil, vinblastine, vincristine |
| P-glycoprotein | ||
| ABCC2/MRP2 | Bilirubin-G, estrone-3-S, glutathione, prostaglandin A2-GS | Adefovir, aflatoxin B1-epoxide-GS, ampicillin, azithromycin, ceftriaxone, cidofovir, cisplatin, cyclophosphamide-GS, dinitrophenyl-GS, doxorubicin, doxorubicin-GS, epirubicin, estradiol 17βD-G, etacrynic acid-GS, etoposide-G, etoposide, hydroxynonenal-GS, hyodeoxycholate-G, indinavir, irinotecan, lopinavir, melphalan-GS, methotrexate, mitoxantrone, nelfinavir, olmesartan, ritonavir, saquinavir, SN-38-G (irinotecan metabolite), valsartan, vinblastine, vincristine |
| ABCC4/MRP4 | Bile salts, conjugated steroids, folate, glycocholate, taurocholate, urate | 6-mercaptopurine, 6-thioguanine, acyclovir, adefovir, cefazolin, ceftizoxime, furosemide, hydrochlorothiazide, leucovorin, methotrexate, olmesartan, PAH, para-methoxy-N-ethylamphetamine, ritonavir, tenofovir, topotecan |
| ABCG2/BCRP | Urate | Canertinib, cimetidine, gefitinib, glyburide, imatinib, irinotecan, lamivudine, methotrexate, mitoxantrone, nilotinib, nitrofurantoin, pantoprazole, prazosin, rosuvastatin, sulfasalazine, topotecan |
| SLC carriers | ||
| SLC22A2/OCT2 | Acetylcholine, berberine, bile acids, choline, creatinine, dopamine, epinephrine, guanidine, histamine, norepinephrine, serotonin | Aflatoxin B1, amantadine, amiloride, cimetidine, cisplatin, D-tubocurarine, ethidium bromide, famotidine, ifosfamide, lamivudine, memantine, metformin, oxaliplatin, pancuronium, paraquat, pindolol, propranolol, ranitidine, varenicline, zalcitabine |
| SLC22A4/OCTN1 | Acetylcholine, carnitine | Doxorubicin, entecavir, ergothioneine, gabapentin, imatinib, metformin, mitoxantrone, oxaliplatin, pregabalin, pyrilamine, quinidine, tiotropium, ipratropium, verapamil |
| SLC22A5/OCTN2 | Carnitine | Cephaloridine, emetine, entecavir, etoposide, imatinib, ipratropium, spironolactone, tiotropium, verapamil |
| SLC22A6/OAT1 | Cyclic nucleotides (cAMP, cGMP), folates, indoksyl sulfate, PGE2, PGF2α, uric acid | Adefovir, cephaloridin, ciprofloxacin, methotrexate, pravastatin, zidovudine |
| SLC22A7/OAT2 | cGMP, creatinine, DHEAS, estrogen sulphate, PGE2, uric acid | 5-fluorouracil, acyclovir, bumetadine, diclofenac, entecavir, ganciclovir, irinotecan, PAH, penciclovir, tetracycline, zidovudine |
| SLC22A8/OAT3 | Uric acid, bile acids, conjugated hormones | Adefovir, cefaclor, ceftizoxime, cephaloridine, ciprofloxacin, conjugated sex steroids, methotrexate, NSAIDs, pravastatin, PGE2, zidovudine |
| SLC47A1/MATE1 | Creatinine, estrone-S, guanidine, thiamine | Acyclovir, cephalexin, cephradine, cimetidine, fexofenadine, ganciclovir, metformin, MPP, paraquat, oxaliplatin, TEA, topotecan |
| SLC47A2/MATE2-K | Creatinine, carnitine, estrone-S, thiamine | Acyclovir, cimetidine, ganciclovir, lamivudine, metformin, oxaliplatin, quinidine, topotecan |
| SLCO4A1/OATP4A1 | ADMA, cAMP, chenodeoxycholate, estrone-3-S, glycoycholate, T3, T4 | Digoxin, ouabain, methotrexate |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drozdzik, M.; Drozdzik, M.; Oswald, S. Membrane Carriers and Transporters in Kidney Physiology and Disease. Biomedicines 2021, 9, 426. https://doi.org/10.3390/biomedicines9040426
Drozdzik M, Drozdzik M, Oswald S. Membrane Carriers and Transporters in Kidney Physiology and Disease. Biomedicines. 2021; 9(4):426. https://doi.org/10.3390/biomedicines9040426
Chicago/Turabian StyleDrozdzik, Marek, Maria Drozdzik, and Stefan Oswald. 2021. "Membrane Carriers and Transporters in Kidney Physiology and Disease" Biomedicines 9, no. 4: 426. https://doi.org/10.3390/biomedicines9040426
APA StyleDrozdzik, M., Drozdzik, M., & Oswald, S. (2021). Membrane Carriers and Transporters in Kidney Physiology and Disease. Biomedicines, 9(4), 426. https://doi.org/10.3390/biomedicines9040426